A Rare Case of Extranodal Natural Killer/T-cell Lymphoma, Nasal Type Associated With Hemophagocytic Lymphohistiocytosis in a Patient With Recurrent Sinusitis
Bryce R Christensen, Chung-ting J Kou, Lauren E Lee

TL;DR
A rare case of nasal-type extranodal natural killer/T-cell lymphoma complicated by hemophagocytic lymphohistiocytosis is described, highlighting diagnostic challenges and successful treatment.
Contribution
This case report highlights the rare association between ENKL nasal-type and HLH, emphasizing the importance of early diagnosis and treatment.
Findings
The patient's symptoms and pathology were initially misinterpreted as invasive sinusitis or fungal disease.
Diagnosis of ENKL nasal-type was confirmed with EBV-encoded RNA staining and histology.
Treatment with M-SMILE regimen and stem cell transplant led to complete remission.
Abstract
We present a rare case of hemophagocytic lymphohistiocytosis (HLH) secondary to nasal-type extranodal natural killer/T-cell lymphoma (ENKL). Nasal-type ENKL is a rare subtype of non-Hodgkin's lymphoma usually associated with Epstein-Barr virus (EBV). The patient was a 19-year-old woman who presented with facial numbness, diminished hearing, and dysgeusia. She was febrile with palatal necrosis, loss of gag reflex, and cranial nerve palsies. Labs revealed neutropenia. Broad-spectrum antimicrobials, including amphotericin, were started. Given concern for invasive fungal disease, she underwent surgical debridement, which revealed inflamed fibrous tissue and extensive necrosis. Pathology showed no fungal elements or malignancy. Lack of clinical improvement and worsening palatal necrosis prompted additional debridement. Histology identified an atypical CD3+/CD56+ cellular infiltrate. Bone…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
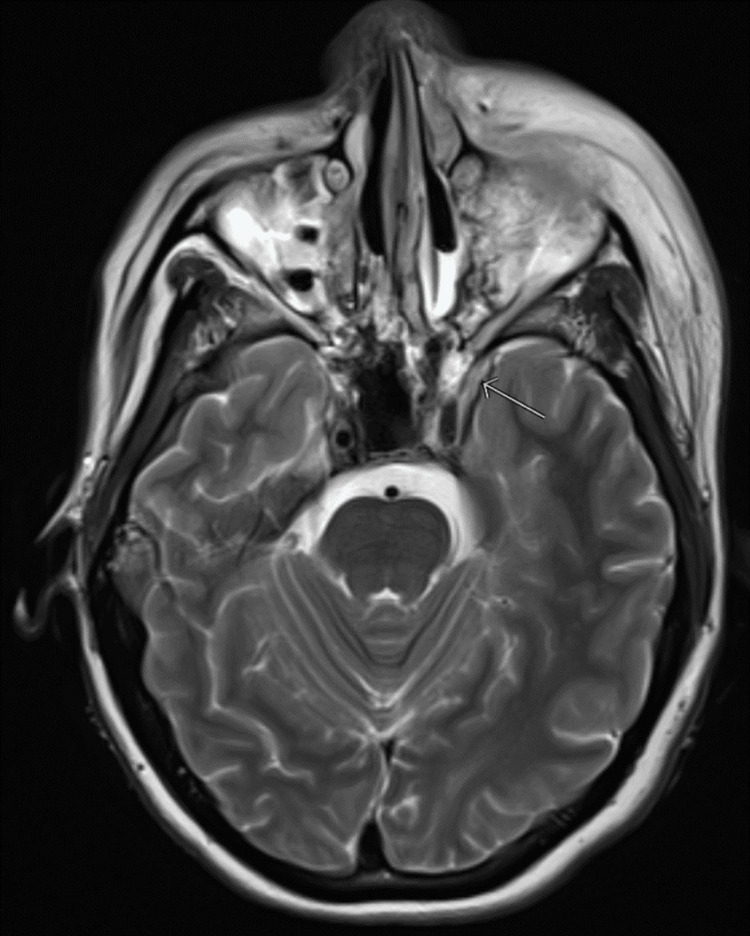 Figure 1
Figure 1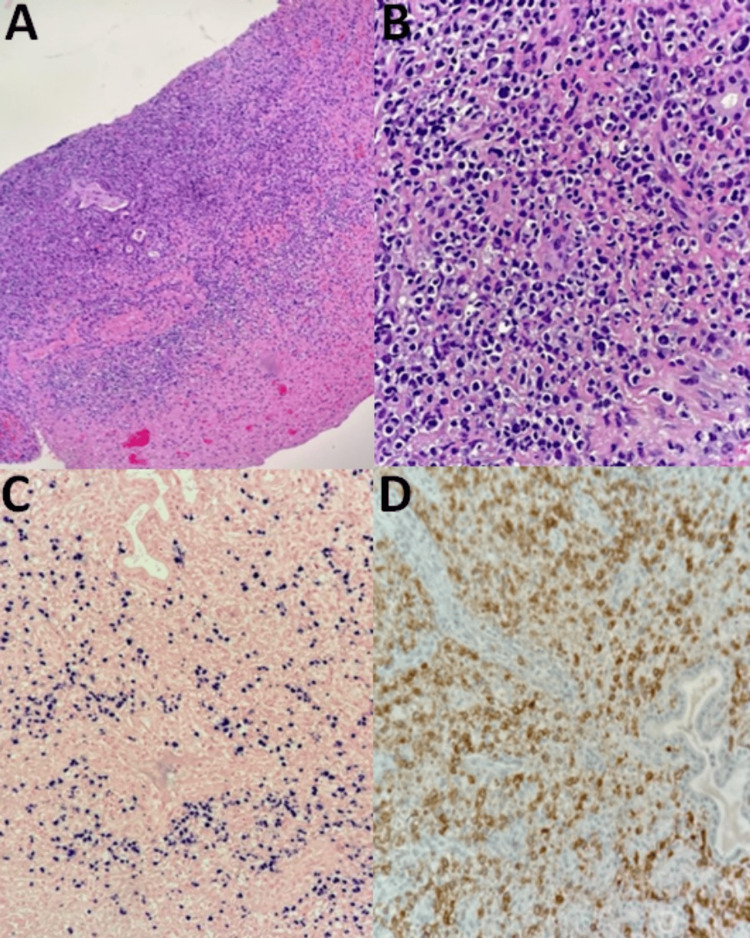 Figure 2
Figure 2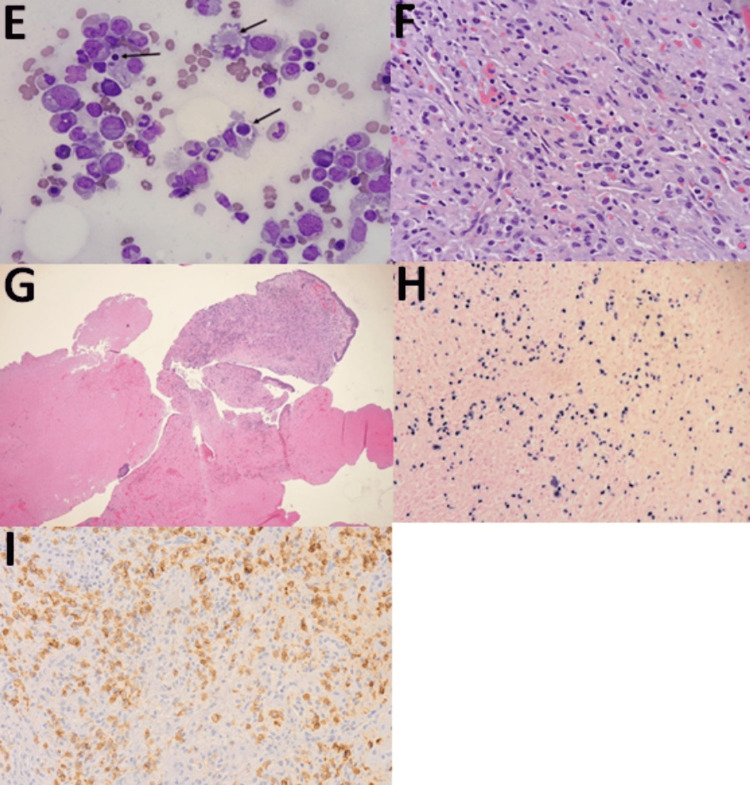 Figure 3
Figure 3| Laboratory study | Value | Reference range |
| White blood cell (WBC) | 1,930 WBC/μL | 4,500 to 11,000 WBCs/μL |
| Absolute neutrophil count (ANC) | 1,310 ANC/μL | 2,500 to 6,000 ANC/μL |
| Platelets | 114,000 platelets/μL | 150,000 to 450,000 platelets/μL |
| Aspartate aminotransferase (AST) | 101 U/L | ≤33 U/L |
| Alanine aminotransferase (ALT) | 112 U/L | 10 to 49 U/L |
| Ferritin | 1,788 ng/mL | 12 to 150 ng/mL |
| Epstein-Barr virus quantitative level | 11,518 IU/mL | 0 IU/mL |
| Original SMILE | Modified SMILE | |
| Medication 1 | Methotrexate 2g/m2 (with leucovorin 15 mg) days 1, 2, 3, and 4 | Methotrexate 2g/m2 (with leucovorin 25 mg) days 1, 2, 3, and 4 |
| Medication 2 | Ifosfamide 1,500 mg/m2 (with mesna 300 mg/m2) days 2, 3, and 4 | Ifosfamide 1,500 mg/m2 (with mesna 300 mg/m2) days 2, 3, and 4 |
| Medication 3 | Dexamethasone 40 mg/d days 2, 3, and 4 | Dexamethasone 40 mg/d days 2, 3, and 4 |
| Medication 4 | Etoposide 100 mg/m2 days 2, 3, and 4 | Etoposide 100 mg/m2 days 2, 3, and 4 |
| Medication 5 | L-asparaginase 6000 U/m2 days 8, 10, 12, 14, 16, 18, and 20 | Peg-asparaginase 1500 U/m2 ~ day 7 (range: from day 5 to 31) |
| Cycle duration | 28 days | 21 days |
| Radiotherapy | None | 45 Gray |
| Expected cycles | 6 | 2 (localized disease), 3-4 (advanced disease) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research · Immune Cell Function and Interaction · Viral-associated cancers and disorders
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare, life-threatening syndrome caused by pathologic activation of the immune system [1]. While most commonly associated with inherited HLH gene mutations in pediatric patients [2], it may also be sporadic and associated with autoimmune disease, infection, or malignancy [3]. Genetic defects for familial HLH frequently map to genes coding for cytotoxic granule formation [4,5]. *PRF1 *- a gene encoding perforin - was one of the first genes discovered to be associated with familial HLH; other associated mutations include UNC13D, STXBP2, Rab27a, STX11, SH2D1A, and *XIAP *[3].
The pathogenesis of HLH involves macrophages, natural killer (NK) cells, and cytotoxic lymphocytes [1]. The disease results from a failure of regulatory pathways that terminate inflammatory responses. The immune system removes cells that are either unnecessary or dangerous. However, the immune system can be tempered to avoid the removal of useful cells [6]. When downregulation of the immune system fails, the resulting hyperinflammatory state can lead to HLH [6]. Symptoms may include splenomegaly, hepatomegaly, and fevers. Cytopenias, elevated triglycerides, and decreased fibrinogen may also be apparent [3]. Hemophagocytosis may be present on bone marrow biopsy, but this is not pathognomonic and may be present in as few as 25% of cases [7].
In a study of 369 patients with HLH, Bergsten et al. [8] showed the presence of fever (95% of patients), splenomegaly (89%), bicytopenia (92%), hypertriglyceridemia or hypofibrinogenemia (90%), hemophagocytosis (82%), ferritin > 500 mcg/L (94%), low to absent NK cell activity (71%), and elevated soluble CD25 elevation (97%). Patients with HLH may present with a sepsis-like picture with fever and performance status deterioration.
The results of the HLH-2004 trial led to the eight diagnostic criteria for HLH: temperature ≥ 38.3°C, splenomegaly, cytopenias (hemoglobin < 9 g/dL for adults, platelets less than 100 x 103/mL, or neutrophils less than 1 x 103/mL) affecting at least two of three lineages in peripheral blood, either hypertriglyceridemia (>265 mg/dL) or hypofibrinogenemia (<150 mg/dL), hemophagocytosis found in the bone marrow, spleen, lymph nodes, or liver, low to absent NK cell activity (<10 lytic units), ferritin > 500 ng/mL, and soluble CD25 ≥2400 U/mL [3,8-10]. Genetic testing may demonstrate homozygous HLH-associated mutations in children or heterozygous HLH-associated mutations with clinical findings of HLH in adults [2,11] in cases of primary HLH. Additionally, CXC chemokine ligand (CXCL) levels have been utilized as novel markers to help distinguish lymphoma-associated HLH from sepsis, as well as markers of response to treatment [12,13].
Unfortunately, some patients with HLH do not meet the above criteria, and untreated HLH carries significant mortality. As such, the diagnosis can sometimes be presumed - and treatment initiated - if clinical suspicion for HLH is high [1]. The Histiocyte Society provided modified HLH diagnostic criteria in 2009, which require any of the following three criteria: molecular diagnosis; three out of the four findings: fever, splenomegaly, cytopenias with a minimum of two cell lines reduced, and/or hepatitis; and presence of at least one of the following: hemophagocytosis, elevated ferritin, elevated CD25, and/or absent or very decreased NK function. The other findings of hypertriglyceridemia, hypofibrinogenemia, and hyponatremia are supportive of HLH diagnosis, but not required [1].
Treatment of HLH dramatically increases survival from only months without treatment, to five or more years with treatment [8,10,14]. Therapeutic regimens often include dexamethasone and etoposide, in addition to allogeneic hematopoietic stem cell transplantation (HSCT) in patients with genetic, relapsing, severe, or persistent disease [8,10,14]. Intrathecal methotrexate and hydrocortisone are also used in patients with involvement of the central nervous system [8,10,14].
The patient presented in this report is one such case where HLH diagnosis was difficult. The suspected culprit initially for her presentation was infection resulting in a delayed diagnosis. Initiation of treatment led to a dramatic improvement in her disease-related symptoms. We present this case to highlight the diagnostic and therapeutic challenges of HLH.
Case presentation
A 19-year-old woman presented to the emergency room with acute-on-chronic sinusitis, nasal obstruction, and left periorbital swelling with purulent discharge. Computed tomography (CT) of the sinuses showed pansinusitis with findings suggestive of preseptal cellulitis. She was admitted for empiric board-spectrum antimicrobials and functional endoscopic sinus surgery with bilateral middle meatal antrostomies.
Due to persistent and progressive symptoms despite completion of appropriate antimicrobial treatment, a repeat CT of the sinus and face was obtained. CT of the sinus and face demonstrated a medial canthus abscess with left periorbital cellulitis and intraorbital myositis. The patient developed left-sided facial and palatal numbness, diminished left-sided hearing, and loss of gag reflex. The patient was transferred to a large military treatment facility for further management. Upon arrival, her vitals were normal. The exam showed significant left eye edema impairing her ability to open her eye independently, impaired sensation to light touch in the left maxillary branch of the trigeminal nerve distribution, diminished left-sided hearing, and a palate that did not elevate. Laboratory assessment (Table 1) revealed a white blood cell (WBC) count of 1,930 per microliter (μL), absolute neutrophil count (ANC) of 1,310 per μL, and thrombocytopenia with platelets of 114,000 per μL. Liver enzymes were elevated, with aspartate aminotransferase (AST) of 101 units per liter (U/L) and alanine aminotransferase (ALT) of 112 U/L. Ferritin was elevated at 1,788 nanograms per milliliter (ng/mL).
Interval CT imaging of the face, chest, abdominal, and pelvis was repeated and was consistent with prior imaging concerning for invasive fungal sinusitis. Brain magnetic resonance imaging (MRI) revealed mucosal non-enhancement in the left maxillary and left sphenoid sinus cavities, with adjacent inflammatory findings suggestive of invasive fungal sinusitis with possible early epidural involvement (Figure 1). Magnetic resonance venography was normal. She underwent extensive debridement by otolaryngology. Broad-spectrum antibiotics as well as voriconazole and amphotericin were initiated for suspected invasive bacterial/fungal infections. A transthoracic echocardiogram demonstrated a mitral valve mobile density concerning for infective endocarditis secondary to a hematogenous infection.
MRI depicting intracranial findings
During the patient’s hospitalization, she developed transfusion-dependent anemia and neutropenia without an identifiable etiology. Hematology/oncology was consulted and a bone marrow biopsy was performed. Neutropenia was initially supported with filgrastim with an adequate response. A maxillary sinus tissue culture from the initial debridement was negative for fungus. However, methicillin-sensitive Staphylococcus aureus and Streptococcus sanguinis were identified on the maxillary sinus tissue culture. The patient’s empiric antimicrobials were de-escalated accordingly based on tissue culture data. Despite these interventions, the patient’s status did not improve, prompting a repeat nasal endoscopy and debridement with the goal of improving access for nasal irrigation and obtaining additional diagnostic tissue. Interval CT of the chest, abdomen, and pelvis showed nonspecific hepatosplenomegaly without lymphadenopathy.
The bone marrow biopsy demonstrated hemophagocytosis and serum interleukin-2 receptor (IL-2R) was elevated. In the clinical context of fevers, hepatosplenomegaly, cytopenias, elevated ferritin, elevated IL-2R, and hemophagocytosis on bone marrow biopsy, high-dose steroids were initiated for suspected HLH secondary to bacterial infection. Germline testing for primary HLH was obtained and negative for genetic HLH. A transesophageal echocardiogram was performed and was negative for vegetation. Although HLH was initially attributed to a bacterial sinus infection, additional operative debridement by otolaryngology was performed given HLH’s association with malignancy and other non-infectious etiologies. Flow cytometry of nasal tissue was non-diagnostic due to inadequate tissue sampling. Flow cytometry of cerebral spinal fluid was negative for malignancy. Immunohistochemistry of the right posterior ethmoid biopsy demonstrated CD56, CD3, and Epstein-Barr virus (EBV) positivity with myogenin and anaplastic lymphoma kinase (ALK) negativity, consistent with extranodal NK/T-cell lymphoma (ENKL), nasal-type (Figures 2, 3).
Histology slides (panel 1 of 2)A: Nasopharynx biopsy: hematoxylin and eosin stain, 10x magnification.B: Nasopharynx biopsy: hematoxylin and eosin stain, 40x magnification.C: Nasopharynx biopsy: Epstein-Barr virus positivity, 20x magnification.D: Nasopharynx biopsy: CD56+, 20x magnification.
Histology slides (panel 2 of 2)E: Bone marrow biopsy: Wright-Giemsa, 1000x magnification. This bone marrow aspirate on oil shows prominent hemophagocytic cells, indicated by arrows.F: Nasopharynx biopsy: hematoxylin and eosin stain, 400x magnification.G: Nasopharynx biopsy: hematoxylin and eosin stain, 40x magnification. Nasal curetting showed bone and respiratory mucosa with prominent necrotic material.H: Nasopharynx biopsy: Epstein-Barr virus positivity, 200x magnification. This is an Epstein-Barr virus in situ hybridization. The neoplastic cells are positive for the Epstein-Barr virus.I: Nasopharynx biopsy: CD56+, 200x magnification.
Staging positron emission tomography (PET)-CT demonstrated abnormal fluorodeoxyglucose (FDG) avidity of the left orbital, paranasal sinuses, nasopharynx, and enlarged level two lymph nodes. Her serum EBV quantitative level was 11,518 international units per milliliter (IU/mL) (Table 1), portending a poorer prognosis. Human T-cell lymphotropic virus (HTLV) was negative, and cerebrospinal fluid assessment results were negative. The patient was staged as advanced disease given skull involvement complicated by neurological deficits and HLH. An anti-neoplastic regimen with dexamethasone, methotrexate, ifosfamide, l-asparaginase, and etoposide (modified-SMILE regimen) was started. Five cycles of modified-SMILE were administered with a total of 5000 centigray over 20 fractions of radiation therapy to the maxillary sinus region. The patient achieved a complete remission based on an undetectable serum EBV, negative nasal biopsy, and resolution of cranial nerve deficits. Surveillance MRI demonstrated a continued response to therapy with a small amount of left anterior extraconal orbital disease, with left interior and medial rectus muscle thickening suspected secondary to inflammation versus scarring. The patient proceeded with high-dose BEAM (carmustine, etoposide, cytarabine, and melphalan) conditioning followed by autologous bone marrow rescue. The patient continued to be in remission over two years after autologous bone marrow transplant.
Discussion
This patient presented with an uncommon disease that can mimic other pathologic processes, making diagnosis challenging. She presented with recurrent sinusitis that failed to improve with debridement and antimicrobial therapy, developed HLH, and was ultimately found to have ENKL, nasal-type.
HLH is a rare entity, with one study estimating an annual incidence of only one in 800,000 persons per year [15]. As previously mentioned, HLH can be primary, but approximately 90% of the time is secondary to infection or malignancy [15,16]. In fact, approximately 1/3 of patients test positive for EBV [16]. Further complicating this case was the fact that the patient fell into an age range where primary HLH was a consideration. Germline testing was negative but should be considered in cases where there is diagnostic uncertainty.
The morbidity and mortality of untreated HLH is high, with long-term survival rates as low as 4% without treatment [14]. Treatment can drastically improve survival, with three to five-year survival rates after treatment greater than 50% [14,16]. The diagnosis of HLH can be difficult, and as demonstrated with our patient, is frequently mistaken for infection [17].
ENKL, nasal-type, is an aggressive T-cell lymphoma, which accounts for 10% of all peripheral T-cell lymphomas in North America. ENKL has an increased incidence among Asian countries [18]. The median survival is 31 months for limited disease and six months for advanced disease based on data from the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER-18) between 2000 and 2018 [19].
Workup of ENKL includes dedicated imaging of nasal sinuses by either MRI or CT, as well as nasal endoscopy to assess for localized versus extra-nasal disease. Multiple biopsies may be necessary to establish a diagnosis, as extensive necrosis can obscure the results. The classic histological presentation of ENKL is extensive angioinvasion with necrosis and positive staining for CD2, CD56, cytoplasmic CD3, but not surface CD3, and cytotoxic markers [20]. Tissue biopsies must demonstrate the presence of EBV-encoded RNA by ISH [20].
Plasma EBV DNA should be obtained at the time of diagnosis and surveilled through treatment given its role in prognosis [20,21]. EBV DNA in the plasma has a higher specificity and sensitivity for EBV-positive disease as compared to EBV in peripheral blood mononuclear cells (PBMC), as demonstrated in a retrospective study of 2142 patients at Johns Hopkins Hospital clinical laboratory by Kanakry et al. [22]. EBV DNA is derived from the plasma in patients with EBV-positive tumors, as the malignant cells can circulate within the plasma, and EBV DNA levels obtained in a PBMC sample only reflect a fraction of circulating EBV tumor cells [22]. A prognostic model for ENKL, called the Prognostic Index for Natural Killer Lymphoma (PINK-E), risk stratifies patients into the following groups: low-risk (no risk factors), intermediate-risk (one risk factor), or high-risk (three or more risk factors) groups. The risk factors are: (1) age older than 60 years, (2) stage III or IV disease, (3) distant lymph node involvement, (4) non-nasal-type disease, and (5) detectable peripheral blood EBV titers [20,23]. The three-year overall survival rates associated with PINK-E low-risk, intermediate-risk, and high-risk categories are 81%, 55%, and 28%, respectively [23].
Due to extensive necrotic tissue mixed with hemorrhagic cells and inflamed respiratory mucosa on the initial nasal biopsy, our patient required two additional biopsies to establish extranodal NK/T-cell lymphoma, nasal-type, complicated by HLH. Our patient was considered low-risk based on the PINK-E model, with her only risk factor being a detectable plasma EBV DNA of 11,518 copies/mL. However, we elected to treat the patient as advanced disease, given her presentation with cranial nerve deficits secondary to mass effect and concomitant HLH.
Treatment of advanced disease consists of chemotherapy, but consolidation with radiation therapy (RT) or autologous HSCT may provide additional benefits [20,24,25]. NK cells express high concentrations of multidrug-resistant P-glycoprotein, making anthracycline-containing regimens ineffective [26]. Chemotherapy options include SMILE (dexamethasone, methotrexate, ifosfamide, l-asparaginase, and etoposide), which is administered every 28 days. Less toxic alternatives such as DDGP (dexamethasone, cisplatin, gemcitabine, and pegaspargase), AspaMETDex, and GemOX have also been studied [20,27]. SMILE treatment was initially evaluated in advanced and relapsed disease, with a high rate of objective response rate (ORR) of 79% in a phase II trial by Yamaguchi et al. of 38 eligible patients after two cycles [28]. However, there were high toxicity rates, with grade four neutropenia being the most common at 92%. Other grade four toxicities include infection (n = 6), hyperbilirubinemia (n = 1), alanine transaminase elevation (n = 2), and encephalopathy (n = 1) [28]. Due to the high risk of morbidity and mortality, SMILE is avoided in patients with poor performance status, lymphopenia (<500/uL lymphocytes), or a high tumor burden [20].
In limited-stage ENKL, RT is associated with improved overall survival (OS), as defined by prospective trials [29,30]. However, the benefit of radiation is less obvious in advanced diseases. Bi et al. demonstrated that post-chemotherapy radiotherapy was associated with a two-year OS of 57.5% vs. 14.5% compared to chemotherapy alone in patients with stage III or IV NK/T-cell lymphoma [24]. A modified SMILE (mSMILE) regimen consisting of dexamethasone, methotrexate, ifosfamide, peg-asparaginase, and etoposide given every three weeks followed by RT with 45 gray (Gy) has been adopted by Memorial Sloan Kettering Center [31]. A 21-day cycle was adopted to provide patients the opportunity to quickly transition to RT after two cycles of mSMILE [31]. Table 2 compares the SMILE and mSMILE regimens.
Ghione et al. demonstrated with mSMILE an ORR of 100% for stage III-IV ENKL, as well as an OS of 100% at the median follow-up at 31 months for both limited and extensive stage ENKL [31]. Our 19-year-old patient completed five cycles of mSMILE with grade two mucositis, oral candidiasis, and Clostridioides difficile infection. She received RT to the maxillary sinus region with 45 Gy over 20 fractions between cycles three and four of SMILE. After completion of chemotherapy and RT, the patient was in complete remission as indicated by an undetectable plasma EBV DNA, negative endoscopic biopsies, and imaging.
The role of HSCT is well established in lymphomas such as relapsed diffuse large B-cell lymphoma and relapsed classic Hodgkin's lymphoma. The role of HSCT in ENKL is less clear. A multinational, multicenter, matched controlled retrospective analysis of 59 patients with ENKL from Japan, Korea, and Hong Kong found a significant survival advantage with autologous HSCT in patients with advanced disease who achieve a clinical remission (CR) at the time of transplantation [32]. A similar retrospective study of ENKL patients in the Western hemisphere by Fox et al. demonstrated better two-year disease-free survival in patients who achieved CR at the time of autologous HSCT compared to patients not in CR [33]. Autologous HSCT is routinely not recommended in limited disease, as the Lee et al. matched analysis demonstrated no improvement in limited disease patients [32]. The American Society of Blood and Marrow Transplantation supports the use of autologous HSCT in relapsed chemo-sensitive localized NK/T-cells [34]. Autologous HSCT (strongly) and allogenic HSCT (weakly) are recommended for disseminated NK/T-cell as front-line consolidation and chemo-sensitive relapsed disseminated disease [34]. Allogenic HSCT is weakly recommended for primary relapsed or relapsed refractory disease [34]. Though our patient’s PET-CT imaging was consistent with contiguous stage IIE disease, she was treated as an advanced disease patient with concurrent cranial nerve deficits and HLH at presentation. The patient underwent BEAM conditioning with autologous HSCT rescue after achieving CR with mSMILE and RT. Two years after mSMILE with RT followed by consolidative autologous HSCT, the patient remained in CR.
Conclusions
We report a case of a patient presenting with recurrent sinusitis who was ultimately diagnosed with ENKL after multiple tissue biopsies with a hematological course complicated by concomitant HLH. Early recognition of HLH and treatment of the underlying etiology is imperative to reducing its high morbidity and mortality. Our case highlights ENKL, a rare subtype of lymphoma caused by EBV, as an etiology of HLH. Pathologic diagnosis of ENKL is challenging due to extensive necrosis, which often necessitates multiple biopsies to establish a diagnosis. Limited-stage ENKL is managed with asparaginase-based chemotherapy with radiation therapy. The optimal treatment for advanced and relapsed ENKL is less defined. Further studies are needed to elucidate the role of hematopoietic stem cell transplantation for advanced and relapsed ENKL.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hemophagocytic lymphohistiocytosis (HLH) and related disorders Hematology Am Soc Hematol Educ Program Filipovich AH 12713120092000819010.1182/asheducation-2009.1.127 · doi ↗ · pubmed ↗
- 2Hypomorphic mutations in PRF 1, MUNC 13-4, and STXBP 2 are associated with adult-onset familial HLH Blood Zhang K Jordan MB Marsh RA 5794579811820112188104310.1182/blood-2011-07-370148 PMC 3228496 · doi ↗ · pubmed ↗
- 3Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO)Pediatr Blood Cancer Jordan MB Allen CE Greenberg J 066201910.1002/pbc.27929 PMC 734008731339233 · doi ↗ · pubmed ↗
- 4A functional analysis of the putative polymorphisms A 91V and N 252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis Blood Voskoboinik I Thia MC Trapani JA 4700470610520051575589710.1182/blood-2004-12-4935 · doi ↗ · pubmed ↗
- 5Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis Am J Hum Genet Göransdotter Ericson K Fadeel B Nilsson-Ardnor S 5905976820011117900710.1086/318796 PMC 1274472 · doi ↗ · pubmed ↗
- 6Histiocytic disorders: recent insights into pathophysiology and practical guidelines Biol Blood Marrow Transplant Filipovich A Mc Clain K Grom A 0916201010.1016/j.bbmt.2009.11.01419932759 · doi ↗ · pubmed ↗
- 7Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients Am J Med Rivière S Galicier L Coppo P Marzac C Aumont C Lambotte O Fardet L 1118112512720142483504010.1016/j.amjmed.2014.04.034 · doi ↗ · pubmed ↗
- 8Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study Blood Bergsten E Horne A AricóM 2728273813020172893569510.1182/blood-2017-06-788349 PMC 5785801 · doi ↗ · pubmed ↗