Identification of Dhx15 as a Major Regulator of Liver Development, Regeneration, and Tumor Growth in Zebrafish and Mice
Irene Portolés, Jordi Ribera, Esther Fernandez-Galán, Elena Lecue, Gregori Casals, Pedro Melgar-Lesmes, Guillermo Fernández-Varo, Loreto Boix, Marco Sanduzzi, Veenu Aishwarya, Maria Reig, Wladimiro Jiménez, Manuel Morales-Ruiz

TL;DR
This study shows that the RNA helicase Dhx15 is crucial for liver development, regeneration, and tumor growth in zebrafish and mice, and its levels are elevated in patients with liver cancer.
Contribution
The study identifies Dhx15 as a key regulator of liver physiology and tumor growth in multiple species and explores its potential as a diagnostic marker.
Findings
Dhx15 deficiency impairs liver development in zebrafish and regeneration in mice.
Dhx15 silencing reduces tumor growth in a hepatocellular carcinoma model.
Elevated Dhx15 levels in blood are observed in patients with liver cancer.
Abstract
RNA helicase DHX15 plays a significant role in vasculature development and lung metastasis in vertebrates. In addition, several studies have demonstrated the overexpression of DHX15 in the context of hepatocellular carcinoma. Therefore, we hypothesized that this helicase may play a significant role in liver regeneration, physiology, and pathology. Dhx15 gene deficiency was generated by CRISPR/Cas9 in zebrafish and by TALEN-RNA in mice. AUM Antisense-Oligonucleotides were used to silence Dhx15 in wild-type mice. The hepatocellular carcinoma tumor induction model was generated by subcutaneous injection of Hepa 1-6 cells. Homozygous Dhx15 gene deficiency was lethal in zebrafish and mouse embryos. Dhx15 gene deficiency impaired liver organogenesis in zebrafish embryos and liver regeneration after partial hepatectomy in mice. Also, heterozygous mice presented decreased number and size of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
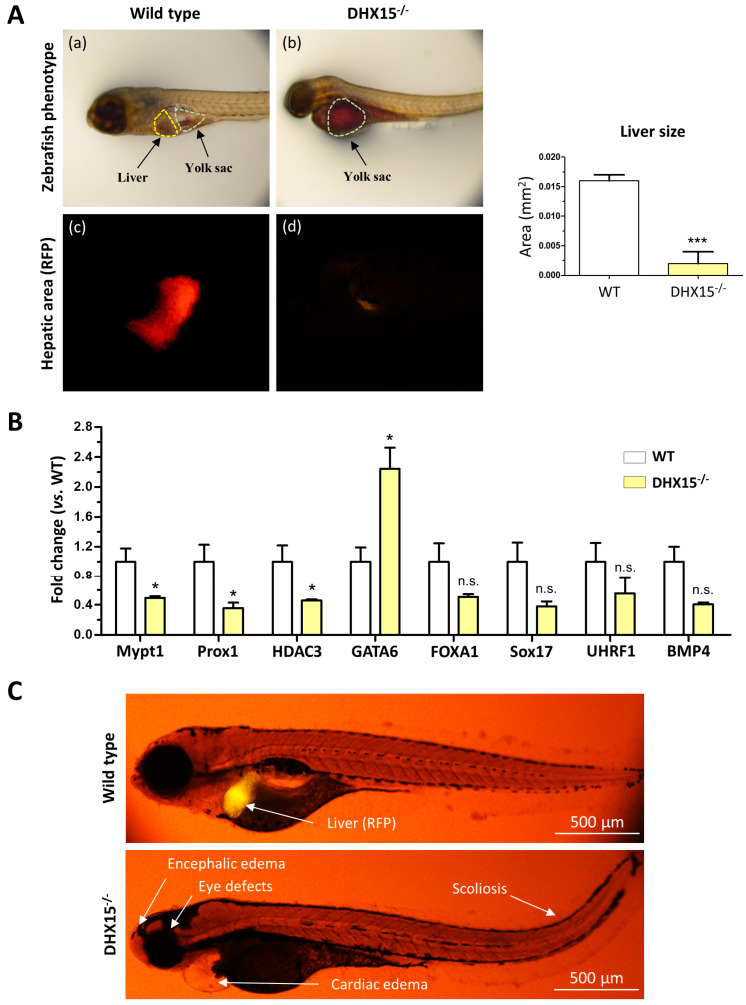 Figure 1
Figure 1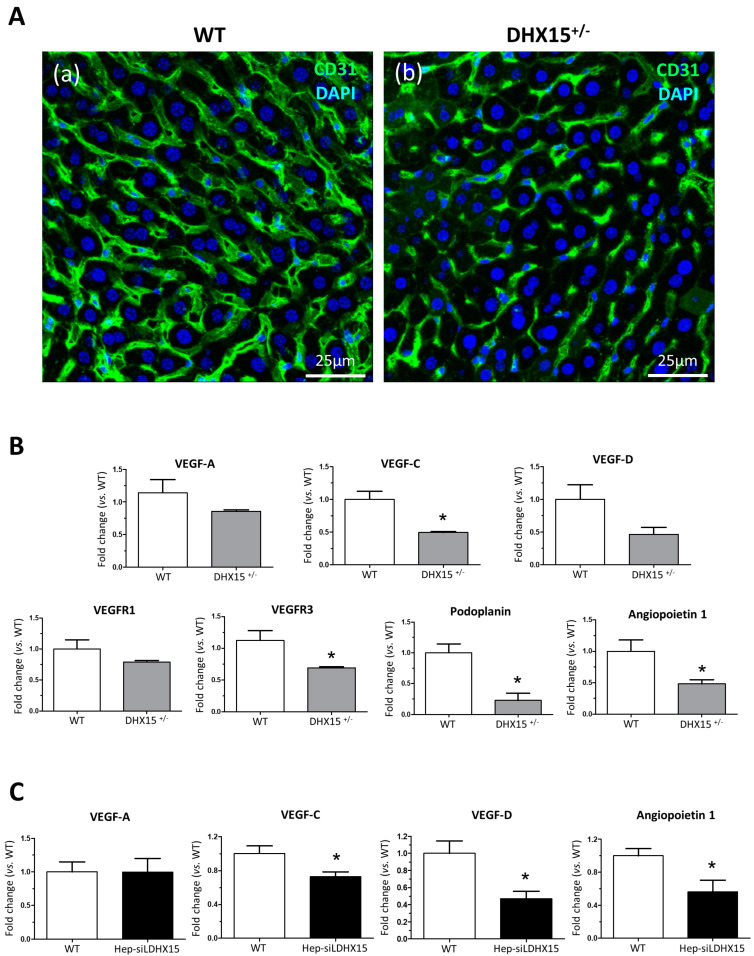 Figure 2
Figure 2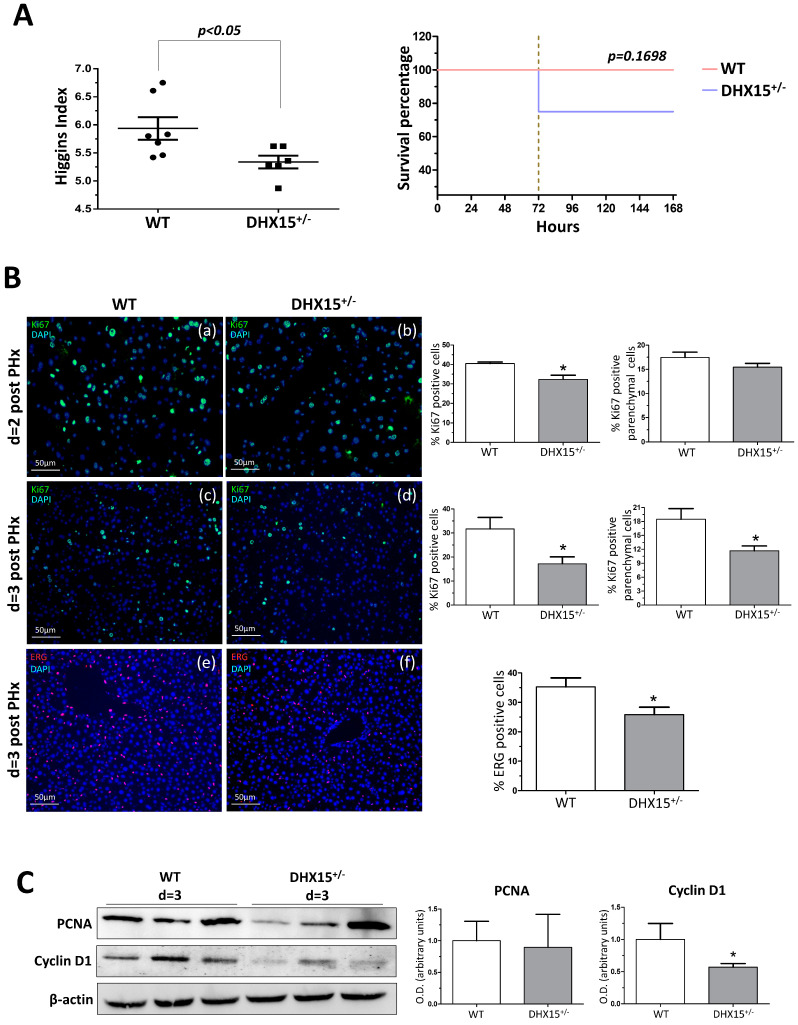 Figure 3
Figure 3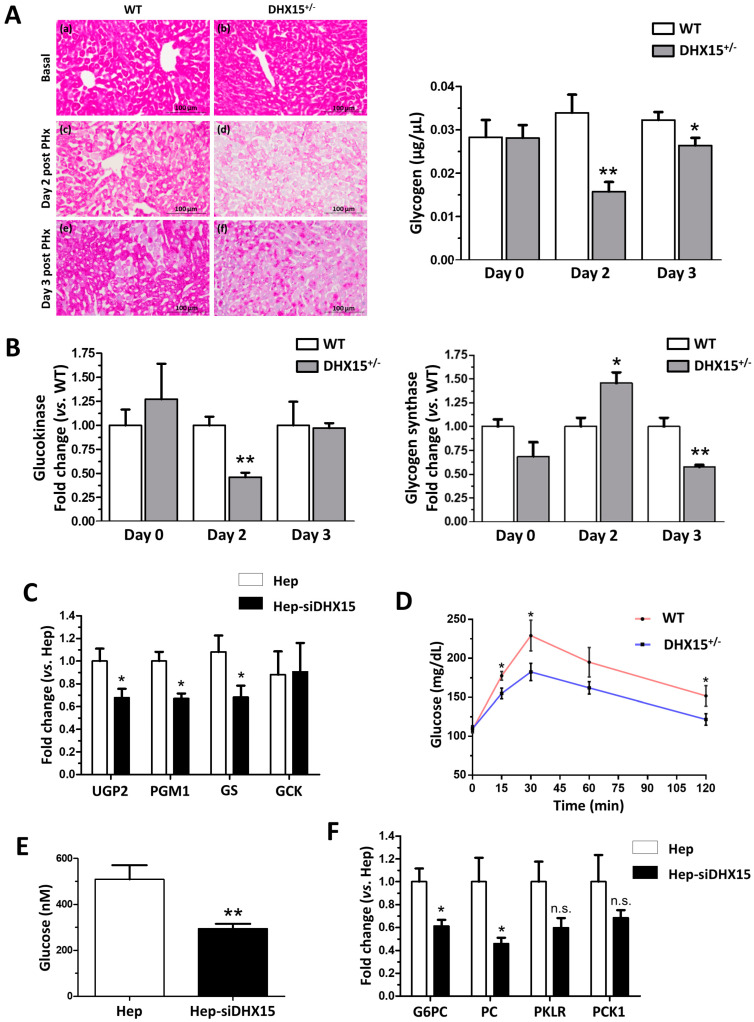 Figure 4
Figure 4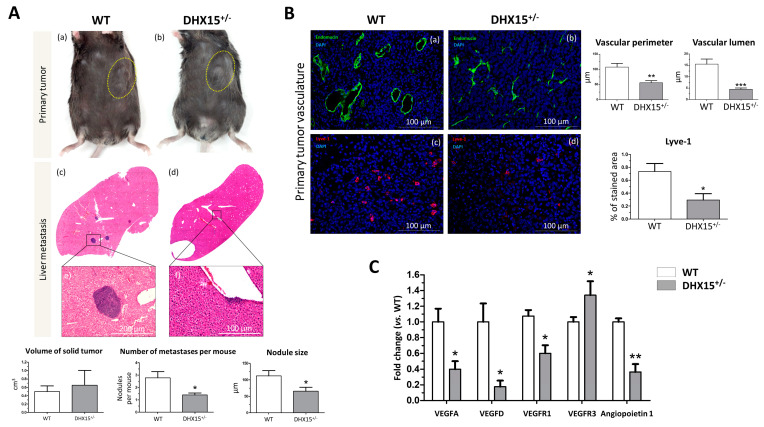 Figure 5
Figure 5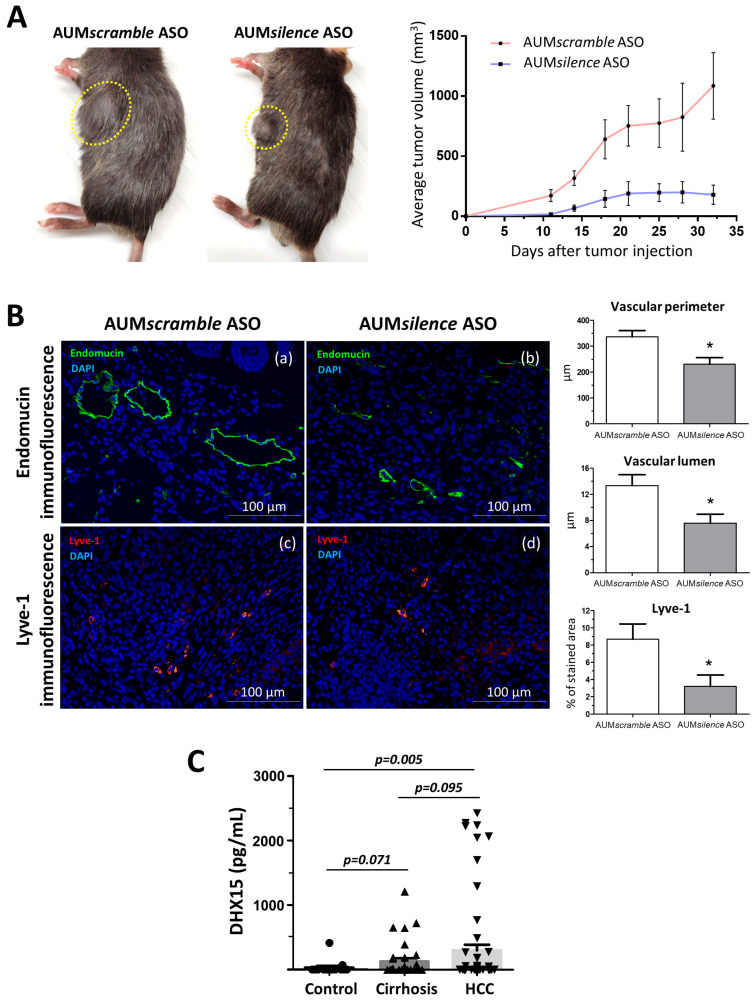 Figure 6
Figure 6- —Agencia Estatal de Investigación
- —Programa Estatal del Plan Estatal de Investigación Científica, Técnica y de Innovación 2021–2023
- —Consolidated Research Group, Departament de Recerca I Universitats de la Generalitat de Catalunya
- —FSE “El FSE invierte en tu futuro”
- —Ramon y Cajal Program
- —Spanish Ministerio de Ciencia, Innovación y Universidades
- —Instituto de Salud Carlos III
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicroRNA in disease regulation · RNA Research and Splicing · Cancer-related molecular mechanisms research
1. Introduction
RNA helicases, mainly encompassing DEAD- and DEAH-box families, are highly conserved enzymes that participate in all processes of RNA metabolism, from transcription to decay, in an ATP-dependent manner. Each RNA helicase displays a specific function in a diverse number of RNA targets; however, many DEAD/DEAH-box helicases lack target specificity per se. For instance, G-patch proteins act as DEAH-box activators by binding and recruiting them to their action sites [1,2,3,4]. Upon target recognition, they exert its ATPase activity to remodel RNA [5].
We previously described that Dhx15 is a downstream target of Akt and that there is a regulatory crosstalk in the expression of both proteins [6,7]. DHX15 expression is not organ-specific and has an ubiquitous variable gene expression. DHX15 participates mainly in mRNA splicing by contributing to the dissociation of the spliceosome subunit U2 upon splicing completion. It is also known to participate in ribosome biogenesis by enhancing small subunit maturation and in viral infections by sensing double RNA strands and stimulating type I IFN and proinflammatory cytokines production [8,9].
Emerging interest has arisen in studying the interplay between helicases and cancer [10]. Mutations in splicing factors typically occur in many cancers; therefore, recent DHX15 studies have focused on elucidating its role in different types of cancer [11]. DHX15 acts as a cancer promoter in breast cancer, prostate cancer, acute myeloid leukemia, and hepatocellular carcinoma (HCC), and as an antitumor factor in glioma due to its growth inhibitory function [12,13,14,15,16]. We also described a relevant function of Dhx15 in lymphatic and blood vasculature development and functioning in vertebrates. In this context, we demonstrated the role of Dhx15 as a regulator of lung metastasis in a syngeneic mouse model of metastasis [7]. Dhx15 gene deficiency resulted in significantly reduced metastasis due to lymphatic vascular defects and impaired endothelial energy metabolism. Further studies analyzing the specificity of DHX15 in cancer and metastasis are necessary, especially as oncologic complications are highly related to metastasis appearance.
Despite the increasing interest in DHX15 and the role played by its upstream regulator Akt in liver function, regeneration, and cytoprotection [17,18,19], this helicase had not been studied in the liver. Recently, two different studies evaluated DHX15 expression in hepatocellular carcinoma patients, showing a differential expression of this helicase. Xie C. et al. described significant overexpression of DHX15 in human primary HCC correlated with poor survival [16]. Later, Zhao M. et al. described DHX15 as an inhibitor of autophagy that was less expressed in HCC tumor tissues [20]. Although both studies show contrasting results, these observations suggest that DHX15 is a protein target for HCC. Thus, it would be necessary to decipher the function of DHX15 in the liver and specifically in the context of HCC. Here, we explore its role in the liver in two different animal models, zebrafish and mouse. We analyze the effects of Dhx15 knockdown in liver organogenesis, liver vasculature, and liver regeneration. Furthermore, we evaluate the functions of Dhx15 in HCC and liver metastasis, in Dhx15 heterozygous mice and using the self-delivering AUMsilence ASO technology, providing evidence for its role in the regulation of metastasis and primary tumor growth.
2. Results
2.1. Impaired Liver Development in Dhx15-Deficient Zebrafish Embryos
As we and others previously described, Dhx15 deficiency in zebrafish embryos depicts phenotypic abnormalities and ultimately results in lethality at day 8 post-fertilization (dpf). Such mutant embryos are characterized by encephalic and cardiac edema, scoliosis, impaired neural/eye growth. and defective pectoral fin and jaw development [7,21]. To determine whether Dhx15 gene deficiency is also associated with liver organogenesis, we generated a Dhx15 knockout zebrafish model in a red fluorescent protein (RFP) background under the liver-specific promoter fab10. At 5 dpf, Dhx15^−/−^ embryos presented evident differences from their wild-type (WT) clutch-mates; at this stage, mutant embryos lack livers (Figure 1A). By means of fluorescent microscopy, we specifically visualized the hepatic area marked with RFP in zebrafish embryos, and we detected residual or no staining tissue in Dhx15^−/−^ embryos (Figure 1A, panels c and d). We also determined the percentage of hepatic yolk retention. Dhx15^−/−^ embryos depicted a 100% yolk retention; in contrast, WT clutch-mates presented a 0–5% yolk retention. This observation suggests that the lack of liver tissue in Dhx15^−/−^ embryos may be the mechanism responsible for yolk retention due to impaired nutrient metabolism, causing metabolite accumulation in the yolk sac (Figure 1A, panel b). Heterozygous embryos were also evaluated and displayed similar characteristics as WT embryos, indicating no evident alterations in heterozygosis (Supplementary Figure S1).
To evaluate whether the absence of liver in Dhx15^−/−^ embryos was caused by genetic alterations induced by Dhx15-deficiency, we evaluated the gene expression of key mediators of liver development, such as, Prox1, Mypt1, Hdac3, Foxa1, Sox17, Uhrf1, and Bmp4. We found that Mypt1, Prox1, and Hdac3 were significantly downregulated in Dhx15^−/−^ embryos, while only Gata6 was significantly upregulated compared to WT embryos. Other genes depicted a nonsignificant tendency towards a reduced expression (Figure 1B).
In Figure 1C, the morphological differences between WT and mutant embryos and the absence of liver in Dhx15^−/−^ larvae are depicted. Such results urged us to elucidate the role of Dhx15 in the liver.
2.2. Altered Liver Vasculature in Dhx15-Partially-Deficient Mice
In a prior study, we described Dhx15-related vascular defects in mutant zebrafish that were also occurring during development [7]. In the present study, we wanted to further expand these previous results by studying the vasculature morphology within the liver. Since our Dhx15^−/−^ zebrafish model presented defects in liver organogenesis, we investigated the impact of the partial Dhx15 deficiency on liver function and hepatic angioarchitecture in adult Dhx15^+/−^ mice, which are viable, compared with Dhx15^−/−^ mice that showed embryonic lethality [7]. Supplementary Figure S2 includes gross liver images, body weight, liver weight, and graphs depicting concentrations of albumin, total bilirubin, and blood urea nitrogen, which are classic parameters for evaluating liver function. Our results indicate that heterozygous Dhx15 deficiency does not negatively impact organ function, as evidenced by the absence of significant changes in these parameters compared with wild-type mice. Regarding the evaluation of hepatic angioarchitecture, we observed differences in the thickness and expansion of the liver vasculature in Dhx15^+/−^ mice, compared to WT (Figure 2A), consisting of the presence of thinner vessels and lower vascular connectivity.
To study the molecular variations associated with the intrahepatic vascular alterations, we quantified RNA expression of different key factors involved in the generation and maintenance of blood and lymphatic vessels. In heterozygous mice, we observed a reduced RNA expression of Vegf-c, Vegfr3, Podoplanin, and Angiopoietin 1 and a nonsignificant tendency towards a lower expression of Vegf-d and Prox1 (Figure 2B). Similarly to the results found in hepatic tissue, we detected a reduced expression of Vegf-c, Vegf-d, and Angiopoietin 1 in a hepatocyte cell line with the Dhx15 gene silenced (Hep-siDhx15), compared to WT hepatocytes (Figure 2C).
2.3. Dhx15 Partial Gene Deficiency Decreases the Regenerative Capacity of the Liver in Mice
To study the role of Dhx15 during regeneration, we performed two-thirds partial hepatectomy (PHx) in WT and Dhx15^+/−^ mice. Mice were sacrificed at 2, 3, and 7 days post-PHx; the wet remnant liver weight together with the total body weight was used to calculate the hepatic regenerative rate known as Higgins Index. Seven days after PHx, Dhx15^+/−^ mice showed decreased regenerative rate associated with a trend of increasing mortality at 72 h after surgery compared with WT mice (Figure 3A), although these differences were not significant.
Liver tissue samples obtained at day 2 and 3 after PHx were used to evaluate cell proliferation by Ki67 immunostaining. In Dhx15^+/−^ mice at day 2 post-PHx, we observed a slight but significant decrease in total cell proliferation compared to WT mice (Figure 3B, panels a and b), although this tendency was not significant for parenchymal cells. At day 3 post-PHx, we observed evident and significant reduction in proliferation in both parenchymal and total cells in Dhx15^+/−^ mice compared with WT mice (Figure 3B, panels c and d). Next, we specifically evaluated the presence of endothelial cells in the regenerating liver at 3 days post-PHx by ETS related gene (Erg) immunostaining. We observed a significant reduction in Erg-positive cells in the heterozygous livers compared to WT (Figure 3B, panels e and f). The reduced proliferation was accordingly associated with a reduced protein expression of Cyclin d1 but not Pcna (Figure 3C).
2.4. Dhx15 Deficiency Alters Glucose Metabolism
Previous RNAseq and proteomic analysis results in Dhx15 silenced endothelial cells led us to conclude that Dhx15 participates in carbohydrate metabolism (Ribera J. 2021 [7]) (Supplementary Figure S3). To further validate these previous published results, we performed functional metabolic analyses in Dhx15^+/−^ mice.
After partial hepatectomy, glycogen storage is used to feed the highly metabolic demand of liver regeneration, finding its lowest peak at 24 h post-PHx, to later recover its normal levels [22,23]. To elucidate whether the defects in regeneration were related to metabolic alterations restricting the energetic demands in the liver, we first quantified glycogen levels in the regenerating livers of our Dhx15-deficient animal experimental model. We observed a significant decrease in glycogen in Dhx15^+/−^ mice two and three days after PHx compared to WT (Figure 4A), indicating that heterozygous mice are unable to normalize glycogen levels post-PHx. We analyzed the expression of glucokinase (Gck), which converts glucose into glucose 6-phosphate, and the expression of glycogen synthase (Gs) which converts UDP-glucose into glycogen, before and after hepatectomy (Figure 4B). We observed that after hepatectomy, Dhx15^+/−^ mice showed significantly lower levels of Gck at day 2, returning to basal levels at day 3. On the other hand, the Gs gene showed differential expression in the context of Dhx15 deficiency with overexpression on day two and gene repression on day three, compared to the wild-type group. Although Gs does not exhibit a defined tendency of differential expression, its consistent transcriptional deregulation together with the differential expression of Gck are concordant with the differences found in glycogen accumulation between the wild-type and the Dhx15^+/−^ groups at different time points (Figure 4A). This may indicate that the deficiency of the Dhx15 helicase is altering the expression of Gck and that Gs expression increases in order to compensate such enzymatic deficiency; however, it is not able to properly restore the glycogen storage levels. Accordingly, in Dhx15 silenced hepatocytes (Hep-siDhx15), we evaluated genes participating in glycogenesis and found lower RNA expression of Phosphoglucomutase 1 and Udp-glucose pyrophosphorylase that participate in the conversion of glucose into glycogen (Figure 4C).
In a previous Dhx15 study we observed decreased mitochondrial activity and ATP production in endothelial cells [7]. We evaluated now if glucose metabolism might be affected by Dhx15 depletion, possibly reducing hepatocyte proliferation during regeneration. Dhx15^+/−^ mice presented a lower glucose production after pyruvate injection compared to WT mice, reaching lower levels of maximal glucose production (Figure 4D). We quantified glucose production in Hep-siDhx15 and observed a significant decreased glucose production caused by Dhx15 deficiency (Figure 4E). To confirm these results, we evaluated the expression of glycolysis-participating enzymes in Hep-siDhx15; we found decreased expression of Glucose-6-phosphatase (G6pc) and Pyruvate carboxylase (Pc) which participate in the gluconeogenic conversion of pyruvate into glucose (Figure 4F), and we detected no differences in Pyruvate kinase (Pklr) and Phosphoenolpyruvate carboxykinase (Pck). These results evidence the metabolic alterations caused by Dhx15 partial deficiency.
2.5. Dhx15-Related Vascular Alterations Derive in Less Hepatic Tumor Nodule Events in an HCC Mouse Model
Recent studies evaluate the role of DHX15 in different cancer types. In hepatocellular carcinoma (HCC), DHX15 was found to be overexpressed in human cancerous livers [16]. Also, Zhao et al. described that DHX15 in HCC has an inhibitor role in HCC proliferation by suppressing autophagy in a hepatoma cell line [20]. Since we previously found lymphatic alterations and reduced metastasis in a lung cancer mouse model in Dhx15 heterozygous mice [7], we now evaluated the role of Dhx15 in HCC and liver metastasis in mice. We benefited from the use of the murine hepatocellular carcinoma cell line named Hepa 1-6 to establish a syngeneic cancer model. Upon subcutaneous injection of Hepa 1-6 cells in the flank of WT and Dhx15^+/−^ mice, we followed primary tumor formation. Five weeks after injection, Dhx15^+/−^ mice exhibited similar tumor size (Figure 5A, panels a and b) and low, nonsignificant expression levels of the HCC marker alpha-fetoprotein (AFP) compared to wild-type mice (Supplementary Figure S4A). We evaluated tumor invasion of the liver by Hepa 1-6 and found small nodules within the livers of Dhx15^+/−^ mice compared to WT mice (65.62 ± 11.77 vs. 111 ± 15.92 µm of nodule size per mouse, respectively; p < 0.05). The liver nodules in WT mice were larger and more numerous (Figure 5A, panels c and d). Dhx15^+/−^ mice presented fewer invasive events in the liver compared to WT mice (1.40 ± 0.16 vs. 2.78 ± 0.49 number of nodules per mouse, respectively; p < 0.05).
We also studied the lymphatic and blood vasculature within the primary tumor to determine if differences in tumor nodule invasion were related to aberrant vasculature structures. In accordance with reduced tumor nodule formation, we observed clear differences in WT and heterozygous mice, depicting vascular abnormalities in Dhx15^+/−^ mice. We studied both endothelial and hematopoietic vasculature by endomucin staining (Figure 5B, panels a and b). In Dhx15^+/−^ tumors, we observed smaller vases with a reduced lumen. We next studied the organization of lymphatic cells by Lyve-1 immunostaining. Comparing WT and Dhx15^+/−^ mice, the heterozygous mice presented a significant decrease in % stained area with Lyve-1 (Figure 5B, panels c and d).
To better analyze lymphatic defects, we quantified the RNA expression of several vascular factors within the primary tumor. We analyzed the expression of Vegf-d which participates in lymphangiogenesis and in endothelial cell growth, being relevant in the development of new lymphatic vasculature in metastasis [24]. We found a significant decrease in Vegf-d and a significant upregulation of its receptor Vegfr3 in the primary tumors of Dhx15^+/−^ mice. We also found a significant decreased expression of Vegf-a, Vegfr1, its receptor, and angiopoietin 1, in the primary tumors of Dhx15^+/−^ mice (Figure 5C). The results suggest that the fewer hepatic tumor nodules observed in Dhx15^+/−^ mice might be due to an impaired growth and development of the vascular network in the primary tumor.
2.6. AUMsilence ASO Mediated Dhx15 Silencing in Mice Reduces Primary Tumor Volume in an HCC Mouse Model
Following previous observations by us and others regarding the function of DHX15 in HCC, we decided to evaluate the potential therapeutic use of Dhx15 deletion in the cancer setting. To do so, we studied the effect of the AUMsilence antisense oligonucleotides (AUMsilence^TM^ ASO) silencing methodology [25] to silence Dhx15 in vivo. Upon a single dose of AUMsilence Dhx15 intravenous injection, we obtained an 80% of Dhx15 silencing in the liver that was maintained up until 72 h post-injection (Supplementary Figure S4B). Dhx15 silencing was also evaluated in the lungs and spleen, where we observed a milder Dhx15 deletion.
To evaluate the effects of a strong Dhx15 inhibition in the mouse liver in a cancerous context, we established the Hepa 1-6 HCC model in AUMsilence ASO-injected mice. We followed primary tumor growth and observed a significantly reduced tumor volume in the AUMsilence Dhx15 injected mice compared to AUMscramble scramble group (AUMscramble ASO) (179.6 ± 80.06 vs. 1085 ± 277.1 mm^3^ primary tumor volume five weeks post-implantation, respectively; p < 0.01; Figure 6A). In agreement with the results found in Dhx15^+/−^ mice, we also observed a reduction in several vascular genes in the livers of AUMsilence ASO Dhx15 injected mice, such as Vegf-a, Vegf-d, Vegfr1, and Vegfr3 (Supplementary Figure S4C).
We also analyzed the vascular angioarchitecture of the primary tumor and, similarly to the HCC model in Dhx15^+/−^ mouse, we observed alterations in the vasculature in terms of a significantly reduced vascular perimeter and lumen of blood vessels detected by endomucin immunostaining and a significant reduction in lymphatic vessels detected by Lyve-1 immunostaining in the AUMsilence ASO Dhx15 group compared to the AUMscramble ASO scramble group (Figure 6B).
Today, the biochemical diagnosis of hepatocellular carcinoma continues to be a challenge. Due to the effect of Dhx15 on tumor growth in our experimental model of HCC, we wanted to evaluate the differential diagnostic utility of this marker in patients. With this purpose, we performed serological evaluation of DHX15 levels by ELISA in a cohort of patients with different liver disease etiologies. As shown in Figure 6C, patients with HCC present significant higher levels of circulating DHX15 compared to healthy subjects (300.3 ± 88.2 vs. 32.4 ± 27.7 pg/mL; p < 0.01; respectively). We also detected a trend of increased circulating values of DHX15 in patients with HCC compared to cirrhotic patients without hepatic tumors, although this trend was not significant (300.3 ± 88.2 vs. 132.0 ± 46.2 pg/mL; p = 0.095; respectively).
3. Discussion
The DEAH-box RNA helicase DHX15 is implicated in diverse biological functions. In this study, we describe a total impairment of liver development caused by the mutation of Dhx15 in our CRISPR/Cas9 zebrafish. To our knowledge, Dhx15 had not previously been described to play a role in liver development; now, we report, for the first time, a lack of the hepatic organ in zebrafish due to Dhx15 knockout. This implies a redundant and noncompensated function of Dhx15 in liver development. Since DHX15 is a splicing factor, we studied whether Dhx15 knockout was affecting genes classically described to be implicated in liver formation in zebrafish. We found reduced expression of Prox1, which is one of the earliest markers for definitive hepatoblasts*, Mypt1,* which participates in bud formation, and Hdac3, which participates in liver budding and differentiation [26], in Dhx15^−/−^ embryos at 4dpf. Knockout experiments in zebrafish have helped to determine the crucial and nonredundant function of several genes in liver development, such as Gata 4 and 6, Hdac3, Hhex, and Mypt1. Single mutation of these genes results in major hepatic development complications [26,27,28,29]. For instance, Huang H. et al. described a liverless phenotype in Mypt1 mutant zebrafish [30]. In that study, Mypt1 mutation caused hepatoblast apoptosis that resulted in blockage of liver bud formation. Here, we added Dhx15 to the list of essential genes in liver development in zebrafish. Furthermore, we observed a regulatory crosstalk between Dhx15 and Mypt1, Prox1, and Hdac3, thus supporting the role of Dhx15 in hepatic organogenesis. More studies are needed to evaluate the contribution of each of these factors in the final effect on organogenesis.
In Dhx15^−/−^ zebrafish embryos, liver absence causes retention of metabolites in the yolk sac. At early embryonic stages, zebrafish energy demands are supplied by the metabolization of nutrients that takes place in the liver [31]. The Dhx15^−/−^ zebrafish mutants die at day 8 post-fertilization, possibly because their development energy demands cannot be met, resulting in embryonic lethality. Some limitations to consider of the zebrafish experiments are that we did not perform rescue experiments to unequivocally demonstrate the specificity of our sgRNA designed to edit the Dhx15 gene by CRISPR/Cas9. However, we believe we can mitigate concerns about off-target effects considering the use of bioinformatically validated gRNAs with minimal off-target potential, as we show in Supplementary Figure S5. Another limitation to consider is the possibility that impaired liver organogenesis could contribute to the observed downregulation of Mypt1, Prox1, and Hdac3. However, the expression patterns of these factors, along with the overexpression of GATA6, which plays a significant role in liver organogenesis, suggest that the absence of the liver is not the primary cause of the observed differential expression of Mypt1, Prox1, and Hdac3 in Dhx15-deficient zebrafish.
In mammals, the embryonic liver is an early hematopoietic organ; therefore, mutations affecting liver or blood development may cause early lethality during embryogenesis [32,33]. In mice, hepatogenesis begins with the formation of the liver bud around gestation day 8.25 [34,35]. As we previously described, our Dhx15^−/−^ mice died prior to the embryonic stage E8.5 [7]. Therefore, we cannot disregard the possibility that embryonic lethality is linked to an impaired liver organogenesis caused by Dhx15 deficiency. Due to early embryonic mortality associated with the loss of Dhx15, this hypothesis could only be tested using Dhx15 conditional KO mice. In mice, RNA helicase knockout often results in embryonic lethality, implying their essential role in developmental processes [36,37,38]. For instance, the loss of Ddx3x, which is associated with cell survival and cell cycle control, causes early post-implantation lethality prior to E6.5 [39]. As far as we know, no other RNA helicase has been described to play an essential role in liver development.
Knowing that the vasculature is crucial in liver development, we studied the vascular phenotype in the liver of heterozygous mice. We had previously observed vascular defects in Dhx15^−/−^ zebrafish during development [7]. Here, we observed differences in thickness and connectivity of liver blood vasculature in Dhx15^+/−^ mice. Additionally, in Dhx15^+/−^ mice, we observed a significant reduction in Angiopoietin 1, Vegf-c, and Podoplanin, which play major roles in the blood and lymphatic vascular growth and maturation. These liver vasculature alterations suggest that Dhx15 might be affecting the development of the hepatic vascular network that is crucial in embryonic stages for the development of the liver and, as a result, altering liver organogenesis, as previously reported by others [40,41].
Vascular alterations within the liver might alter hepatic functionality. One of the main characteristics of the liver is its capacity to regenerate owing to the proliferative potential of quiescent hepatocytes. It was previously described that loss of Akt hinders hepatic regeneration by reducing cell proliferation, cell hypertrophy, glycogenesis, and lipid droplet formation [18]. Since Dhx15 is a downstream target of Akt 1 [6], we evaluated the effects of Dhx15 depletion in liver regeneration. In this context, we observed decreased overall regeneration and cellular proliferation in Dhx15^+/−^ mice. However, the slightly decreased proliferation 48 h after hepatectomy was not linked to a decreased expression of proliferative genes such as Pcna or Cyclin d1. We also observed a lower mice survival after 72 h post-PHx. One of the reasons behind heterozygous mouse mortality at 72 h post-PHx might be correlated with a significantly reduced proliferation that is linked to a decreased expression of Cyclin d1.
We also studied an alternative mechanism to explain impaired function after partial hepatectomy. One of the major functions of the liver is to act as a “glucostat”. It has been demonstrated how glucose supplementation in liver regeneration mouse models increases survival in different gene deficiency models [42]. Furthermore, we had previously observed, by -omic analysis, that endothelial cells with a Dhx15-deficient background showed impaired glucose metabolism [7]. Therefore, we analyzed if Dhx15 partial deficiency was promoting metabolic alterations. First, we observed that heterozygous mice were not able to normalize glycogen levels after partial hepatectomy. Such impaired glycogen restorage was linked to alterations in the expression of the enzymes of the glycogenic pathway such as glucokinase and glycogen synthase. During the regenerative process, glycogen is consumed and used as source of energy to meet with the high metabolic demands of regeneration [22,23]. This may indicate that the observed liver regeneration alterations may also be the outcome of a decreased energetic availability, hindering proper proliferation. In Dhx15^+/−^ mice, we also observed a lower glucose production. Accordingly, we show a significantly decreased glucose production in silenced hepatocytes that may be related to a reduced expression of G6pc, Pklr, and Pc, which are key enzymes needed for the release of free glucose into blood circulation. The metabolic defects caused by Dhx15 deficiency, together with the reduced cellular expression of regulators of the cell cycle progression, such as Cyclin d1, may explain the lower cell proliferation of hepatocytes found in Dhx15^+/−^ livers. These results combined, with the endothelial cell defects that surge at 72 h post PHx, result in an impaired liver regeneration derived from Dhx15 partial deficiency.
Next, we evaluated the role of Dhx15 in the context of hepatocellular carcinoma (HCC) and liver metastasis. Recently, several studies have analyzed the role of DHX15 in different cancer types. DHX15 contributes as a cancer promoter in breast cancer, prostate cancer, acute myeloid leukemia, and hepatocellular carcinoma, and as an antitumor factor in glioma due to its growth inhibitory function [12,13,14,15,16]. In the present study, we evaluated the potential predictive use of DHX15 serologic analysis as a biomarker of HCC. Dhx15 in the serum of the HCC cohort was noticeably higher than that of healthy individuals. However, we did not detect significant differences in the Dhx15 circulating levels when we compared the HCC and the cirrhosis groups, despite detecting a trend of greater concentration of Dhx15 in patients with HCC. One important limitation of our results is the design of this observational clinical study. Therefore, and considering the urgent need of accurate biomarkers for HCC in the clinical laboratory, we guaranteed further prospective validation studies to confirm the diagnostic utility of DHX15 as a noninvasive biomarker for HCC in comparison with other liver tumors.
In a murine HCC xenograft model, we observed the formation of similar-volume primary tumors in WT and Dhx15^+/−^ mice. However, significantly smaller, and fewer tumoral nodules implanted in the liver were linked to Dhx15 depletion. In agreement with the reduced tumoral liver invasion, we observed a dysfunctional lymphatic vasculature in Dhx15^+/−^ mice. In a previous study modeling pulmonary metastasis in mice, we detected a significant metastasis reduction due to Dhx15 depletion [7]. Considering the role played by lymphatic vessels in tumor invasion [43], we suggest, as a model, that the impaired lymphatic growth within the primary tumor associated with Dhx15 deficiency limited cancer cell invasion into other organs, including the liver. These new findings highlight the role of Dhx15 as a potential target in metastasis.
To start evaluating the potential therapeutic benefits of Dhx15 inhibition in cancer and metastasis, we designed specific Dhx15-inhibiting oligonucleotides for in vivo use. We first analyzed the silencing potential of the AUMsilence oligonucleotides in mouse and observed a successful Dhx15 inhibition in the liver and other organs. AUMsilence ASOs are third-generation chemically modified oligos which have the capability of self-delivery (gymnosis) without the use of any delivery reagents or formulations. In our experience, AUMsilence oligos are highly sequence specific to their target and show no toxicity. We have shown optimal delivery to the liver using AUMsilence ASOs. Such attributes make AUMsilence ASOs ideal for target discovery and preclinical studies. Then, we established the HCC Hepa 1-6 model in AUMsilence ASO-injected mouse to target Dhx15 expression and we observed a significant reduction in average tumor growth. Evaluation of chronic toxicity, routes of administration, and dosage are still pending to establish robust conclusions about the biosafety of the AUMsilence ASO Dhx15 treatment. Also, liver-specific targeting of Dhx15 silencing and the generation of a conditional knockout to restrict the Dhx15 deficiency to the liver are needed to ensure the safety of an anti-Dhx15 therapeutic strategy in the context of tumor treatment and to prevent side effects in other organs. These experiments are currently underway in our laboratory.
We believe the presence of the other Dhx15 allele in heterozygous mice prevents us from seeing a net effect on the decrease in the primary tumor volume. We base this rationale on the observation that silencing Dhx15 in mice with the AUMsilence ASO Dhx15 treatment does have a clear impact on reducing the size of the primary tumor, since these specific anti-Dhx15 oligo sequences further reduce the presence of Dhx15 compared to the levels of Dhx15 expression found in the Dhx15^+/−^ mice. Therefore, the absence of Dhx15, either in heterozygosis or due to specific Dhx15 silencing, translates into an antitumor effect when analyzing tumor invasion or the size of the primary tumor. These concordant results achieved using two different experimental situations (gene deficiency and Dhx15 silencing) robustly support our hypothesis that a potential antitumor strategy can be achieved by suppressing the activity of Dhx15. These results, in turn, agree with the role of DHX15 in tumor promotion discussed in the introduction [12,13,14,15,16] since, in these situations, there is a complete allelic expression of DHX15.
In summary, our study provides insights into an essential role for Dhx15 in the development of liver in zebrafish. Dhx15 knockout in zebrafish results in a liverless phenotype and early embryonic lethality. An impaired liver development could be the consequence of the observed blood and lymphatic vascular defects together with the reduced expression of hepatogenic enzymes. Also, Dhx15 depletion resulted in hepatic regeneration and metabolic alterations. The observed alterations in liver regeneration may be caused by a reduced proliferation linked to an aberrant glucose metabolism acting together with endothelial defects, impeding the de novo formation of hepatic vasculature and impaired liver regeneration. Also, Dhx15 deficiency led to reduced hepatic tumor invasion and tumor growth in a murine HCC model. Regarding the potential use of DHX15 as a diagnostic marker for liver disease, HCC patients showed increased levels of DHX15 in blood samples compared with subjects without hepatic affectation. Therefore, our results support the potential role of DHX15 as a diagnostic and therapeutic target in liver disease, as well as a major regulator of liver regeneration and organogenesis.
4. Materials and Methods
4.1. Mouse-Induced Tumor Model
HEPA 1-6 cells (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum, 50 U/mL penicillin, and 50 µg/mL streptomycin in humified atmosphere at 37 °C and 5% CO_2_. Syngeneic Hepa 1-6 tumor cells (5 × 10^6^) were subcutaneously injected into the flank of Dhx15^+/−^ and wild-type mice (n = 10). Primary tumor growth was controlled during the first 5 weeks. Tumor growth was monitored by measuring volumes using a digital slide-caliper. Tumor volume was calculated by the following formula: V = 4/3 × π × (length × depth × width). Primary tumors were fixed in 4% PFA and cryopreserved in tissue-tek O.C.T. compound (Sakura, Flemingweg, The Netherlands). The post-surgical metastasis model was performed as follows: five weeks post-injection, Hepa 1-6-injected mice were sacrificed, and the liver was extracted to perform metastasis analyses. Tile scan images of hematoxylin–eosin (H&E)-stained paraffin liver sections were visualized using an Olympus BX51 microscope equipped with DP71 camera (Olympus Europa SE & CO.KG., Hamburg, Germany), and the percentage of hepatic metastatic area as percent of total hepatic area was measured with ImageJ software (ImageJ version 1.52b; National Institutes of Health, Bethesda, MD, USA).
4.2. In Vivo Knockdown Experiments
Knockdown experiments were performed using AUMsilence^TM^ antisense oligonucleotides (AUMsilence^TM^ ASOs) that were designed and provided by AUM BioTech, LLC, Philadelphia, PA, USA. For general knockdown, the oligos were intravenously injected into the mouse tail vein at a dose of 10 mg/kg/day every third day. In vivo knockdown efficiency was determined by Dhx15 Western blot determination in different vital organs (liver, spleen, kidney, lungs, and heart), achieving an 80% knockdown in the liver. To study the impact of Dhx15 depletion in the Hepa 1-6 HCC model, Hepa 1-6 cells (5 × 10^6^ cells, subcutaneously injected) were implanted subcutaneously into the flank of the mice (n = 10) and allowed to grow for five weeks in previously AUMsilence ASO-injected mouse. An unrelated AUMscramble ASO SCR was used as a control. Tumor growth was monitored by measuring volumes using a digital slide-caliper.
4.3. Patients
A prospective cohort of consecutive patients treated at Hospital Clinic de Barcelona was evaluated. Informed consent was obtained from all patients involved in the study that was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Hospital Clínic de Barcelona.
Included population consisted of patients with (1) HCC diagnosed according to AASL guidelines; (2) cirrhosis associated with hepatitis C virus infection (HCV) without HCC; and healthy volunteers. The demographic and clinical characteristics of the patients included in the study are shown in Supplementary Tables S1 and S2. This study included a total of 24 serum samples from control subjects without neoplastic or liver disease. The samples were collected, anonymized, and stored according to the ethical rules of the Hospital Clinic.
4.4. Statistical Analysis
Quantitative data were analyzed using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA), and statistical analysis of the results was performed using unpaired Student’s t-tests and ANOVA models (with Tukey’s post hoc test) with normally distributed data. Partial hepatectomy mortality scores were analyzed by log-rank test and survival curves were generated using the product limit method of Kaplan and Meier. For other type of data, the Mann–Whitney U-test was used. Correlations between variables were evaluated using Spearman’s rho or Pearson’s r, when appropriate. Differences were considered significant at a p-value < 0.05. The data are presented as the mean ± standard error of the mean.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Studer M.K. IvanovićL. Weber M.E. Marti S. Jonas S. Structural basis for DEAH-helicase activation by G-patch proteins Proc. Natl. Acad. Sci. USA 20201177159717010.1073/pnas.191388011732179686 PMC 7132122 · doi ↗ · pubmed ↗
- 2Silverman E. Edwalds-Gilbert G. Lin R.J. D Ex D/H-box proteins and their partners: Helping RNA helicases unwind Gene 200331211610.1016/S 0378-1119(03)00626-712909336 · doi ↗ · pubmed ↗
- 3Sloan K.E. Bohnsack M.T. Unravelling the Mechanisms of RNA Helicase Regulation Trends Biochem. Sci.20184323725010.1016/j.tibs.2018.02.00129486979 · doi ↗ · pubmed ↗
- 4Robert-Paganin J. Réty S. Leulliot N. Regulation of DEAH/RHA helicases by G-patch proteins Bio Med Res. Int.2015201593185710.1155/2015/93185725692149 PMC 4322301 · doi ↗ · pubmed ↗
- 5Abdelkrim Y.Z. Banroques J. Tanner N.K. RNA Remodeling Proteins: Methods and Protocols. Chapter 3: Known Inhibitors of RNA Helicases and Their Therapeutic Potential Methods in Molecular Biology Humana New York, NY, USA 2021355210.1007/978-1-0716-0935-4_333201461 · doi ↗ · pubmed ↗
- 6Lee M.Y. Luciano A.K. Ackah E. Rodriguez-Vita J. Bancroft T.A. Eichmann A. Simons M. Kyriakides T.R. Morales-Ruiz M. Sessa W.C. Endothelial Akt 1 mediates angiogenesis by phosphorylating multiple angiogenic substrates Proc. Natl. Acad. Sci. USA 2014111128651287010.1073/pnas.140847211125136137 PMC 4156707 · doi ↗ · pubmed ↗
- 7Ribera J. Portolés I. Córdoba-Jover B. Rodríguez-Vita J. Casals G. la Presa B.G.-D. Graupera M. Solsona-Vilarrasa E. Garcia-Ruiz C. Fernández-Checa J.C. The loss of DHX 15 impairs endothelial energy metabolism, lymphatic drainage and tumor metastasis in mice Commun. Biol.20214119210.1038/s 42003-021-02722-w 34654883 PMC 8519955 · doi ↗ · pubmed ↗
- 8Lu H. Lu N. Weng L. Yuan B. Liu Y.-J. Zhang Z. DHX 15 Senses Double-Stranded RNA in Myeloid Dendritic Cells J. Immunol.20141931364137210.4049/jimmunol.130332224990078 PMC 4108507 · doi ↗ · pubmed ↗