Macular hypoplasia and high myopia in 48, xxyy syndrome: a unique case of 48, xxyy syndrome that presents with high myopia and macular dysplasia
Aohan Hou, Xinyu Liu, Limei Sun, Xiaoyan Ding

TL;DR
A rare case of 48, XXYY syndrome is reported with high myopia and severe macular dysplasia, a previously unrecorded combination.
Contribution
This is the first documented case of macular dysplasia in a patient with 48, XXYY syndrome.
Findings
A 6-year-old boy with 48, XXYY syndrome exhibited high myopia and grade 3 macular hypoplasia.
The case expands the known phenotypic spectrum of 48, XXYY syndrome to include ocular abnormalities.
Karyotype and whole-exome sequencing confirmed the diagnosis of 48, XXYY syndrome.
Abstract
Among sex chromosome aneuploidies, 48, XXYY syndrome is a rare variant. This condition is marked by the existence of an additional X and Y chromosome in males, leading to a diverse range of physical, neurocognitive, behavioral, and psychological manifestations. Typical characteristics include a tall stature and infertility. Other phenotypes include congenital heart defects, skeletal anomalies, tremors, obesity, as well as the potential for type 2 diabetes and/or peripheral vascular disease. A 6-year-old boy, who had been experiencing progressive vision deterioration in both eyes for the past two years, presented with a history of poor vision, delayed motor skills. The patient was diagnosed with micropenis in the pediatric outpatient clinic. Sparse hair, an unusually tall stature and craniofacial dysmorphology characterized by ocular hypertelorism, depressed nasal bridge, and epicanthic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
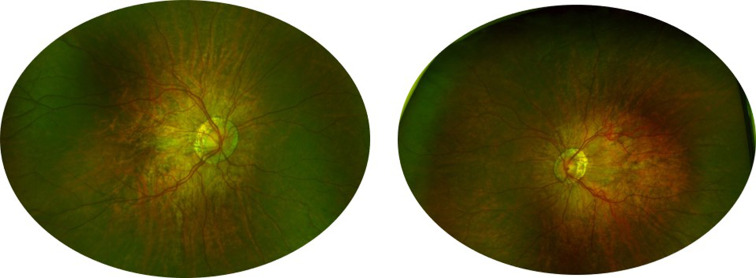 Figure 1
Figure 1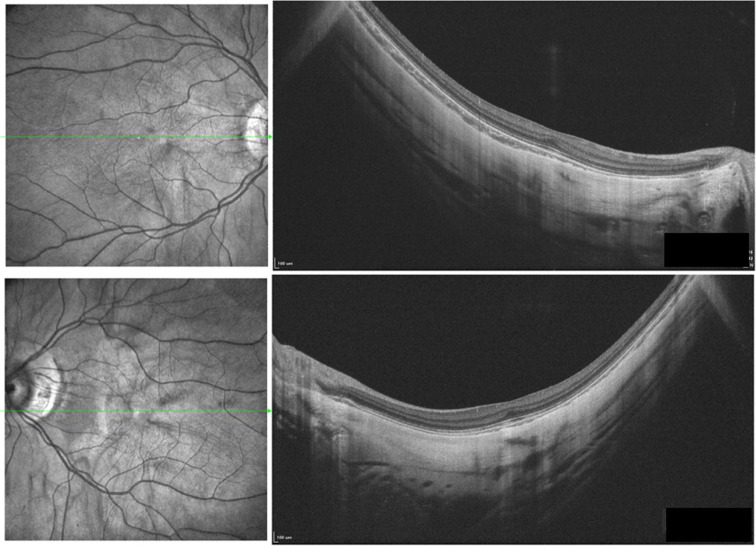 Figure 2
Figure 2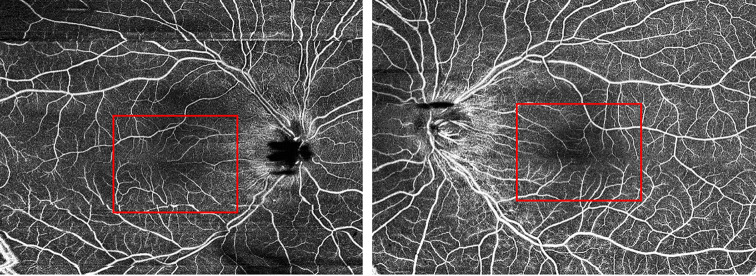 Figure 3
Figure 3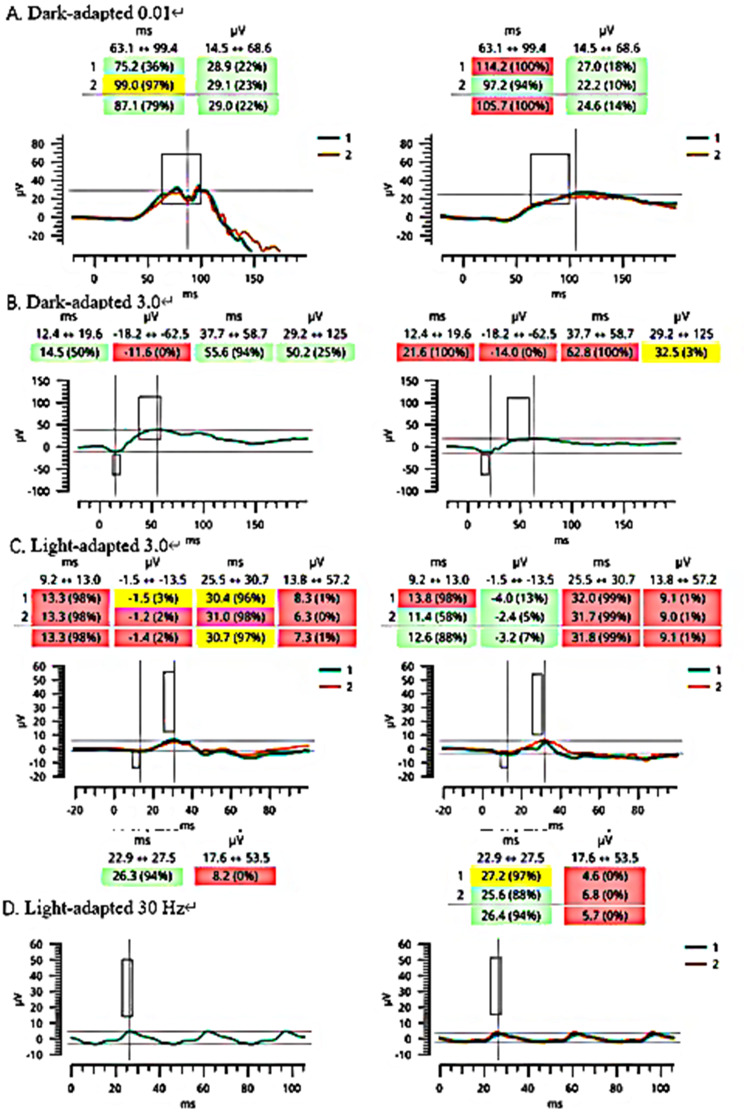 Figure 4
Figure 4- —Construction Project of High-Level Hospitals in Guangdong Province
- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
- —http://dx.doi.org/10.13039/501100021171Basic and Applied Basic Research Foundation of Guangdong Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · Genomic variations and chromosomal abnormalities · Chromosomal and Genetic Variations
Introduction
Among sex chromosome aneuploidies, 48, XXYY syndrome is a rare variant. This condition is marked by the existence of an additional X and Y chromosome in males, leading to a diverse range of physical, neurocognitive, behavioral, and psychological manifestations. Typical characteristics include a tall stature and infertility. Other phenotypes include congenital heart defects, skeletal anomalies, tremors, obesity, as well as the potential for type 2 diabetes and/or peripheral vascular disease [1]. Notably, the syndrome’s phenotypic presentation does not typically include ocular abnormalities. In this report, however, we introduce an exceptional case of 48, XXYY syndrome that presents with high myopia and macular dysplasia.
Case report
A 6-year-old boy suffered from progressive deterioration of vision bilaterally during the previous 2 years. His past ophthalmic history included poor vision since early childhood and motor development delays (holding head up at 6–7 months old, walking at 1.5 years old). The patient was diagnosed with micropenis in the pediatric outpatient clinic. Both of his parents were emmetropic without any visual or systemic medical disorders. No other family history was noted. Whole Exome Sequencing was conducted on the patient and their parents, with chromosomal karyotype analysis being exclusively performed on the patient. No significant abnormalities were observed in the parents’ results.
Physical examination revealed tall stature (127 cm) compared to the normal value of 117.4 ± 5.0 for 6-year-old boys in Han [3]. The patient is present with craniofacial dysmorphology characterized by ocular hypertelorism, depressed nasal bridge, and epicanthic folds. Sparse hair was observed in the occipital region. Cycloplegic refraction revealed Bilateral high myopia and astigmatism (RE: -9.75DS/-3.50DC177°, LE: -12.50DS/-3.50DC12°). Best corrected visual acuity (BCVA) was LogMAR 0.4 (Snellen 20/50) in the right eye and LogMAR 0.54 (Snellen 20/70) in the left, respectively. Anterior segment was unremarkable. Vitreous Liquification and tessellated fundus were noted. The optic disc was surrounded by a ring of chorioretinal atrophy. In addition, multiple thin emanating vessels and posterior staphyloma centered with the optic disc were observed. (Fig.1).
Grade 3 macular hypoplasia, in detail, absence of foveal pit, extrusion of the inner retina layer and lengthening of the outer segment, was noted (Fig.2) [12]. Choroidal atrophy and staphyloma were noted in both eyes. Subfoveal choroidal thickness was 40 μm in the right eye and 38 μm in the left eye. Optical coherence tomography angiography (OCTA) showed increased vessel density with reduction of the foveal avascular zone (Fig.3). Axial lengths were 28.88 mm (OD) and 29.59 mm (OS). Significant reduced amplitudes were observed on light-adapted electroretinography (ERG) and light-adapted 30 Hz ERG bilaterally (Fig.4).
Karyotype analysis were performed in accordance with the ISCN 2016, revealed an abnormal karyotype with two X chromosomes and two Y chromosomes in all cell lines consistent with 48, XXYY syndrome.
The ocular phenotype and systemic presentations were not consistent with other causes of foveal hypoplasia and high myopia, such as optic nerve hypoplasia, familial exudative vitreoretinopathy, stickler syndrome, retinopathy of prematurity, and albinism.
Discussion
48, XXYY syndrome, as a rare sex chromosome disorder, was first described in 1964. It is estimated to occur in 1:18,000–1:40,000 male births. It presents with a wide variety of physical, psychosocial, and neurocognitive findings. Most of the literature currently focuses on the physiological and psychological disorders of the 48, XXYY phenotype. The wide spectrum of symptom variability is considered to be related to sex chromosome dosage and skewed X-inactivation, but further investigation is still needed to understand these underlying genetic variations and their connection to clinical signs and symptoms [1].
There are few reports of ocular abnormalities in association with 48, XXYY syndrome with only three known cases to date that detail ocular complications, including strabismus, Duane’s syndrome, and retinal dysfunction [5, 11, 13]. Only a single case has been reported wherein high myopia was associated with 48, XXYY syndrome [5]. In that case, bilateral poor visual acuity (BCVA LogMAR 1.0, Snellen 20/200) was found, which was align with the findings from our patient. Notably, although our patient did not exhibit the bilateral extinguished ERG response documented in the previous report [4, 7]. there was a significantly reduced amplitude in light-adapted and flicker ERG.
In this study, we present the case to describe macular dysplasia in a patient with 48, XXYY syndrome. The absence of the foveal pit, protrusion of the inner retinal layer, and elongation of the outer segment were noted on optical coherence tomography with increased vessel density with reduction of the foveal avascular zone on OCTA. Chen etc. reported a case of macular dysplasia with 48, XXYY syndrome. In their study, the degree of macular hypoplasia in patients was less severe than in our case, with better visual acuity, and there was no description of the refractive status [2]. The severity of macular dysfunction and poor BCVA was correlated with the degree of macular dysplasia and high myopia observed. Posterior staphyloma and pronounced choroidal atrophy were also noted. Although both conditions could occur as secondary changes in high myopia patients, such alterations usually require extensive periods to manifest. Given these findings, we propose that fundus changes, notably macular hypoplasia, may represent a hitherto feature of 48, XXYY syndrome [6], Table 1).
In 2010, Ottesen et al. examined the influence of sex chromosome genes and the gene dosage effect, potentially accounting for the increased stature [9]. A similar mechanism may apply to the genes linked to myopia, such as MYP1, ARR3, which are located on the X chromosome, suggesting a parallel between the genetic determinants of height and myopia [8, 10, 14].
In summary, we have documented the first reported macular dysplasia in a patient with 48, XXYY syndrome. Our report further expands the clinical spectrum of this disease. The patient’s decreased visual acuity could be attributed to the combined effects of high myopia and macular dysplasia. We postulate that these macular changes, particularly macular hypoplasia, could represent a previously unidentified aspect of 48, XXYY syndrome.
Fig. 1. Fundoscopy showed tessellates fundus, peripapillary and posterior staphyloma.
Fig. 2OCT showed grade 3 macular hypopiasia, extreme choroidal atrophy and staphyloma formation bilaterally.
Fig. 3. Optical coherence tomography (OCTA) showed increased vessel density with reduction of the foveal avascular zone bilaterally.
Fig. 4. Dark-adapted 0.01 ERG and Dark-adapted 3.0 ERG ahowed allghtiy peak time delay on the right eye (A-B). Light-adapted 3.0 ERG and Light-adapted 30 Hz ERG showed algnificantli reduced amplltude bllaterally, while peak time delay on the right eye (C-D).
Table 1. Ocular clinical characteristics of 48, XXYY syndrome in previous studiesPatientOcular findingReference12y,MaleDuane syndrome and mild myopiaWeis et al., 20118m,MaleAlternating exotropia, foveal hypoplasia and reduction of the foveal avascular zoneChen et al., 202328y,MaleLightly pigmented retinal pigment epithelium and choroid, peripapillary atrophy, pattern visual evoked potentials and pattern electroretinogram were undetectable bilaterally and high myopiaKarampelas et al., 2013
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Blumling AA Martyn K Talboy A Close S Rare sex chromosome variation 48,XXYY: an integrative review Am J Med Genet C 2020184238640310.1002/ajmg.c.3178932501621 · doi ↗ · pubmed ↗
- 2Chen C, Guo S, Huang Z, Fu T, Jiang L, Chen FK. Foveal hypoplasia in a Chinese adolescent with 48, XXYY syndrome. Ophthalmic Genet. 2023;1–4. 10.1080/13816810.2023.2291669.10.1080/13816810.2023.229166938087495 · doi ↗ · pubmed ↗
- 3Coordinating Study Group of Nine Cities on Physical Growth and Development of Children & Capital Institute of Pediatrics. [A national survey on growth of children under 7 years of age in nine cities of China, 2005]. Zhonghua Er Ke Za Zhi = Chin J Pediatr. 2007;45(8):609–14.18021536 · pubmed ↗
- 4Flitcroft DI Adams GGW Robson AG Holder GE Retinal dysfunction and refractive errors: an electrophysiological study of children Br J Ophthalmol 2005894484810.1136/bjo.2004.04532815774929 PMC 1772604 · doi ↗ · pubmed ↗
- 5Karampelas M Gardner J Holder G Hardcastle A Webster A Retinal dysfunction and high myopia in association with 48,XXYY syndrome Doc Ophthalmol Adv Ophthalmol 20131273245710.1007/s 10633-013-9406-x 24048723 · doi ↗ · pubmed ↗
- 6Kobayashi K Ohno-Matsui K Kojima A Shimada N Yasuzumi K Yoshida T Futagami S Tokoro T Mochizuki M Fundus characteristics of high myopia in children Jpn J Ophthalmol 20054943061110.1007/s 10384-004-0204-616075331 · doi ↗ · pubmed ↗
- 7Mc Culloch DL Kondo M Hamilton R Lachapelle P Messias AMV Robson AG Ueno SISCEV extended protocol for the stimulus-response series for light-adapted full-field ERG Doc Ophthalmol Adv Ophthalmol 201913832051510.1007/s 10633-019-09685-830929108 · doi ↗ · pubmed ↗
- 8Metlapally R Michaelides M Bulusu A Li Y-J Schwartz M Rosenberg T Hunt DM Moore ATZüchner S Rickman CB Young TL Evaluation of the X-linked high-grade myopia locus (MYP 1) with cone dysfunction and color vision deficiencies Investig Ophthalmol Vis Sci 20095041552810.1167/iovs.08-245519098318 PMC 3934550 · doi ↗ · pubmed ↗