A Residual N-Terminal Peptide Enhances Signaling of Depalmitoylated Hedgehog to the Patched Receptor
Sophia F. Ehlers, Dominique Manikowski, Georg Steffes, Kristina Ehring, Fabian Gude, Kay Grobe

TL;DR
This study shows that a specific N-terminal peptide is needed for the signaling of a modified form of the Sonic hedgehog protein to its receptor.
Contribution
The study reveals that a residual N-terminal peptide compensates for the loss of N-palmitate in Hedgehog signaling.
Findings
ShhC, a cholesterol-modified variant of Sonic hedgehog, remains bioactive without N-palmitate.
Removing N-terminal peptides longer than eight amino acids inactivates depalmitoylated ShhC.
The findings suggest a new mechanism for Hedgehog signaling involving the N-terminal peptide.
Abstract
During their biosynthesis, Sonic hedgehog (Shh) morphogens are covalently modified by cholesterol at the C-terminus and palmitate at the N-terminus. Although both lipids initially anchor Shh to the plasma membrane of producing cells, it later translocates to the extracellular compartment to direct developmental fates in cells expressing the Patched (Ptch) receptor. Possible release mechanisms for dually lipidated Hh/Shh into the extracellular compartment are currently under intense debate. In this paper, we describe the serum-dependent conversion of the dually lipidated cellular precursor into a soluble cholesteroylated variant (ShhC) during its release. Although ShhC is formed in a Dispatched- and Scube2-dependent manner, suggesting the physiological relevance of the protein, the depalmitoylation of ShhC during release is inconsistent with the previously postulated function of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
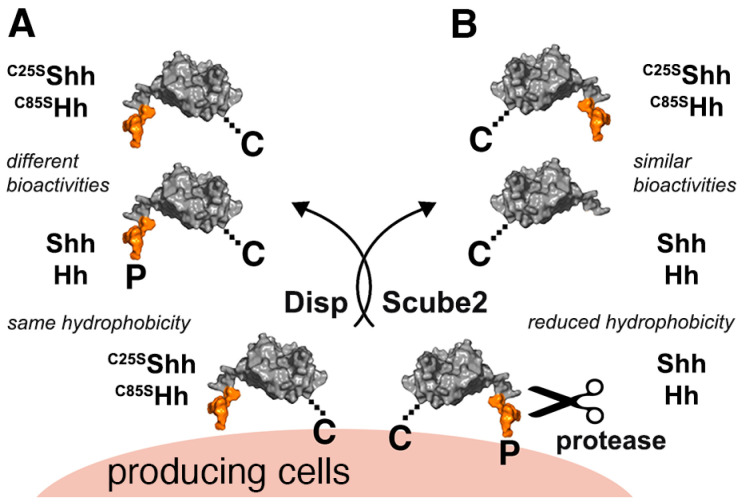 Figure 1
Figure 1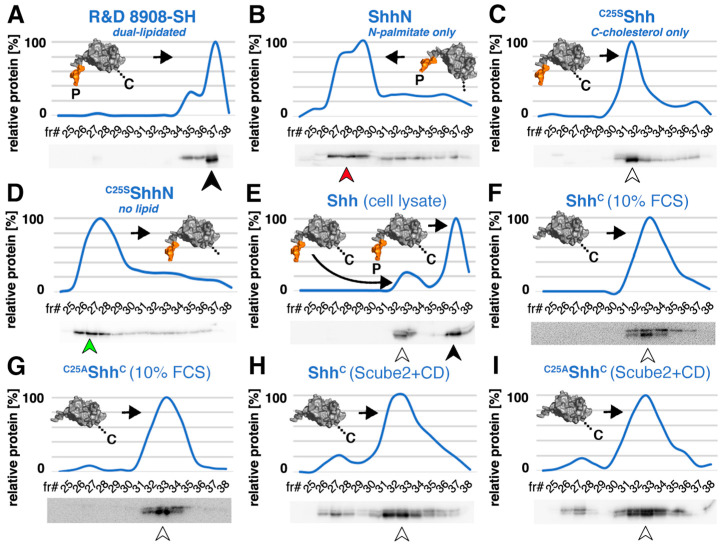 Figure 2
Figure 2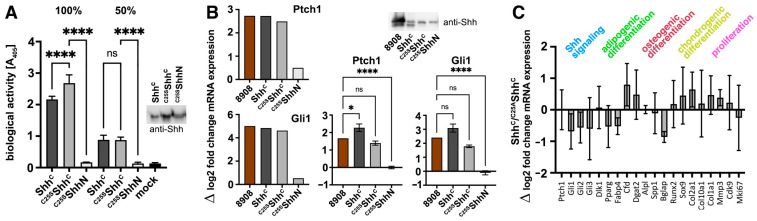 Figure 3
Figure 3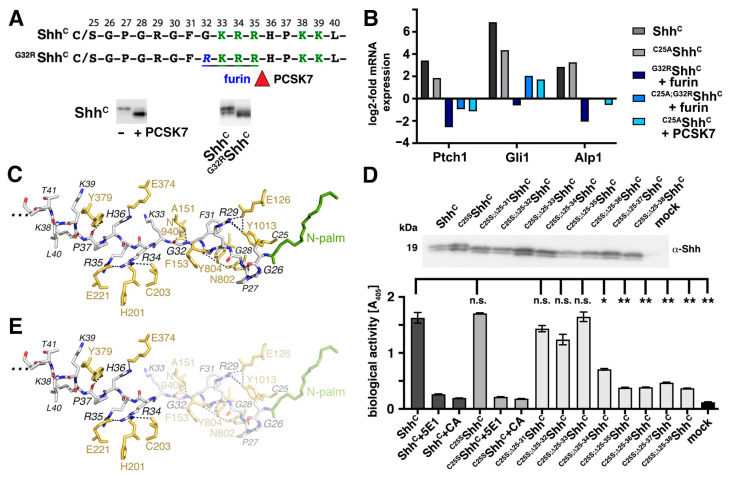 Figure 4
Figure 4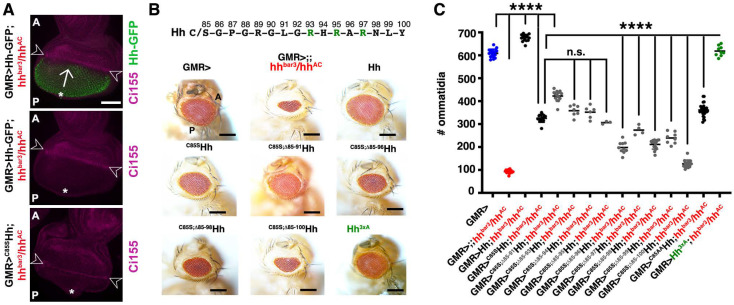 Figure 5
Figure 5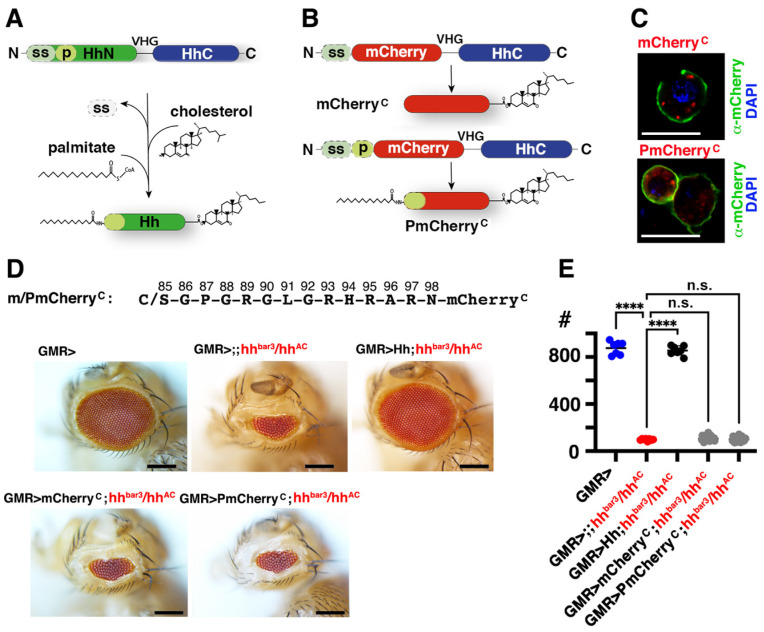 Figure 6
Figure 6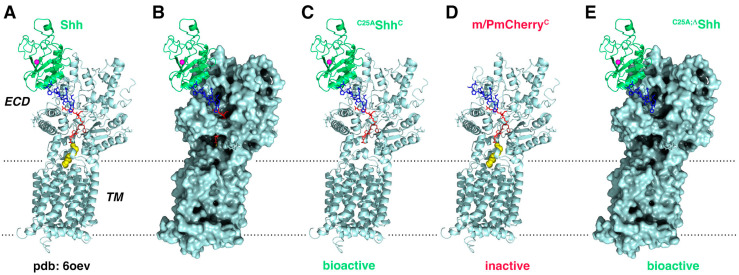 Figure 7
Figure 7- —DFG (German Research Council)
- —Medical Faculty of the University of Münster
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHedgehog Signaling Pathway Studies · Ocular Disorders and Treatments · Developmental Biology and Gene Regulation
1. Introduction
The Hedgehog (Hh) family of morphogens is essential for animal development and plays a major role in stem cell maintenance and adult tissue regeneration [1,2,3,4,5]. Known activities of the vertebrate Hh ortholog Sonic hedgehog (Shh) include patterning of the embryonic ventral neural tube and posterior limb bud, the somites, brain, foregut, lung, and face (reviewed in [6]). Paracrine signaling in Shh-expressing cancers is also well established [7]. In Drosophila, Hh acts as a signaling molecule during embryonic development and later in the imaginal eye discs, which can therefore be used as a robust and reliable readout of Hh biofunction. Hhs also control many other important processes in development and in cancer, including cell proliferation and migration. The underlying mechanisms that allow such functional versatility are poorly understood, but are likely to involve a highly conserved set of unusual post-translational modifications during Hh biosynthesis in the ER and in the Golgi apparatus. The first unusual modification of all vertebrate and invertebrate Hh family members begins with the autocatalytic cleavage of a 45 kDa precursor molecule by its C-terminal cholesterol transferase domain [8]. This reaction yields 19 kDa cholesteroylated vertebrate Shh and Drosophila Hh. Next, a membrane-bound acyltransferase (called Hh acyltransferase (Hhat) in vertebrates or Skinny Hedgehog (Ski) in invertebrates) adds a palmitate to conserved N-terminal cysteines (C85 in fly Hh or C25 in mouse Shh) [9,10,11]. A well-established role for both lipids is to attach nascent vertebrate and invertebrate Hhs to the outer plasma membrane leaflet of producing cells (Figure 1). However, it is less clear whether the well-established release factors Dispatched (Disp, a 12-pass transmembrane protein [12,13]) and Scube2 (signal peptide, cubulin domain, epidermal growth factor-like protein 2, a soluble glycoprotein expressed only in vertebrates [14,15]) remove Hhs together with both lipid anchors from the plasma membrane (Figure 1A), or whether Hh release requires proteolytic processing of the lipidated terminal peptides to render the Hh/Shh signaling domain soluble (Figure 1B). To address this question, previous studies have constructed, expressed, and analyzed artificial Hh proteins lacking one or both terminal lipids. These studies have shown that the replacement of the N-terminal cysteine acceptor with serine or alanine (C25→A/S in ^C25A/S^Shh, C85→A/S in Drosophila ^C85A/S^Hh) specifically blocks palmitate addition and impairs Hh biofunction in vitro and in vivo [9,10,16,17,18,19,20], although to varying degrees [17,18,20]. The current interpretation of these published results is that the retained N-terminal Hh lipid directly contributes to maximal Hh/Shh signaling to the Ptch receptor and that the solubilized protein always remains dually lipidated [21,22,23,24].
However, another possibility to explain the impaired biofunction of ^C25A/S^Shh and ^C85A/S^Hh is that the lack of N-palmitoylation during biosynthesis loosens Hh tethering to the plasma membrane of producing cells and thereby perturbs Disp- and Scube2-mediated Hh solubilization in an unpredictable manner. Conceivably, the lack of spatiotemporal control of Hh solubilization could also lead to variably impaired Hh biofunction, as observed [9,10,16,17,18,19,20]. In this study, we analyzed Shh solubilization using cell culture conditions that mimic physiological conditions in fly hemolymph or vertebrate interstitial fluid. To this end, we expressed dually lipidated Shh in the presence of 10% serum. Using reverse-phase (RP) high-performance liquid chromatography (HPLC) to compare proteins based on their hydrophobicity, we identified high levels of soluble Shh variants that retained their C-cholesteroylated peptide but lacked their N-terminal palmitoylated peptides as a consequence of proteolytic processing at the plasma membrane [25]. Throughout this study, we refer to this monolipidated serum-released Hh variant as Hh^C^/Shh^C^. Importantly, despite the observed N-processing and depalmitoylation during its release, Shh^C^ induced Shh target gene transcription and differentiation in Hh reporter cells. Consistent with this result, engineered, unpalmitoylated ^C25S^Shh^C^ induced similar transcriptional and differentiation profiles when compared to Shh^C^ in vitro. Both results demonstrate that the N-terminal lipid is not essential for Ptch receptor activation in the presence of serum and that the dual-lipidated cellular Hh/Shh precursor may undergo different post-translational modifications upon release to enhance the versatility of Hh signaling. We also observed that the additional deletion of N-terminal amino acids from the unpalmitoylated protein abolished most of its biofunction in vitro and in vivo. This suggests that Hh^C^/Shh^C^ biofunction requires a short retained N-peptide, limiting proteolytic processing of the Shh N-terminus to sites most proximal to the plasma membrane.
2. Materials and Methods
2.1. Fly Lines
Ectopic expression of Hh variants or mCherry fusion proteins in the morphogenetic furrow of the eye disc was performed by crossing the following fly lines: UAS-Hh variant/Cherry*/CyO^WeeP^;hh^AC^/Tm6B* and GMR-GAL4/GMR-Gal4;hh^bar3^/Tm6B. The resulting UAS-Hh variant/Cherry*/GMR-GAL4;hh^bar3^/hh^AC^* flies were analyzed using a Nikon SMZ25 microscope (Nikon, Tokyo, Japan). w^−^;+/+;hh^bar3^/hh^AC^ flies served as negative controls; GMR-GAL4/+ flies served as positive controls. Hh constructs were inserted into the 51C1 landing site and mCherryC constructs into the 68E landing site (BestGene, Chino Hills, CA, USA) by using germline-specific PhiC31 integrase [26]. mCherryC on the third chromosome was first recombined with hh^AC^ before being crossed with GMR-Gal4;hh^bar3^/Tm6B to obtain GMR-Gal4;hh^bar3^/UAS-mCherryC hh^AC^ flies. Flies were crossed at 18 °C to achieve moderate protein expression, following an established protocol [27].
2.2. Generation of Recombinant Proteins
Shh expression constructs were generated from murine cDNA (NM_009170: nucleotides 1–1314, corresponding to amino acids 1–438 and human Hhat cDNA (NM_018194). Both cDNAs were cloned into pIRES (Clontech) for their coupled expression from bicistronic mRNA to achieve near-quantitative Shh palmitoylation. Non-cholesteroylated ShhN (nucleotides 1–594, corresponding to amino acids 1-198) and non-palmitoylated ^C25A/S^Shh cDNA (amino acids 1–438) were generated by site-directed mutagenesis (Stratagene) and inserted into pcDNA3.1 (Invitrogen, Carlsbad, CA, USA). We also expressed N-truncated Shh versions and amino acid exchange versions as indicated. Co-expressed human Scube2 constructs were a kind gift from Ruey-Bing Yang (Academia Sinica, Teipei, Taiwan), furin, PACE4, and PCSK5/7 expressing cDNA were a kind gift from Nabil Seidah (Montreal Clinical Research Institute, Montréal, QC, Canada). Hh cDNA (nucleotides 1–1416, corresponding to amino acids 1–471 of D. melanogaster Hh) and HhN cDNA (nucleotides 1–771, corresponding to amino acids 1–257) were used. Two variants of the mCherry construct were inserted into the pUAST vector background and injected. Upstream of mCherry, the Drosophila Hh export signal sequence (codons 1 to 84) was followed by the Drosophila Hh extended N-terminal peptide with a functional palmitoylation acceptor cysteine (p, codons 85 to 98). C-terminal to the mCherry fluorophore, the three most C-terminal amino acids of the Hh signalling domain (amino acids VHG) were followed by the Drosophila Hh C-terminal domain (HhC), which is required for covalent cholesteroylation of the VHG glycine. HhC extended from codon 258 to 472, with codon 472 being the Hh stop codon. The resulting construct was named PmCherryC. The mCherry control lacked the Hh N-terminus. The constructs were inserted into the Drosophila 3L 68E1 landing site by BestGene (Chino Hills, CA, USA).
2.3. Protein Expression
Bosc23 cells (a HEK293 derivative (Research Resource Identifier (RRID): CVCL_4401) obtained from D. Robbins, University of Miami, Coral Gables, FL, USA) were seeded into six-well plates and transfected with 1 µg Shh constructs, with or without 0.5 µg Scube2 constructs, by using Polyfect (Qiagen, Hilden, Germany). Cells were grown for 2 days at 37 °C with 5% CO_2_ in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS) and penicillin–streptomycin (100 µg/mL). Shh-conditioned media (referred to as +10% FCS throughout this study) was aliquoted, shock frozen in liquid N_2_, and stored at −80 °C for subsequent use. To produce the media called -FCS in this work, cells were washed and serum-free DMEM added for 6 h, the DMEM harvested, centrifuged at 300× g for 10 min to remove debris, the batch aliquoted, shock frozen in liquid nitrogen, and stored at −80 °C. In some experiments, serum-free medium was supplemented with 600 µg/mL methyl-β-cyclodextrin (CD, a cyclic oligosaccharide with a hydrophobic core to accommodate cholesterol and extract the sterol from the plasma membrane) for 6 h before harvesting. For Western blot analysis and protein quantification, frozen aliquots were thawed on ice and samples without FCS were immediately incubated with 10% trichloroacetic acid (TCA) for 30 min on ice, followed by centrifugation at 13,000× g for 20 min to precipitate the proteins. For protein pulldown (PD) from serum-containing media, 40 μL of heparin–sepharose beads were added to Shh-containing media (500 μL media per PD) and incubated overnight on a rotator at 4 °C. All precipitates and pulldowns were analyzed by reducing SDS-PAGE and immunoblotting by using goat-α-Shh antibodies (AF464, R&D Systems, Minneapolis, MN, USA), followed by incubation with horseradish-peroxidase-conjugated secondary antibodies and chemiluminesce detection (Pierce, Carlsbad, CA, USA). Where indicated, known amounts of dual-lipidated, HEK293-derived human Shh (R&D Systems, 8908-SH) were used as a size control and to quantify Bosc23-expressed, TCA-precipitated proteins on the same blot. Proteins were quantified on Western blots using ImageJ (NIH, Bethesda, MD, USA) and the values obtained were used to adjust the flash-frozen protein amounts for subsequent assays, such as C3H10T1/2 differentiation, to ensure that similar amounts of proteins from the frozen batches were used in all subsequent assays. C3H10T1/2 cells (RRID: CVCL_0190) were obtained from ATCC (CCL-226). For 5E1 immunoprecipitation and quantification of ^C25S^Shh^C^ and its N-truncated variants, 10 µg of monoclonal 5E1 antibody (DSHB) was coupled to 2.5 mg Protein A–sepharose beads (Sigma, St. Louis, MO, USA) per immunoprecipitation (IP). Five hundred microlitres of medium per IP was incubated overnight on a rotator. S2 cells (RRID: CVCL_Z232) were cultured in Schneider’s medium (Invitrogen) supplemented with 10% fetal calf serum and 100 μg/mL penicillin–streptomycin. The cells were obtained from C. Klämbt, University of Münster, Münster, Germany. For visualization of mCherry on the surface of S2 cells (CVCL_Z232, cells were obtained from C. Klämbt, University of Münster, Germany), S2 cells were transfected with constructs encoding mCherry^C^ or PmCherry^C^ together with a vector encoding actin–GAL4 via Effectene (Qiagen) and cultured in Schneider’s medium for 48 h before fixation. Cells were fixed with 4% PFA and incubated with polyclonal anti-mCherry antibodies (Invitrogen PA5-34974) under non permeabilizing conditions. mCherry^C^ or PmCherry^C^ was visualized with secondary anti-rabbit IgG (Dianova) using a Zeiss LSM700 confocal microscope (Zeiss, Oberkochen, Germany). All cell lines were tested negative for mycoplasma.
2.4. Chromatography
Reverse-phase HPLC separation depends on the binding of molecules to immobilized hydrophobic ligands attached to the stationary phase and their elution in order of increasing molecular hydrophobicity. RP-HPLC was performed as described [10] using a C4-300 column (Tosoh, Tokyo, Japan) and an Äkta Basic P900 protein purifier (Cytiva, Uppsale, Sweden). Briefly, Bosc23 cells were transfected with expression plasmids for Shh and monolipidated or unlipidated mutants as described previously. Two days after transfection, cells were lysed in RIPA buffer containing a complete protease inhibitor cocktail (Roche, Basel, Switzerland) on ice, ultracentrifuged, and the soluble total cell extract was precipitated with acetone. Media obtained after Shh expression was treated in the same way. Protein precipitates were resuspended in 35 µL of (1,1,1,3,3,3)-hexafluoro-2-propanol and solubilized with 70 µL of 70% formic acid, followed by sonication. Reverse-phase HPLC was performed using a 0–70% acetonitrile/water gradient with 0.1% trifluoroacetic acid for 30 min at room temperature. Elution samples were vacuum-dried, resolubilized in reducing sample buffer, and analyzed by SDS-PAGE and immunoblotting. The signals were quantified using ImageJ (NIH, Bethesda, MD, USA). HEK293-derived human Shh (R&D Systems, 8908-SH) was used as a double-lipidated control protein.
2.5. Verification of Mouse Mesenchymal Stromal Cells
To confirm the multipotency of the reporter cells routinely used in our laboratory to quantify Shh biofunction, we used a mouse mesenchymal stem cell functional identification kit (R&D, SC010) according to the manufacturer’s instructions. Briefly, C3H10T1/2 [28] mesenchymal stromal cells were cultured in MEM basal medium supplemented with 10% FCS and antibiotics (for adipogenesis) or DMEM supplemented with 10% FCS and antibiotics (for osteogenesis and chondrogenesis), and differentiation was induced by media supplements to induce adipogenesis (hydrocortisone, isobutylmethylxanthine, and indomethacin), osteogenesis (ascorbate phosphate, β-glycerolphosphate, and BMP-2) or chondrogenesis (dexamethasone, ascorbate phosphate, proline, pyruvate, and TGF-β3). Cells were cultured for 14 days (adipocytes and osteocytes) and 21 days (chondrocytes), with old media removed and fresh media added every 3–4 days, fixed, and the differentiation status confirmed using goat anti-mouse FABP-4 polyclonal antibody (adipocytes), sheep anti-mouse collagen II polyclonal antibody (chondrocytes), and goat anti-mouse osteopontin polyclonal antibody (osteocytes). Cells were stained with Alexa Fluor 488 NL-557-conjugated donkey anti-goat or anti-sheep IgG secondary antibodies and nuclei were counterstained with DAPI (blue) to confirm their multipotent status.
2.6. Shh Reporter Assays
C3H10T1/2 cells were grown in DMEM supplemented with 10% FCS and antibiotics. Twenty-four hours after seeding, Shh-conditioned media was mixed 1:1 with DMEM containing 10% FCS and antibiotics, and applied to C3H10T1/2 cells in 15 mm plates. In some cases, 0–6 ng of double-lipidated, HEK293-derived human Shh (R&D Systems, 8908-SH) served as positive activity controls during the assays. To some samples, 2.5 μM cyclopamine, a specific inhibitor of Shh signaling, and 1 µg/mL of Shh neutralizing antibody 5E1 [29] were added to confirm the specificity of the assay. In general, due to variable expression levels, mutant and wild-type proteins were adjusted to similar levels prior to the induction of C3H10T1/2 differentiation, as determined by Western blotting of an aliquot of the same stock. Cells were lysed 5–6 days after induction (20 mM Hepes, 150 mM NaCl, 0.5% TritonX-100, pH 7.4) and osteoblast-specific alkaline phosphatase (Alp1) activity was measured at 405 nm after the addition of 120 mM p-nitrophenol phosphate (Sigma) in 0.1 M glycine buffer, pH 9.5. Assays were always performed in triplicate. Cells were authenticated and tested negative for mycoplasma.
2.7. qRT-PCR
C3H10T1/2 cells were stimulated with recombinant Shh^C^/^C25A^Shh^C^ in triplicate and the media was changed every 3–4 days. TriZol reagent (Invitrogen) was used to extract RNA from C3H10T1/2 cells after 14 days. A first-strand DNA synthesis kit and random primers (Thermo, Schwerte, Germany) were used for cDNA synthesis before a control PCR was performed with murine β-actin primers. Amplification with Rotor-Gene SYBR-Green on a BioRad CFX 384 machine was performed in triplicate according to the manufacturer’s protocol. Primer sequences are provided in the Supplementary Table S1. Cq values of technical triplicates were averaged, the difference to β-actin mRNA levels calculated using the ∆∆Ct method, and the results expressed as the log2-fold change if compared to the internal control of C3H10T1/2 cells stimulated with mock-transfected media.
2.8. Confocal Microscopy of Drosophila Eye Discs
Eye discs were dissected from L3 larvae, fixed, permeabilized, and mounted. Samples were stained with α-Ci155 antibodies (2A1 rat, DSHB, 1:100, overnight) and Alexa647-conjugated goat-α-rat antibodies (1:300) (Invitrogen). Stage-matched discs were immunolabeled by using the same antibody batch and dilution always following the same procedure. Images were captured on an LSM 700 Zeiss confocal microscope using ZEN software (Version 3.6), always with the same settings.
2.9. Bioanalytical and Statistical Analysis
All statistical analyses were performed in GraphPad Prism by using one-way analysis of variance tests (ANOVA) (parametric, post-test as indicated, confidence interval 95%). Ommatidia from male and female flies were counted and statistically analyzed in the same way. qRT-PCR results were analyzed and visualized in GraphPad Prism MacOS version 9.4.1.
2.10. Molecular Modeling
The structure of Ptch1 with double-lipidated Shh has been visualized using the PyMol molecular graphics system, version 2.3.0, from Schrödinger LLC, Boston, MA, USA.
3. Results
3.1. Serum Promotes the Release of Selectively Depalmitoylated ShhC from the Dual-Lipidated Cellular Precursor
To date, most studies have examined Shh release from expressing cells in serum-free or serum-depleted media [30,31,32,33]. However, serum starvation is known to affect the amounts of secreted proteins and their secretion pathways [34]. Furthermore, in vivo, Shh-expressing cells are immersed in interstitial fluid that circulates between cells and tissues. Interstitial fluid is a filtrate of blood serum through the capillary walls and therefore contains >20 g/L of proteins and low-mass lipoprotein particles from the serum [35]. Therefore, to test Shh solubilization under experimental conditions that better mimic the cellular microenvironment in vivo, we expressed several Shh variants together with the physiological release factors Disp [12,13] and Scube2 [14,15] in the presence of 10% FCS. We then asked whether Shh is released with both lipids still present or whether the protein undergoes (partial) delipidation during its solubilization in the serum-containing medium. To this end, we compared secreted Shh from serum-containing media with the cellular precursor or with commercially available dual-lipidated control proteins by reverse-phase high performance liquid chromatography (RP-HPLC). RP-HPLC is a powerful technique for separating proteins based on their hydrophobicity. The instrument was first calibrated with commercially available recombinant human R&D 8908-SH Shh. This protein is produced by detergent extraction from the plasma membrane of transfected HEK293 cells and therefore represents the dual lipidated Shh pre-release state upstream of Disp and Scube2 function. As shown in Figure 2A, R&D 8908-SH eluted predominantly in the late fraction #37 (black arrowhead). In contrast, an artificial monolipidated (N-palmitoylated) ShhN standard elutes in the earlier fractions #27–29 (Figure 2B, red arrowhead) and monolipidated (C-cholesteroylated) ^C25S^Shh elutes predominantly in and around fraction #32 (Figure 2C, white arrowhead). Non-lipidated artificial ^C25S^ShhN elutes in the earliest fractions #26–28 from the C4 column (Figure 2D, green arrowhead). We used the same C4 column and buffer stocks for the subsequent RP-HPLC characterization of cellular dual-lipidated Shh precursors and soluble proteins generated from these precursors.
As expected from its established dual lipidation during biosynthesis, we found that the majority of pre-release Shh from Bosc23 cell lysates eluted in fraction #37 (Figure 2E, black arrowhead), just like the dual lipidated R&D 8908-SH control. We also noticed a fraction of less hydrophobic cellular proteins (Figure 2E, fraction #33, white arrowhead) that eluted similarly to non-palmitoylated ^C25S^Shh control proteins. This fraction likely represents overexpressed recombinant proteins that escaped Hhat-mediated N-palmitoylation during biosynthesis. Importantly, the same shift in hydrophobicity was observed when dually lipidated Shh was released from producing cells into media containing 10% FCS (Figure 2F, white arrowhead). This result indicates that the N-palmitate, but not the C-cholesterol, is removed during the release process. We call the resulting soluble depalmitoylated variant Shh^C^ to distinguish it from the cellular, dually lipidated Shh precursor. Non-palmitoylated but cholesteroylated artificial ^C25S^Shh^C^ control proteins eluted in the same fractions from the C4 column (Figure 2G, white arrowhead), confirming selective delipidation of the Shh^C^ N-terminus during release.
Notably, the replacement of serum with 600 μg/mL of the pharmacological cholesterol chelator methyl-β-cyclodextrin (CD, an oligosaccharide that complexes and shields cholesterol, thereby rendering it soluble) solubilized Shh^C^ and ^C25A^Shh^C^ with their C-terminal cholesteroylated peptides intact, yet again with the N-terminal palmitate removed (Figure 2H,I, fractions #32–34 represent N-processed Shh^C^ and ^C25A^Shh^C^ (white arrowhead), Shh^C^ and ^C25A^Shh^C^ fractions #27–28 represent small amounts of dually delipidated soluble Shh [32,33,36]). Taken together, these results suggest that physiological or pharmacological sterol acceptors likely interact with the C-terminal cholesteroylated Shh peptide to render it soluble, which in turn may protect it during release and limit the proteolytic processing of Shh to the plasma membrane-anchored N-peptide [25].
3.2. ShhC Induces In Vitro Differentiation of C3H10T1/2 and NIH3T3 Cells
As described previously, it has been established that Shh/Hh expression in the dual lipidated form is required for unimpaired Hh biofunction in vivo [17,37,38,39,40] and that unpalmitoylated ^C25S^Shh is less bioactive than Shh [9,17]. To test whether selectively N-terminally processed Shh^C^ released in the presence of serum is still functional, we used the Ptch1-expressing multipotent fibroblastic C3H10T1/2 cell line as a reporter. We first tested the multipotency of our C3H10T1/2 cell line to differentiate into osteoblasts [28], chondrocytes [41], or adipocytes [42]. To this end, C3H10T1/2 cells were cultured in the presence of adipogenic, chondrogenic, and osteogenic supplements for different time periods and their responsiveness was confirmed based on the phenotype and the expression of cell surface markers (as shown in [25]). We then expressed Shh in serum-containing media in the presence of the physiological release regulators Scube2 [15,43] and Disp [12,13], normalized the proteins by Western blotting (inset), and added similar protein amounts to the C3H10T1/2 cells. As a control for the possible saturation of Ptch1 receptors on these cells, we also added the proteins at 50% concentration. As shown in Figure 3A, both Shh^C^ and ^C25S^Shh^C^ induced robust and concentration-dependent differentiation of C3H10T1/2 progenitor cells into alkaline phosphatase-producing osteoblasts (Shh^C^ at 100%: 2.16 ± 0.1 arbitrary units (au) (n = 3) and ^C25S^Shh^C^: 2.67 ± 0.26 au (n = 3), p < 0.0001, Shh^C^ at 50%: 0.88 ± 0.14 au (n = 3) and ^C25S^Shh^C^: 0.87 ± 0.09 au (n = 3), p > 0.999). As a negative control, ^C25S^ShhN bioactivity was strongly reduced (0.17 ± 0.005 au (100% protein, n = 3) and 0.13 ± 0.04 au (50% protein, n = 3), similar to the the mock (empty vector) medium control (0.13 ± 0.02 au (n = 3)). We confirmed the bioactivity of ^C25S^Shh^C^ by quantitative reverse transcription–polymerase chain reaction (qPCR) analysis of Ptch1 and Gli1 expression in C3H10T1/2 cells (Figure 3B, left two graphs). Ptch1 is known to be upregulated by the ligand [44], and Gli1 is a zinc finger transcription factor that operates downstream of Ptch1 and that is also transcribed in an Shh-dependent manner [45]. As a positive control, we added R&D 8908-SH Shh (labeled 8908 in Figure 3B) to fresh serum-containing medium to compare its biofunction with that of solubilized N-truncated Shh^C^ and unpalmitoylated ^C25S^Shh^C^ (inset). Again, ^C25S^ShhN served as a negative control. qPCR confirmed similar increases in the mRNA expression of Ptch1 by 8908-SH (we observed a 2.7-fold transcriptional increase), Shh^C^ (a 2.6-fold increase), and ^C25S^Shh^C^ (a 2.1-fold increase, ^C25S^ShhN: a 0.5-fold increase). Gli1 mRNA expression increased by 5-fold, 4.8-fold, 4.6-fold, and 0.5-fold, respectively. This suggests that N-terminal delipidation of Shh^C^ does not reduce its biofunction below that of the dually lipidated R&D 8908-SH control, at least when Shh^C^ and ^C25S^Shh^C^ are expressed in the presence of 10% serum. We confirmed this finding in the Shh-responsive NIH3T3 cell line (Figure 3B, right graphs). Again, 8908-SH and ^C25S^Shh^C^ induced Ptch1 mRNA expression to a similar extent (8908-SH induced a 1.67 ± 0.001-fold transcriptional increase, ^C25S^Shh^C^ a 1.39 ± 0.2-fold increase, ^C25S^ShhN: a −0.003 ± 0.12-fold increase, and Shh^C^ a 2.27 ± 0.37-fold increase). Gli1 transcription increased 2.4 ± 0.001-fold (8908-SH), 1.78 ± 0.16-fold (^C25S^Shh^C^), 3.1 ± 0.49-fold (Shh^C^), and −0.11 ± 0.25-fold (^C25S^ShhN). It is important to note that widely used alternative assays, such as the NIH3T3-Light2 assay for Gli1 transcription, require greatly reduced serum levels and often measure the activity of proteins expressed in the absence of serum. Differences between our results and previously published results [9,17] may therefore be explained by the different conditions used for protein expression and activity determination.
We next used qPCR of Hh target gene expression in multipotent C3H10T1/2 cells to test whether equivalent amounts of soluble Shh^C^ and ^C25A^Shh^C^ in serum-containing media induce similar or different expression profiles in other differentiation pathways of these cells (Figure 3C). We found that ^C25A^Shh^C^ and Shh^C^ not only induced proportional transcriptional changes in key targets of Hh signaling (Ptch1, Gli1-3) as previously shown, but also did not differentially affect the expression of regulators of adipogenesis (Dlk1, Pparγ, Fabp4, Cfd, Dgat2), osteogenesis (alkaline phosphatase (Alp1), also Spp1, Bglap, Runx2), chondrogenesis (Sox9, Col2a1, Col10a1, Col1a1, Mmp3), and proliferation (Cdk9, Mki67).
3.3. N-Terminal Amino Acids Contribute to ShhC Biofunction In Vitro
Previously, it was shown that aberrant processing of the most N-terminal 11 Shh amino acids by furin-like proteases prevents truncated Shh binding to Ptch1 [46]. Furin (also called proprotein convertase subtilisin/kexin type (PCSK) 3 or PACE) is one of seven members of the PCSK family that cleave their substrates at single or paired basic residues, mostly in the secretory pathway. Other members of this family are PCSK5 (also called PC5), PCSK6 (also called Pace4), and PCSK7. Initially, we expected that their cleavage specificity for single or paired lysines or arginines would make these proteases strong candidates to cleave the Shh N-terminal peptide at the highly conserved, polybasic HS-binding site (the so-called Cardin–Weintraub (CW) sequence K-R-R-x-x-K-K [47], where x is any amino acid) (Figure 4A, labeled in green). We screened all PCSK family members for their ability to cleave the CW sequence and found that only PCSK7 processed this site (Figure 4A, bottom inset, the right lane shows processed Shh^C^). Furin cleaves at a site following the minimal motif R-x-x-R or at the preferred motif R-x-K/R-R in the secretory pathway. Therefore, a G-to-R exchange just upstream of the CW site (generating ^G32R^Shh^C^ and ^C25A;G32R^Shh^C^) renders the molecules susceptible to furin cleavage (Figure 4A, inset). To test whether furin- and PCSK7 cleavage inactivated the solubilized proteins, we incubated C3H10T1/2 cells with similar amounts of recombinant furin-cleaved ^G32R^Shh^C^ and ^C25A; G32R^Shh^C^ and PCSK7-cleaved ^C25A^Shh^C^ (Figure 4B). In this assay, Shh^C^ and ^C25S^Shh^C^ served as positive controls. Indeed, furin/PCSK7 processing downstream of the CW site strongly reduced or abolished Shh biofunction, as indicated by strongly reduced or abolished induction of Ptch1, Gli1, and Alp1 mRNA expression (Figure 4B) [46]. In some cases, the transcription of Hh target genes was even suppressed below unstimulated control (mock) levels (transcription of all three target genes was most strongly suppressed by ^G32R^Shh^C^ + furin). We conclude from these results that proteolytic removal of a significant portion of the Shh^C^ N-terminus abolishes Hh-induced C3H10T1/2 cell differentiation, even when the proteins were expressed in the presence of serum. Note that reduced protein function after processing was not entirely dependent on the N-palmitate, as cleavage also reduced the activitiy of ^C25A^Shh^C^ (as observed for (incompletely) furin-cleaved ^C25A; G32R^Shh^C^ and for ^C25A^Shh^C^ in the presence of PCSK7).
The observed differences in Shh^C^/^C25A^Shh^C^ biofunctions in the presence or absence of subtilisin/kexin-type convertases suggested to us that the N-terminal Shh^C^ peptide contributes to Ptch1 binding and signaling. Such functions of the Shh N-peptide are supported by two recent cryogenic electron microscopy (cryo-EM) structures [22,23] (Figure 4C). These structures suggest multiple hydrogen bonds and salt bridges between the N-terminal palmitoylated peptide and Ptch1 to connect the most N-terminal Shh amino acids P^27^, R^29^, G^30^, and F^31^ with residues N^802^, Y^804^, N^940^, and Y^1013^ of the second extracellular domain of Ptch1, and to connect more than 10 residues of the first extracellular domain of Ptch1 with the Shh residues G^32^, R^34^, R^35^, H^36^, and P^37^. To test the putative functional role of these interactions (independent of postulated palmitate functions in Hh signaling), we expressed ^C25S^Shh^C^ and unpalmitoylated variants that additionally lacked increasing numbers of N-terminal amino acids. All solubilized proteins were pulled down using heparin–sepharose to control for their comparable production and secretion (Figure 4D, top), and media aliquots containing 10% FCS were added to C3H10T1/2 progenitor cells to induce their Hh-dependent differentiation into osteoblasts (in this experiment, Alp1 protein biofunction again served as a functional readout for Shh-induced differentiation). As observed previously, Shh^C^ and ^C25S^Shh^C^ induced C3H10T1/2 differentiation into Alp1-producing osteoblasts to a similar extent (Figure 4D, bottom). The teratogen cyclopamine (CA) [48] and the monoclonal antibody 5E1, which blocks Shh/Ptch1 interactions [49], both inhibited Shh-induced C3H10T1/2 differentiation, demonstrating the specificity of the assay (Shh^C^: 1.5 arbitrary units (au) ± 0.13 au, Shh^C^ + 5E1: 0.26 ± 0.01 au, Shh^C^ + CA: 0.19 ± 0.003 au, p ≤ 0.0001, n = 3; ^C25S^Shh^C^: 1.71 ± 0.03 au, ^C25S^Shh^C^ + 5E1: 0.21 ± 0.01 au, ^C25S^Shh^C^ + CA: 0.18 ± 0.003 au, p ≤ 0.0001, n = 3). Negative control media obtained from mock transfected Bosc23 cells were also inactive, as expected (0.12 ± 0.001 au). However, C3H10T1/2 differentiation induced by truncated ^C25S^Shh^C^ variants was variable: While ^C25S;Δ26–31^Shh^C^, ^C25S;Δ26–32^Shh^C^ and ^C25S;Δ26–33^Shh^C^ remained bioactive (^C25S;Δ26–31^Shh^C^: 1.41 ± 0.05 au, p = 0.64 when compared to Shh^C^; ^C25S;Δ26–32^Shh^C^: 1.26 ± 0.08 au, p = 0.01 when compared to Shh^C^ and ^C25S;Δ26–33^Shh^C^: 1.65 ± 0.27 au, p = 0.31 when compared to Shh^C^), the bioactivities of all proteins that were truncated beyond ^C25S^Shh^C^ amino acid 33 were strongly reduced (^C25S;Δ26–34^Shh^C^: 0.7 ± 0.04 au; ^C25S;Δ26–35^Shh^C^: 0.38 ± 0.002 au; ^C25S;Δ26–36^Shh^C^: 0.39 ± 0.003 au; ^C25S;Δ26–37^Shh^C^: 0.46 ± 0.002 au; ^C25S;Δ26–38^Shh^C^: 0.36 ± 0.01 au, n = 3 and p < 0.0001 for all forms when compared to Shh^C^). These results suggest that N-terminal Shh peptide interactions with the first, but not with the second, extracellular domain of Ptch1 contribute to Shh^C^ biofunction (Figure 4E) and that Shh^C^ processing likely occurs upstream of the amino acids R34/R35 to maintain the bioactivity of the solubilized protein.
3.4. The N-Terminal Hh Peptide Contributes to Morphogenetic Furrow Progression in the Drosophila Eye Disc
We next tested whether required interactions between the N-terminal amino acids of the ligand and Ptch1 receptors are conserved between vertebrates and invertebrates, e.g., whether the Hh-N-terminal peptide also contributes to Hh biofunction in vivo. For this purpose, we used Drosophila eye development as a model. The Drosophila eye consists of a honeycomb matrix of photoreceptors (ommatidia) that develop in a wave of differentiation that moves from the posterior to the anterior of the eye disc, the so-called morphogenetic furrow (Figure 5A). Here, cells located anterior to the furrow respond to Hh secreted by cells posterior to the furrow by producing the same protein. This creates a cyclic Hh signaling mode that drives the furrow across the entire disc [50] and determines the number of ommatidia in the adult eye. As a genetic background, we analyzed hh^bar3^ eye discs in trans with hh^AC^, a homozygous lethal null mutation. These combined mutations impair endogenous Hh production and furrow progression in the disc, resulting in small kidney-shaped eyes consisting of 100 ± 8 ommatidia/eye instead of 610 ± 18 ommatidia/eye in the wild type (hh^bar3^/hh^AC^ versus GMR>, n = 16 male eyes were analyzed, Figure 5B,C) [51]. To restore the phenotype, we used an established eye disc-specific GMR (glass multimer reporter)-Gal4 driver [52] to express Hh or Hh-GFP [53] to visualize the protein (Figure 5A, top), non-palmitoylated ^C85S^Hh, and truncated variants thereof from the same attP 51C landing site to ensure similar protein expression levels [26]. We found that GMR-GAL4-driven Hh expression in discs made deficient in endogenous Hh synthesis restored most eye development (676 ± 15 ommatidia/eye, positive control), whereas ^C85S^Hh restored a smaller part of the compound eye (325 ± 17 ommatidia/eye, n = 16, and 361 ± 25 ommatidia/eye, n = 31, respectively, Figure 5B,C). Mechanistically, Figure 5A suggests that the reduced number of ommatidia resulted from a delay in furrow progression across the disc: In stage-matched larvae, the furrow driven by ^C85S^Hh expresses Ci155 as a consequence of signaling activation in anterior receiving cells (bottom), demonstrating ^C85S^Hh biofunction (as previously observed for ^C25S^Shh (Figure 3 and Figure 4)), but it lags behind the furrow driven by Hh (shown in the middle)). An important finding in this system is that the additional deletion of six N-terminal amino acids (resulting in the N-terminal peptide S-G^92^-R^93^-H^94^-R^95^-A^96^-R^97^-Hh, Figure 5B) significantly increased ^C85S;∆85–91^Hh biofunction (423 ± 23 ommatidia, n = 16). This finding suggested to us the possibility of preferential physiological N-terminal processing at this site during Hh release. ^C85S;∆85–93^Hh and ^C85S; ∆85–94^Hh activities were similar to ^C85S^Hh (359 ± 24 ommatidia/eye and 352 ± 27 ommatidia/eye, respectively), as were ^C85S;∆85–95^Hh activities (306 ± 6 ommatidia/eye). However, additional truncations reduced the activities of the ^C85S^Hh variants to varying degrees (^C85S; ∆85–96^Hh: 198 ± 25 ommatidia/eye; ^C85S;∆85–97^Hh: 274 ± 21; ^C85S;∆85–98^Hh: 211 ± 20; ^C85S;∆85–99^Hh: 239 ± 25; ^C85S;∆85–100^Hh: 127 ± 16, n = 16 and p < 0.001 for all forms when compared to ^C85S^Hh), consistent with the abolished activities of extensively truncated ^C25S^Shh^C^ variants in vitro (Figure 4A). We note that the strong loss-of-function of truncated ^C85S^Hh variants was not caused by partial or complete deletion of the three basic CW amino acids present in fly Hh (Figure 5B, top) [54], because GMR-GAL4-driven expression of Hh^3xA^, which has this site functionally deleted, restored most eye development (619 ± 22 ommatidia/eye, n = 9). Taken together, our results show that the N-terminal peptide contributes to Hh biofunction in vitro and in vivo. This led us to ask whether short palmitoylated N-peptides alone could regulate Ptch function in vivo, as previously suggested [55].
3.5. Isolated Palmitoylated or Unpalmitoylated N-Terminal Peptides Are Not Active In Vivo
Using the same Drosophila eye developmental model, we tested whether isolated, palmitoylated, or unpalmitoylated N-terminal Hh peptides are sufficient for Ptch receptor binding and signaling in vivo. To this end, we replaced the N-terminal signaling domain of Drosophila Hh (HhN, amino acids 99–195, Figure 6A) with an mCherry tag (Figure 6B). This strategy ensured quantitative cholesteroylation of the C-terminal amino acid during mCherry secretion to the cell surface. We then generated two UAS-regulated expression constructs from the cholesteroylated mCherry backbone: One with its N-terminal Hh peptide palmitoylated by endogenous Ski palmitoyltransferase expression (termed PmCherry^C^), and an mCherry^C^ control lacking the N-terminal lipid and peptide (Figure 6B). Expression of both constructs under an actin–GAL4 control in S2 cells confirmed their similar biosynthesis, secretion, and cell surface association (Figure 6C). For subsequent in vivo assays, both constructs were inserted into the attP 68E landing site on the third fly chromosome and expressed under GMR-GAL4 control in hh^bar3^/hh^AC^ eye discs (Figure 6D,E). Again, Hh expression as a positive control restored eye development (855 ± 40 ommatidia/eye, n = 7; negative control hh^bar3^/hh^AC^ eye discs: 98 ± 8 ommatidia/eye, n = 8; wild type: 876 ± 56 ommatidia/eye, n = 7). In contrast, both mCherry^C^ and PmCherry^C^ were both inactive (105 ± 18 ommatidia/eye, n = 28 and 100 ± 19 ommatidia/eye, n = 27). We conclude that interactions between the palmitoylated Hh N-peptide and the receptor alone are not sufficient to induce signaling and that physical linkage of the N-peptide to the globular Hh domain is a minimal requirement for the generation of bioactive protein, at least in the Drosophila eye model. However, we note that another explanation for our result is that the interactions of the N-peptide may have been blocked by the attached mCherry tag, a possibility that we cannot rule out.
4. Discussion
Recently published cryo-EM studies have suggested several possible modes of Shh binding to its receptor Ptch1—without lipids, with one lipid, or with both lipids involved. The latter possibility was supported by Qi et al. who reported high-resolution structures of Ptch1 together with dually lipidated R&D 8089-SH/CF [22,23]. In these structures (Figure 7A, shown in yellow; Figure 7B shows the Ptch1 surface), the palmitoylated Shh N-terminus inserts deep into a Ptch1 “conduit” to block Ptch1-mediated cholesterol transport, which is also thought to pass through this conduit [57]. This proposed mode of Ptch control may represent one point in a conceivable spectrum of Ptch activity. However, the regulation of Ptch1 activity by monolipidated Shh variants carrying only N-palmitate [58] or only C-cholesterol [25] has also been demonstrated. Support for these alternative signaling modes comes from the in vivo finding that the unpalmitoylated protein is active in developing tissues, such as the mouse limb bud, because it leads to a spectrum of phenotypes similar to that induced by Shh [17]. Based on these findings, dual- or monolipidated Shh signaling to Ptch1 is not mutually exclusive but may be used differently in different tissues and developmental contexts to increase the versatility of the Hh pathway.
We support this hypothesis by demonstrating that the dual-lipidated, cell surface-associated precursor is converted into C-terminally monolipidated Shh^C^ by N-terminal proteolytic processing. Support for proteolytic processing during release is provided by the increased electrophoretic mobility of the solubilized protein, as determined by SDS-PAGE/immunoblotting, and its decreased hydrophobicity, as determined by RP-HPLC. The proteolytic conversion of Shh during release is physiologically important because it is strictly dependent on the co-expression of the Shh release proteins Disp and Scube2 [25] (supporting the release mechanism shown in Figure 1B). The physiological importance of the proteolytic conversion of Shh is further supported by the functionality of the solubilized protein product in vitro and in vivo, although we note that the bioactivity of unpalmitoylated proteins was somewhat reduced in NIH3T3 cells and in the Drosophila eye disc. Our results show that this residual activity depends on Shh^C^ binding to Ptch1 via a protein–protein interface together with the N-terminal peptide to enhance signaling (Figure 7C). However, in contrast to the amino acids proximal to the globular domain, the most N-terminal amino acids of Shh^C^ (Figure 7D, shown in red) are not essential for the regulation of Ptch1 activity (Figure 7E). This result is consistent with the published finding that furin-mediated removal of eleven amino acids from the N-terminus of a mutated Shh, including amino acids that we determined to be critical for receptor activation, severely reduces Ptch binding and signaling [46]. The result also suggests that furin resistance of the wild-type N-terminal CW sequence, although rich in basic amino acids, is critical for protecting Shh during its secretion to the plasma membrane and during furin-mediated Disp activation at the cell surface [59,60] to maintain its biofunction.
We also show that Shh^C^ release from the plasma membrane requires the presence of serum. This result supports a model in which lipoproteins serve as soluble vehicles for lipid-linked morphogens in vitro and in vivo [58,61]. Notably, the retained presence of the C-cholesterol moiety in serum-released Shh^C^ is consistent with the proposed molecular mechanism by which Disp is thought to release Hh/Shh from the plasma membrane of producing cells. It is well known that all vertebrate and invertebrate Disp family members contain a sterol sensing domain (SSD) that is conserved in proteins that bind, transport, or respond to cellular sterols, such as SREBP cleavage activating protein (SCAP) and NPC1 [62,63,64]. The SSD extends into a hydrophobic surface channel that has been proposed to function as an open “slide” for lipophiles [65] for subsequent transfer to an acceptor [59]. This acceptor is likely to be a serum lipoprotein, as supported by the published concept that the fly lipoprotein called lipophorin carries cholesteroylated Hh in vivo [58,61,66] and a previous report showing that C-cholesterol is necessary and sufficient for Disp-mediated protein export [30]. Finally, our results show that the transfer is terminated by proteolytic processing of the palmitoylated N-peptide at a membrane-proximal position, allowing the globular domain of Hh/Shh and the N-terminal “stub” of lipoprotein-associated Hh^C^/Shh^C^ to interact with the receptor Ptch on the surface of the signal-receiving cell (Figure 7E).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1De Luca A. Cerrato V. Fuca E. Parmigiani E. Buffo A. Leto K. Sonic hedgehog patterning during cerebellar development Cell Mol. Life Sci.20167329130310.1007/s 00018-015-2065-126499980 PMC 11108499 · doi ↗ · pubmed ↗
- 2Huang J. Kalderon D. Coupling of Hedgehog and Hippo pathways promotes stem cell maintenance by stimulating proliferation J. Cell Biol.201420532533810.1083/jcb.20130914124798736 PMC 4018789 · doi ↗ · pubmed ↗
- 3Jiang J. Hui C.C. Hedgehog signaling in development and cancer Dev. Cell 20081580181210.1016/j.devcel.2008.11.01019081070 PMC 6443374 · doi ↗ · pubmed ↗
- 4Peng T. Frank D.B. Kadzik R.S. Morley M.P. Rathi K.S. Wang T. Zhou S. Cheng L. Lu M.M. Morrisey E.E. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration Nature 201552657858210.1038/nature 1498426436454 PMC 4713039 · doi ↗ · pubmed ↗
- 5Vervoort M. Hedgehog and wing development in Drosophila: A morphogen at work?Bioessays 20002246046810.1002/(SICI)1521-1878(200005)22:5<460::AID-BIES 8>3.0.CO;2-G 10797486 · doi ↗ · pubmed ↗
- 6Groves I. Placzek M. Fletcher A.G. Of mitogens and morphogens: Modelling Sonic Hedgehog mechanisms in vertebrate development Philos. Trans. R. Soc. Lond B. Biol. Sci.20203752019066010.1098/rstb.2019.066032829689 PMC 7482217 · doi ↗ · pubmed ↗
- 7Yauch R.L. Gould S.E. Scales S.J. Tang T. Tian H. Ahn C.P. Marshall D. Fu L. Januario T. Kallop D. A paracrine requirement for hedgehog signalling in cancer Nature 200845540641010.1038/nature 0727518754008 · doi ↗ · pubmed ↗
- 8Porter J.A. Young K.E. Beachy P.A. Cholesterol modification of hedgehog signaling proteins in animal development Science 199627425525910.1126/science.274.5285.2558824192 · doi ↗ · pubmed ↗