Data reduction when aggregating information about harms associated with medical interventions
Edoardo Giuseppe Ostinelli, Toshi A Furukawa

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
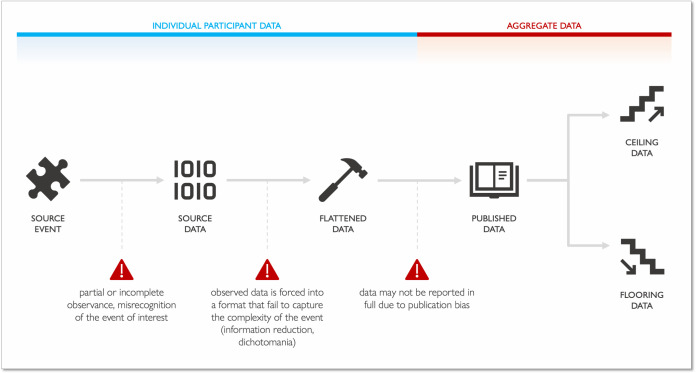 Figure 1
Figure 1- —NIHR Oxford Health Biomedical Research Centre
- —http://dx.doi.org/10.13039/501100023233National Institute for Health and Care Research Applied Research Collaboration Oxford and Thames Valley
- —http://dx.doi.org/10.13039/100010368Brasenose College, University of Oxford
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPharmacovigilance and Adverse Drug Reactions · Statistical Methods in Clinical Trials · Health Systems, Economic Evaluations, Quality of Life
In interpreting and aggregating data in published reports, readers and authors must be aware that some data loss and transformation are inevitable in the process (figure 1).1 Kamp and colleagues recently examined the beneficial and adverse event (AE) profiles of tricyclic antidepressants in a systematic review of available evidence from randomised controlled trials. The authors identified 103 trials randomising 10 590 participants, concluding that in the short term these medications may reduce depressive symptoms (mean difference on the 17-item Hamilton Rating Scale for Depression of −3.77, 95% CIs −5.91 to –1.63; 17 studies; low certainty of evidence) and increase the chances of ‘serious AEs’ (SAEs) (OR 2.78, 95% CI 2.18 to 3.55; 35 trials; very low certainty of evidence) compared with placebo.2
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), the European Medicines Agency, and the Food and Drug Administration define AEs as ‘any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have to have a causal relationship with this treatment’. An AE is considered serious and thus cause regulatory implications when it ‘results in death, is life-threatening, requires inpatient hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability/incapacity, or is a congenital anomaly/birth defect’, with each criterion being evaluated at a patient and event level.3–5 For instance, ‘the term life-threatening in the definition of serious refers to an event in which the patient was at risk of death at the time of the event, rather than an event which hypothetically might have caused death if it were more severe’.
In their systematic review, Kamp and colleagues applied their own judgement in categorising specific AEs as non-serious or serious, ultimately consisting in a worst-case scenario when severity details were considered inadequately reported by the original authors.2 For instance, ‘taste alteration/perversion’ was considered an SAE occurring in 26 out of 677 participants enrolled in four studies (figure S18, Kamp and colleagues).2 Moreover, they had access only to aggregate data to evaluate the seriousness of AEs. Not all the AEs that, on average, are associated to additional care are SAEs at an individual level (eg, not all individuals experiencing blurred vision will require hospitalisation or will be in a life-threatening condition; figure S16, Kamp and colleagues).2 The widely accepted definition of SAE appeared in the mid-1990s.3 As 11 out of the 103 studies contributing to the primary outcomes were published after 2000, it is expected that the original investigators did not report the exact numbers of SAEs as currently understood.
When observed clinical information (source event) is translated into source data at the collection site, how data are measured will set implications downstream (figure 1). This is where data flattening can occur, a process where data are simplified via reduction of their number of dimensions (eg, instead of measuring a variable as continuous, it is categorised into an ordinal variable or dichotomised). This may happen voluntarily to reduce the amount of information stored or to avoid collecting data that are considered not relevant. After data are flattened, restoration of lost information is not possible, with imputation being the only possible solution.6 External researchers are limited to flattened aggregate data reported by original authors (published data). When seriousness of AEs is not clearly reported, researchers can (1) renounce to use that data, (2) consider all the events as non-serious (flooring, best-case scenario) or (3) consider all as serious (ceiling) based on the average outcome (ie, a specific AE usually results in hospitalisation) or the worst outcome (ie, a specific AE may worsen and result in hospitalisation, worst-case scenario). Any of these assumptions might generate deviations from the truth and should be carefully examined and discussed.7
It is truly important to understand the absolute and relative frequencies of AEs and potential harms associated with medical interventions.8 We applaud the authors’ efforts towards this goal. Access to individual participant data can overcome reporting bias but, to limit information loss, what data should be collected and in which format should follow rigorous criteria widely established across the regulatory and scientific communities (standardisation). This is increasingly important given their emerging role in shared decision-making processes and patient decision aids aimed at identifying who may be at higher risk of experiencing harms.9
For the very same reason, it is essential to rely on a lingua franca of research on medical interventions when referring to aggregate data on harms.^ 9 ^ Additionally, uniformity on characterisation and format of safety data would translate into dataset harmonisation across studies, countries and sponsors, introducing several benefits10: federated analyses would bypass data sharing agreement while preserving individual patient’s privacy, increasing access to data and maximising transparency11; linking harmonised data to pharmacovigilance repositories would be facilitated, allowing real-time contribution of multiple data sources into a synchronous environment.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ioannidis JPA , Evans SJW , Gøtzsche PC , et al . Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med 2004;141:781–8. 10.7326/0003-4819-141-10-200411160-00009 15545678 · doi ↗ · pubmed ↗
- 2Kamp CB , Petersen JJ , Faltermeier P , et al . Beneficial and harmful effects of tricyclic antidepressants for adults with major depressive disorder: a systematic review with meta-analysis and trial sequential analysis. BMJ Ment Health 2024;27:e 300730. 10.1136/bmjment-2023-300730 PMC 1080686939093721 · doi ↗ · pubmed ↗
- 3International conference on Harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH). In: Clinical safety data management: definitions and standards for expedited reporting (E 2A). ICH, 1994. Available: https://database.ich.org/sites/default/files/E 2A_Guideline.pdf [accessed 26 Jan 2024].10.1111/j.1365-2125.1994.tb 05705.x PMC 13648938054244 · doi ↗ · pubmed ↗
- 4European medicines Agency (EMA). guideline for good clinical practice E 6(R 2). EMA; 2016. Available: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-good-clinical-practice-e 6r 2-step-5_en.pdf [Accessed 26 Jan 2024].
- 5Food and Drug Administration (FDA) . What is a serious adverse event? FDA; 2023. Available: https://www.fda.gov/safety/reporting-serious-problems-fda/what-serious-adverse-event [Accessed 26 Jan 2024].
- 6Ioannidis JP , Lau J . Completeness of safety reporting in randomized trials: an evaluation of 7 medical areas. JAMA 2001;285:437–43. 10.1001/jama.285.4.437 11242428 · doi ↗ · pubmed ↗
- 7Chou R , Helfand M . Challenges in systematic reviews that assess treatment harms. Ann Intern Med 2005;142(12 Pt 2):1090–9. 10.7326/0003-4819-142-12_part_2-200506211-00009 15968034 · doi ↗ · pubmed ↗
- 8Ziegler DK , Mosier MC , Buenaver M , et al . How much information about adverse effects of medication do patients want from physicians Arch Intern Med 2001;161:706–13. 10.1001/archinte.161.5.706 11231703 · doi ↗ · pubmed ↗