A Case of a Metastatic Pancreatic Neuroendocrine Tumor: A Surgical Conundrum Wrapped in Functionality's Embrace
Adam Mylonakis, Michail Vailas, Panagiotis Sakarellos, Lysandros Karydakis, Eleandros Kyros, Spyridon Davakis, Alexandros Papalampros, Evaggelos Felekouras

TL;DR
A rare case of a non-functioning pancreatic tumor turning into a functioning one is described, showing how surgery can help manage symptoms temporarily.
Contribution
The case highlights a rare transformation of a non-functioning tumor into a functioning one and the potential role of surgery in managing symptoms.
Findings
A non-functioning pNET transformed into a functioning one, causing hypoglycemia and diarrhea.
Surgical intervention temporarily resolved symptoms but disease re-emerged after seven months.
The case underscores the need for vigilance in monitoring non-functioning pNETs for possible transformation.
Abstract
This case study reports a rare case of a non-functioning metastatic pancreatic neuroendocrine tumor (pNET) transforming into a functioning pNET. A 59-year-old male, previously treated with distal pancreatectomy, splenectomy, lymph node dissection, liver metastasectomy, and pharmacotherapy, presented with weakness, hypoglycemia, and daily episodes of watery diarrhea. A functioning neuroendocrine liver metastasis expressing insulin and gastrin was identified. Surgical intervention, including left lateral hepatectomy and microwave ablation of multiple intrahepatic lesions, resulted in symptom resolution and uneventful recovery. However, metastatic liver disease re-emerged seven months post-surgery, necessitating chemotherapy. This case highlights the importance of vigilance for symptom development in non-functioning pNETs, signaling potential disease relapse and phenotype transformation,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
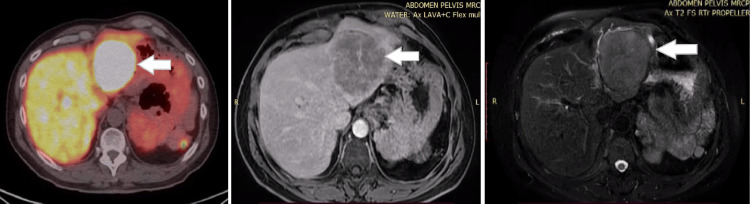 Figure 1
Figure 1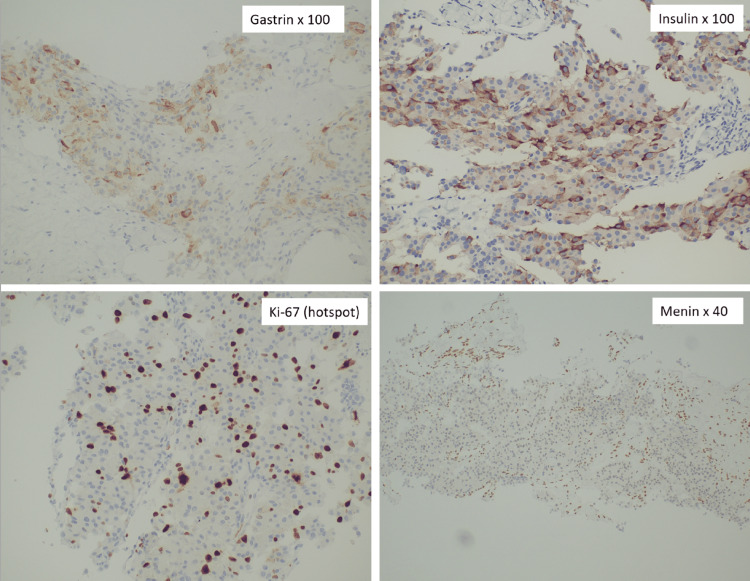 Figure 2
Figure 2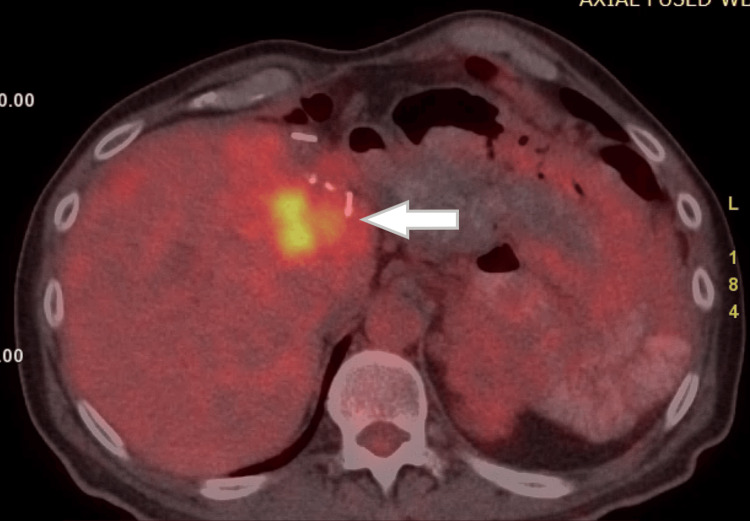 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeuroendocrine Tumor Research Advances · Neuroblastoma Research and Treatments · Pancreatic and Hepatic Oncology Research
Introduction
Pancreatic neuroendocrine tumors (pNETs) are a subgroup of neuroendocrine neoplasms (NEN) originating from endocrine cells of the pancreas. They exhibit unique biological behavior and require clinical approaches that differ significantly from those used for pancreatic adenocarcinoma [1].
Based on the clinical evidence of syndromes caused by excessive tumor hormone secretion, pNETs are divided into two types, namely, functioning and non-functioning. Functioning pNETs, comprising about 35% of pNETs, are further categorized based on the specific hormones they secrete, including insulinomas, gastrinomas, and glucagonomas, among others [2]. Non-functioning pNETs, on the other hand, do not secrete hormones or produce clinical symptoms related to hormone hypersecretion. It is extremely rare, but documented, that non-functioning pNETs can transform into functioning pNETs. However, the mechanism underlying this transformation remains unclear and is a subject of ongoing research.
In this study, we report the case of a 59-year-old male patient who was referred to our Surgical Department for the management of a functioning pNET. This presentation occurred nine years after the surgical resection of a previously diagnosed non-functioning, metastatic pNET. Additionally, we provide a review of our considerations regarding this rare clinical scenario.
Case presentation
A 59-year-old man was referred to our Surgical Department due to daily episodes of watery diarrhea and multiple hypoglycemia episodes leading to syncope, resulting in five hospital admissions over the previous year. His medical history revealed that he had undergone a distal pancreatectomy, splenectomy, lymph node dissection, and liver metastasectomy for a non-functioning pNET nine years prior to the current hospitalization. The tumor was then pathologically staged as pT3N1M1 with a Ki-67 index of 4%. Postoperatively, the patient initially received somatostatin analogs for two years, which were later switched to everolimus due to progressive liver disease. However, everolimus treatment was discontinued due to intolerance, and the patient was subsequently treated with peptide receptor radionuclide therapy (PRRT). He underwent four cycles of PRRT two years after the initial diagnosis, followed by an additional four cycles six years post-diagnosis.
Upon admission, the diagnosis of a relapsed functioning pNET was highly suspected based on laboratory findings. The gastrin level was 41.1150 pg/ml (normal range <110 pg/ml), and chromogranin A (CgA) was markedly elevated at 210 nmol/l (normal range <4 nmol/l). Due to protracted hypoglycemia, the patient was treated with octreotide, diazoxide, and a continuous infusion of dextrose in water (D/W) at 35%. Upper gastrointestinal (GI) endoscopy revealed multiple ulcerations in the gastric and duodenal mucosa. Additionally, a colonoscopy was performed and ruled out other primary sites. Magnetic resonance imaging (MRI) of the abdomen identified a significant 8.1 cm lesion in the left lobe of the liver and multiple smaller lesions in liver segments VI and VII. A 68Ga/64Cu-DOTA-somatostatin analog (SSA) positron emission tomography-computed tomography (PET-CT) scan demonstrated increased uptake in the liver, epiphrenic lymph nodes, and the right thyroid lobe. The diagnostic workup also included an MRI scan of the brain, which revealed the presence of a non-functioning pituitary adenoma (Figure 1).
Axial PET-CT and MRI images of the abdomenThe white arrow demonstrates a left lobe liver lesion.PET-CT: positron emission tomography-computed tomography; MRI: magnetic resonance imaging
Following a multidisciplinary team review, a percutaneous liver biopsy of the left lobe of the liver was performed under ultrasound guidance. The histological analysis identified a neuroendocrine tumor while immunohistochemical staining demonstrated strong-diffuse insulin expression and moderate-diffuse gastrin expression. The Ki-67 proliferation index was 31%, leading to the classification of the tumor as a Grade 3 neuroendocrine tumor (NET G3) while menin was not expressed in the tumor cells (Figure 2).
Immunohistochemical staining of liver biopsy for gastrin (x100), insulin (x100), Ki-67 (hotspot x100) and menin expression (x40)
Due to the persistence of symptoms indicative of a functioning pNET and resistance to medical therapy, the patient was referred for surgical intervention. No intervention was planned for the epiphrenic lymph nodes or the thyroid lesion identified through imaging, with the surgical efforts concentrated on areas most significantly contributing to the patient's symptomatic hypoglycemia and diarrhea. He underwent a left lateral hepatectomy with intraoperative ultrasound guidance, resection of a lesion in segment V, and microwave ablation of multiple remaining intrahepatic lesions. Intraoperatively, the patient’s glucose levels normalized, eliminating the need for intravenous infusion of dextrose-containing fluids. Postoperatively, the patient experienced cessation of diarrhea and both gastrin and glucose levels normalized. He made an uneventful recovery and was discharged on the eleventh postoperative day with prescriptions for a proton pump inhibitor and somatostatin analog therapy.
Pathological examination of the resected liver specimens revealed metastases from well-differentiated pNET with two distinct histological patterns. The lesion from segment V showed a Ki-67 proliferation index of 3%, classifying it as a Grade 2 neuroendocrine tumor (NET G2). The tumor from the left liver lobe exhibited a Ki-67 index of 25%, classifying it as a Grade 3 neuroendocrine tumor (NET G3).
The patient was scheduled for a series of follow-up PET-CT scans. Seven months after the hepatectomy, a new hepatic disease was detected (Figure 3). Subsequently, the patient was treated with a combination therapy of the CAPTEM regimen (capecitabine 750 mg/m^2^ per os twice daily on days 1-14, temozolomide 200 mg/m^2^ per os daily on days 10-14, repeated every 28 days), and somatostatin analogs (SSAs). As of five years post-hepatectomy, the patient remains alive with stable hepatic disease and no evidence of a functioning neuroendocrine syndrome.
Axial PET-CT image of the abdomenThe white arrow demonstrates liver disease recurrence.PET-CT: positron emission tomography-computed tomography
Discussion
NETs are epithelial neoplasms with predominant neuroendocrine differentiation. Their recognition has increased due to advancements in diagnostic technologies and heightened awareness of this rare entity [3]. Specifically, pancreatic NETs (pNETs) have a clinical detection rate of approximately 1 in 100,000 people, accounting for 1-2% of pancreatic neoplasms [4]. pNETs are classified into two types, functioning and non-functioning, based on the presence or absence of clinical syndromes resulting from excessive hormone secretion by the tumor cells.
While rare, there have been documented cases where non-functioning pNETs transform into functioning tumors [5-8]. This phenotypic change is often associated with liver metastasis. The mechanisms behind this transformation are not fully understood, but hypotheses include the influence of the liver tissue microenvironment, drug-induced changes [5], or the existence of a pluripotent progenitor cell [9].
The primary curative treatment for pNETs, when feasible, is surgical resection. At diagnosis, 40-80% of patients present with metastatic disease, predominantly in the liver (40-93%), followed by bone (12-20%) and lung involvement (8-10%) [10]. Treatment options for metastatic disease are broadly categorized into surgical resection, liver-directed therapies, radionuclide therapy, and medical therapy.
In patients with metastatic disease, surgical intervention is often pursued to relieve symptoms caused by excessive hormone secretion and to improve quality of life [11]. Tumor debulking has also been shown to potentially enhance overall survival in selected cases [12]. Liver-directed therapies include techniques such as ablation, transarterial chemoembolization, or radioembolization [13]. Radionuclide therapy involves the use of radiolabeled somatostatin analogs to deliver targeted radiation to pNETs expressing somatostatin receptors, offering a high therapeutic index [14]. Finally, medical therapy, which may include targeted therapy or chemotherapy in combination with somatostatin analogs, remains a viable option for patients with metastatic NETs [15].
In this report, we discuss a patient with a metastatic non-functional pNET. Initial treatment involved surgery (distal pancreatectomy, splenectomy, lymph node dissection, liver metastasectomy) and medical treatment escalating from a somatostatin analog to everolimus, then to peptide receptor radionuclide therapy (PRRT). Nine years later, the tumor became symptomatic and functional, causing severe hypoglycemia and diarrhea. A left lateral hepatectomy, metastasectomy in liver segment V, and microwave ablation were performed, leading to symptom remission. Seven months post-surgery, a PET-CT scan revealed recurrent liver disease, treated with capecitabine and temozolomide. Despite the recurrence of hepatic disease, the patient displayed no evidence of clinical syndromes related to the neuroendocrine tumor post-resection.
The distinctive aspect of this case lies in the transformation of a metastatic non-functioning pancreatic neuroendocrine tumor (pNET) into a functioning tumor, occurring nine years post the initial diagnosis. Such cases have been rarely documented and are often associated with new or concurrent distant metastases. Notably, this case represents the fifth case, to our current knowledge, where a patient has undergone liver metastasectomy or ablation as a successful therapeutic measure to alleviate symptoms associated with this condition [6-8,16].
Another remarkable trait of this case is the simultaneous expression of two hormones by the tumor, leading to two distinct clinical syndromes: severe hypoglycemia and gastrin-related symptoms. Notably, both syndromes regressed following the surgical resection of the liver metastases. This dual hormone expression and the subsequent effective surgical resolution of the resulting clinical syndromes add further uniqueness to this already rare clinical presentation and underscore the importance of continual monitoring and adaptability in treatment strategies for these patients.
Conclusions
The delayed transformation of a non-functioning metastatic pNET into a functioning pNET represents an exceedingly rare clinical entity. The emergence of symptoms in patients with previously non-functioning pNETs should prompt a clinical suspicion of disease relapse and potential phenotypic transformation. Based on the findings from this case, we propose that surgical treatment could be a viable consideration in similar scenarios, considering the patient's clinical profile, the severity of symptoms, and the expertise of the treating physician.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pancreatic neuroendocrine tumors: a review of serum biomarkers, staging, and management World J Gastroenterol Ma ZY Gong YF Zhuang HK 230523222620203247679510.3748/wjg.v 26.i 19.2305 PMC 7243647 · doi ↗ · pubmed ↗
- 2One hundred years after "carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States J Clin Oncol Yao JC Hassan M Phan A 306330722620081856589410.1200/JCO.2007.15.4377 · doi ↗ · pubmed ↗
- 3Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States JAMA Oncol Dasari A Shen C Halperin D 13351342320172844866510.1001/jamaoncol.2017.0589 PMC 5824320 · doi ↗ · pubmed ↗
- 4Endocrine tumours of the pancreas Best Pract Res Clin Gastroenterol Oberg K Eriksson B 7537811920051625389910.1016/j.bpg.2005.06.002 · doi ↗ · pubmed ↗
- 5Transformation of nonfunctioning pancreatic neuroendocrine carcinoma cells into insulin producing cells after treatment with sunitinib Endocrinol Metab (Seoul) Ohn JH Kim YG Lee SH Jung HS 1491522820132439667010.3803/En M.2013.28.2.149PMC 3811707 · doi ↗ · pubmed ↗
- 6Non-functioning neuroendocrine pancreatic tumors transforming to malignant insulinomas - four cases and review of the literature Neuro Endocrinol Lett Juhlin CC Skoglund S Juntti-Berggren L Karlberg M Calissendorff J 175183402019 https://pubmed.ncbi.nlm.nih.gov/32087093/32087093 · pubmed ↗
- 7Metastatic insulinoma presenting 14 years after benign tumour resection: a rare case and management dilemma Endocrinol Diabetes Metab Case Rep Keen F Iqbal F Owen P Christian A Kumar N Kalhan A 2020202010.1530/EDM-20-0065 PMC 757666233434180 · doi ↗ · pubmed ↗
- 8Transformation of a non-secretory neuroendocrine tumor to insulinoma after treatment with Sunitinib: a case report and review of the literature J Oncol Pharm Pract Clover T Abdelkader A Guru Murthy GS 151615192520193008943210.1177/1078155218791309 · doi ↗ · pubmed ↗