Genetic underpinnings explored: OPA1 deletion and complex phenotypes on chromosome 3q29
Ethan Hung-Hsi Wang, Pei-Hsuan Lin, Pei-Liang Wu, Eugene Yu-Chuan Kang, Laura Liu, Lung-Kun Yeh, Kuan-Jen Chen, Meng-Chang Hsiao, Nan-Kai Wang

TL;DR
A patient with optic atrophy and a brain aneurysm was found to have a rare 3q29 deletion involving the OPA1 gene, highlighting the importance of detecting copy-number variations in genetic disorders.
Contribution
This case highlights the underestimation of OPA1 deletions due to sequencing limitations and emphasizes the need for targeted CNV analysis in ADOA patients.
Findings
A de novo 960 kb deletion on 3q29 encompassing OPA1 was identified in a patient with optic atrophy and a brain aneurysm.
The optic atrophy is conclusively linked to the OPA1 deletion, but the aneurysm is likely coincidental.
Sequencing technology limitations may lead to underdiagnosis of OPA1 deletions in Autosomal Dominant Optic Atrophy.
Abstract
Copy number variations (CNVs) have emerged as significant contributors to the elusive genetic causality of inherited eye diseases. In this study, we describe a case with optic atrophy and a brain aneurysm, in which a de novo CNV 3q29 deletion was identified. A 40-year-old female patient was referred to our department after undergoing aneurysm transcatheter arterial embolization for a brain aneurysm. She had no history of systemic diseases, except for unsatisfactory best-corrected visual acuity (BCVA) since elementary school. Electrophysiological tests confirmed the findings in retinal images, indicating optic nerve atrophy. Chromosomal microarray analysis revealed a de novo deletion spanning 960 kb on chromosome 3q29, encompassing OPA1 and six neighboring genes. Unlike previously reported deletions in this region associated with optic atrophy, neuropsychiatric disorders, and obesity,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
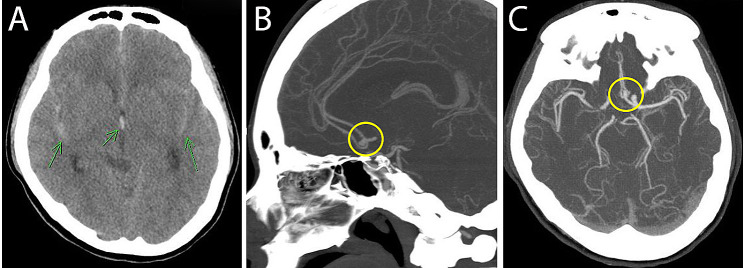 Figure 1
Figure 1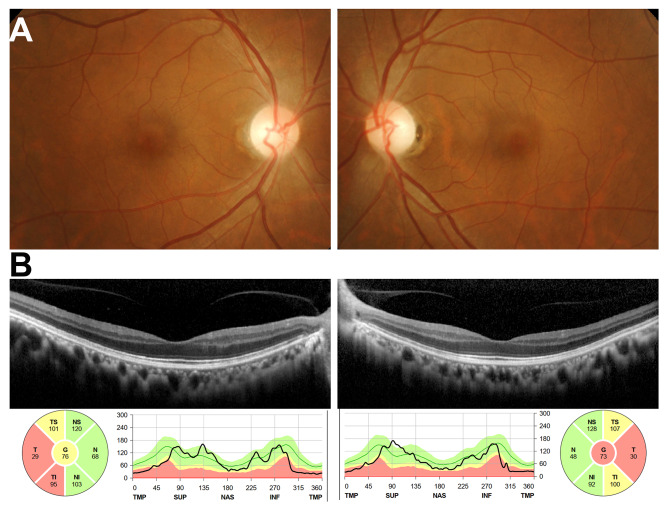 Figure 2
Figure 2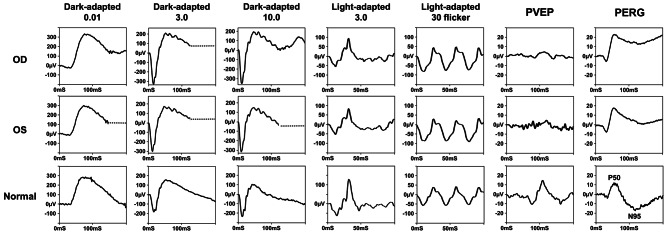 Figure 3
Figure 3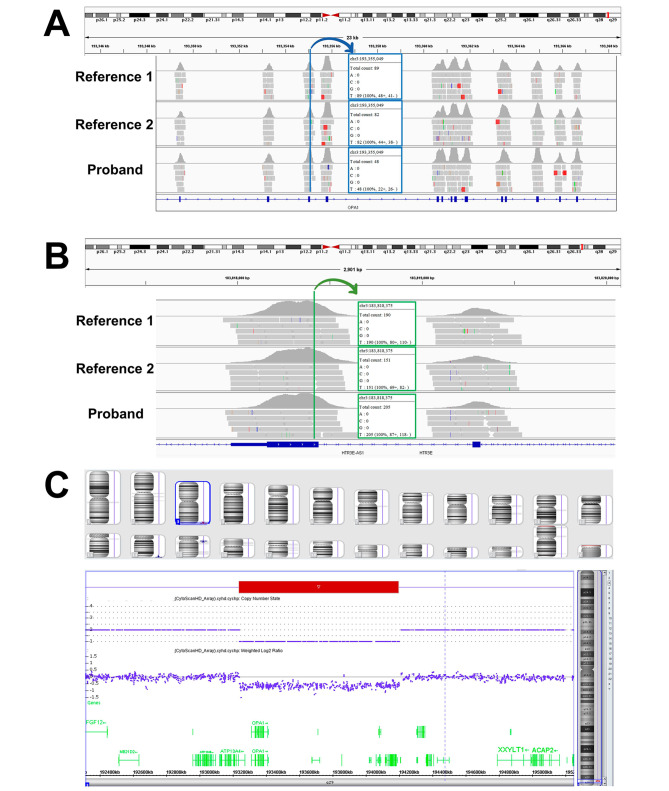 Figure 4
Figure 4- —NIH
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsConnective tissue disorders research · Congenital heart defects research · Genomics and Rare Diseases
Background
The nuclear OPA1 gene encodes for a dynamin-like GTPhase protein (OPA1 Protein) located in the inner mitochondrial space, responsible for mitochondrial dynamics (inner membrane fusion), cristal integrity, energetics, and mitochondrial DNA (mtDNA) maintenance. Variants in the OPA1 gene will disrupt mitochondrial functions, causing a mitochondrial disease known as Autosomal Dominant Optic Atrophy (ADOA). ADOA is characterized by the selective degeneration of retinal ganglion cells (RGC), which leads to progressive visual loss. Up to 20% of OPA1 variant carriers reported having ADOA plus, which includes extra ocular clinical features of myopathy, peripheral neuropathy, ataxia, encephalopathy, sensorineural hearing loss, and chronic progressive external ophthalmoplegia [1].
Exome Sequencing is considered cost-effective compared to Genome Sequencing because it specifically targets the protein-coding regions of the genome, known as the exome. Exome Sequencing is a valuable tool for identifying disease-causing variants in genetic disorders, especially when sequencing specific regions of interest. The techniques used in Exome Sequencing are capable of detecting low-level genetic variants that may be missed by traditional Sanger sequencing [2]. However, Exome Sequencing has its limitations. It does not cover variants in non-coding regions of the genome. In such cases, Sanger sequencing or Genome Sequencing may be more suitable alternatives. Moreover, some genes may have inadequate coverage in Exome Sequencing due to highly repetitive sequences, such as those found in the open-reading frame 15 (ORF15) region of the RPGR gene or in mitochondrial DNA (mtDNA). In these instances, Sanger sequencing or targeted panels for mtDNA are more advantageous. Exome Sequencing also falls short in identifying structural variations like deletions, duplications, translocations, and inversions. It is worth noting that copy number variation (CNV), a common genetic variation in the human genome [3], has recently emerged as a significant contributor to the genetic basis of inherited eye diseases [4].
In this case, we report a de novo 960 kb deletion in 3q29 that was presented with both optic atrophy and a brain aneurysm. Within this deletion, the haploinsufficiency of OPA1, associated with autosomal dominant optic atrophy, is likely responsible for the ophthalmological anomalies. The presence of the aneurysm could be coincidental.
Case presentation
A 40-year-old female without previous medical history was brought to the emergency room at Chang Gung Memorial Hospital, Taiwan, after suffering from severe headache and vomiting. Brain computed tomography (CT) revealed a diffuse subarachnoid hemorrhage (SAH) with a hematoma measuring one mm, diffuse brain swelling and hydrocephalus (green arrows Fig. 1A). The CT angiography also showed a small aneurysm with lobulated contour, 4.6 mm in dimension noted in anterior communicating artery (yellow circles Fig. 1B and C). Aneurysm transcatheter arterial embolization was performed following the discovery. The patient was then reported to the Department of Ophthalmology for postoperative examination.
In ophthalmic clinic, the patient claimed to have poor vision prior to the brain aneurysm, with a best corrected visual acuity of 20/60 ever since she was in elementary school. She also reported a central scotoma in her visual field. None of the patient’s parents nor related family members showed similar symptoms. Upon further ophthalmological examinations, the dilated fundus exam showed no abnormal pigmentation on the retina and macula. However, temporal optic nerve pallor was revealed (Fig. 2A). The spectral-domain optical coherence tomography (SD-OCT) revealed a normal retina structure, with thinning of the retina nerve fiber layer around the optic nerve head, depicting optic atrophy (Fig. 2B). The full field electroretinogram (ERG) test results were within normal range, however the pattern electroretinogram (PERG) and pattern visual evoked potential (PVEP) results were not. The PERG results have missing negativities at approximately 95 ms (N95) for both eyes. For the PVEP, the positives at 100 ms (P100) were reduced in amplitudes and delayed (Fig. 3). The results from the electrophysiological test (Fig. 3) supported the findings in the retinal images (Fig. 2), which indicated optic nerve atrophy.
Informed consent
for genetic testing was obtained from the patient and both parents, and the study was conducted according to the declaration of Helsinki and approved by the Institutional Review Board of Chang Gung Memorial Hospital (No. 201601569B0C602). The patient’s DNA was extracted from peripheral blood. Exome capture was performed using xGen Exome Research Panel v2 (Integrated DNA Technologies, Coralville, Iowa, USA) and sequencing was performed using NovaSeq 6000 (Illumina, San Diego, CA, USA). In total, 9,162,817,743 bases of sequence were generated and uniquely aligned to the Genome Reference Consortium Human Build 37 (GRCh37) and Revised Cambridge Reference Sequence (rCRS) of the mitochondrial genome, generating 141.19 mean depth-of-coverage within the 34,366,188 bases of the captured region, which is approximately 99.3% of the RefSeq protein coding region. Approximately 98.80% of the targeted bases were covered to a depth of ≥ 20x. In total, 67,480 single nucleotide variants (SNV) and 11,123 small insertions and deletions (indel) were identified. Due to limitations in the coverage and depth of mtDNA using exome sequencing, additional Sanger sequencing was conducted to confirm the absence of three hotspots (m.11778G > A, m.14484T > C, or m.3460G > A) associated with Leber Hereditary Optic Neuropathy in our patient. The Exome Sequencing results did not identify any disease-causing variants related to optic atrophy. Through further pipeline analysis, a deletion spanning the OPA1 gene was suspected. Approximately 50% fewer reads were observed in the OPA1 gene in the proband’s sample compared to two other unrelated reference samples (Fig. 4A). Meanwhile, the reads in the neighboring genes showed similar patterns between the proband and the two unrelated reference samples (Fig. 4B). Chromosomal microarray analysis (CMA) was performed with the CytoScan HD array (Affymetrix, Santa Clara, CA) according to the manufacturer’s instructions on the GeneChip™ Scanner 3000 7G platform (ThermoFisher Scientific, Waltham, US), which contains more than 2.67 million markers for copy number analysis, including 750,000 SNPs and 1.9 million nonpolymorphic probes. CMA data were visualized and analyzed with Chromosome Analysis Suite (ChAS) software package (Affymetrix, Santa Clara, CA). The results showed a 960 kb deletion on chromosome 3q29 (arr[GRCh37] 3q29(193,238,393_194,198,653)x1, Fig. 4C). This large heterozygous deletion encompasses 7 RefSeq coding-genes, namely ATP13A4, OPA1, HES1, CPN2, LRRC15, GP5, and ATP13A3 genes. The Autosomal Dominant Optic Atrophy diagnosis has been confirmed by the OPA1 heterozygous deletion identification. We have examined the asymptomatic parents who showed no signs of ocular abnormalities (as indicated in Supplement Fig. 1). Parental analysis by CMA concluded that the deletion was de novo.
Discussion and conclusions
Currently, the majority of OPA1 disease-causing variants are single nucleotide variants in exon, and OPA1 large deletions are not commonly found [5]. To further investigate the molecular etiology, sequencing technologies (including next generation sequencing and Sanger sequencing) are commonly performed for individuals with ADOA. Fuhrmann et al. has found that genomic rearrangements in OPA1 are frequent in patients with autosomal dominant optic atrophy (8/42), and therefore the frequency of OPA1 copy-number variations may have been underestimated due to the sequencing technology limitations [6]. Clinical geneticists working in this field should be familiar with the mutational spectrum of OPA1 and may use alternative methods to detect possible OPA1 copy-number variations, such as multiplex ligation-dependent probe amplification (MLPA) and microarray [7]. Large deletions encompassing multiple genes often result in more extensive and severe phenotypes. This phenomenon is well exemplified by deletions involving the NF1 and RB1 genes [8, 9]. Individuals with the NF1 deletion often develop a severe form of the disease with frequent cognitive impairment and an increased risk of tumors, in addition to typical NF1 characteristics. Likewise, individuals with RB1 deletion often have developmental abnormalities and facial dysmorphisms, in addition to typical retinoblastoma features. In that regard, it’s important for clinicians to remain attentive to “additional phenotypes” that might manifest in patients with optic atrophy, including conditions like aneurysms. In such cases, the utilization of microarray testing could be instrumental in detecting large OPA1 deletions that involve neighboring genes.
We have identified a 0.96 Mb deletion centromeric to the established 3q29 deletion syndrome [10]. While this deletion does not directly overlap with the well-known 3q29 deletion, it is noteworthy that it aligns with a 1.36 Mb deletion previously reported by Biamino et al. [11]. In their study, this deletion exhibited features including optic atrophy, autism, intellectual disability, psychiatric disorders, and obesity. The intriguing alignment of our findings with those reported by Biamino et al. adds a layer of complexity to our understanding of genetic variations in this region. While our patient and those in Biamino’s study both experienced optic atrophy due to the deletion of the OPA1 gene, a notable contrast arises as our patient did not exhibit symptoms or signs related to neuropsychiatric disorders or obesity. This discrepancy may be attributed to the larger deletion in Biamino’s cases, encompassing three additional genes—TMEM44, FAM34A, and LSG1—compared to our patient’s deletion. Furthermore, the absence of brain aneurysms in the cases described by Biamino et al. suggests that this phenotype may not be directly associated with the deletion.
As mentioned previously, the patient presented in this case suffered from a brain aneurysm as well as optic atrophy, two seemingly unrelated conditions that happened coincidentally. Subarachnoid hemorrhages, life-threatening conditions resulting from blood accumulation between the arachnoid and pia mater [12], have a global incidence of 7.9 per 100,000 person-years, with women having a higher risk ratio of 1.3 [13]. This trend is mirrored in the U.S., where the incidence was 11.4 per 100,000 person-years between 2007 and 2017, with a higher incidence in women [14]. Around the world, Japan and Finland report higher cases of subarachnoid hemorrhage [13]. The incidence of SAH increases with age worldwide, particularly in women over 55 years [13]. In the U.S., individuals aged 65 or older had over five times the incidence compared to those aged 20–44 years, with women consistently having a higher incidence in each age group [14]. A study by de Rooij NK et al. further supports these findings, showing a higher incidence of SAH in men than women in the 25–45 years age group, but a reversal of this trend in the 55–85 years age group [15]. According to the microarray results, the 960 kb deletion encompasses not only the OPA1 gene, but also neighboring genes, including ATP13A4, HES1, CPN2, GP5, LRRC15, and ATP13A3. It has been observed that among the 29 individuals reported in Decipher with deletions that overlap the 3q29 deletion identified in our study, none of them displayed brain aneurysms. This observation supports the idea that the occurrence of brain aneurysms is not directly associated with the identified deletion. However, it is important to acknowledge that the possibility of a somatic two-hit event remains a valid hypothesis. We regret that biopsy tissue testing was not conducted to explore this hypothesis further. It is plausible that the brain aneurysm phenotype found in our case is coincidental; nonetheless, we discovered that two genes, HES1 and CPN2, are associated with vascular function.
Carboxypeptidase N2 (CPN2) is a type of plasma metalloprotease that has the ability to cleave basic amino acids from the C-terminal of peptides and proteins. Its primary function involves regulating vasoactive peptide hormones, growth factors, and cytokines by specifically cleaving their C-terminal basic residues [16]. Vasoactive peptide hormones are crucial signaling molecules that play a pivotal role in regulating blood vessel tone and blood pressure. They also influence the contraction or relaxation of smooth muscle cells in blood vessels, playing an essential role in maintaining hemodynamic stability and cardiovascular homeostasis. While there is currently no direct association between the function of CPN2 and aneurysms, any dysregulation of vasoactive, any dysregulation of vasoactive peptide hormones may contribute to conditions such as vascular diseases.
HES1 belongs to a family of basic helix-loop-helix (bHLH) proteins essential for various biological processes, including neurogenesis, myogenesis, hematopoiesis, and sex determination. It serves as a transcriptional repressor for numerous genes but can also function as a transcriptional activator [17]. In the developing mouse embryo, using in situ hybridization, Hes1 was found to be expressed in almost all endothelial cells (ECs) of the developing internal carotid artery at embryonic day 10.5 (E10.5). Notably, in almost half of Hes1 knockout embryos, hemorrhage was observed in the cerebrospinal nervous system, indicating that Hes1 plays a role in regulating vascular remodeling and arterial fate specification of endothelial cells during brain development. Furthermore, Hes1 is identified as a critical transducer of Notch signals in brain vascular development [18]. Further functional study using a mouse model to knock out HES1 gene could be helpful in elucidating the role of HES1 in aneurysm formation.
Conclusion
In summary, we present a unique case featuring a de novo 960 kb deletion on chromosome 3q29 in a patient exhibiting optic atrophy and a brain aneurysm. The optic atrophy is conclusively attributed to the OPA1 deletion, and the aneurysm could be a coincidental association. This case highlights the difficulties faced in interpreting CNV in syndromic cases. Clinical geneticists who specialize in this area should have knowledge of the mutational spectrum of OPA1 and may consider using alternative techniques, such as MLPA and microarray analysis, to identify possible OPA1 CNVs.
Fig. 1. Brain computed tomography (CT). (A) Brain CT revealed a diffuse subarachnoid hemorrhage (green arrows). (B&C) The CT angiography also showed a small aneurysm with lobulated contour, 4.6 mm in dimension noted in anterior communicating artery (yellow circles**)**
Fig. 2. Retinal images. (A) Fundus photo showed temporal optic nerve pallor. (B) The spectral-domain optical coherence tomography revealed a normal retina structure, with thinning of the retina nerve fiber layer around the optic nerve head, depicting optic atrophy
Fig. 3. Ocular electrophysiology. The full field electroretinogram (ERG) showed normal scotopic and photopic responses. The pattern electroretinogram (PERG) revealed reduced in amplitudes and delayed at the positives 100 ms (P100) and pattern visual evoked potential (PVEP) showed missing negativities at approximately 95 ms (N95) for both eyes
Fig. 4. Exome sequencing and Chromosomal microarray analysis. (A) Approximately 50% fewer reads were observed in the OPA1 gene in the proband’s sample compared to two other unrelated reference samples. (B) The reads in the neighboring genes showed similar patterns between the proband and the two unrelated reference samples. (C) Chromosomal microarray analysis showed a deletion of 960 kb on chromosome 3q29
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1Supplementary Material 1
Supplementary Material 2
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lenaers G Neutzner A Le Dantec Y Juschke C Xiao T Decembrini S Dominant optic atrophy: culprit mitochondria in the optic nerve Prog Retin Eye Res 20218310093510.1016/j.preteyeres.2020.10093533340656 · doi ↗ · pubmed ↗
- 2Chang YH Kang EY Liu L Jenny LA Khang R Seo GH Maternal mosaicism in SSBP 1 causing optic atrophy with retinal degeneration: implications for genetic counseling Orphanet J Rare Dis 202318113110.1186/s 13023-023-02748-937259171 PMC 10233871 · doi ↗ · pubmed ↗
- 3Zarrei M Mac Donald JR Merico D Scherer SWA copy number variation map of the human genome Nat Rev Genet 20151631728310.1038/nrg 387125645873 · doi ↗ · pubmed ↗
- 4Zampaglione E Kinde B Place EM Navarro-Gomez D Maher M Jamshidi F Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations Genet Med 202022610798710.1038/s 41436-020-0759-832037395 PMC 7272325 · doi ↗ · pubmed ↗
- 5Fuhrmann N Alavi MV Bitoun P Woernle S Auburger G Leo-Kottler B Genomic rearrangements in OPA 1 are frequent in patients with autosomal dominant optic atrophy J Med Genet 20094621364410.1136/jmg.2008.06257019181907 · doi ↗ · pubmed ↗
- 6Weisschuh N Schimpf-Linzenbold S Mazzola P Kieninger S Xiao T Kellner U Mutation spectrum of the OPA 1 gene in a large cohort of patients with suspected dominant optic atrophy: identification and classification of 48 novel variants P Lo S ONE 2021167 e 025398710.1371/journal.pone.025398734242285 PMC 8270428 · doi ↗ · pubmed ↗
- 7Pacot L, Vidaud D, Sabbagh A, Laurendeau I, Briand-Suleau A, Coustier A et al. Severe phenotype in patients with large deletions of NF 1. Cancers (Basel). 2021;13(12).10.3390/cancers 13122963 PMC 823197734199217 · doi ↗ · pubmed ↗
- 8Marshall AE, Roes MV, Passos DT, De Weerd MC, Chaikovsky AC, Sage J et al. RB 1 deletion in retinoblastoma protein pathway-disrupted cells results in DNA damage and Cancer progression. Mol Cell Biol. 2019;39(16).10.1128/MCB.00105-19PMC 666460331138663 · doi ↗ · pubmed ↗