Draft genome sequence of Vibrio diabolicus isolated from the starlet sea anemone Nematostella vectensis
Quinton Krueger, Adam Reitzel

TL;DR
This paper presents the complete genome sequence of a Vibrio diabolicus strain isolated from a sea anemone, aiding in understanding host-bacteria interactions.
Contribution
The paper provides the first full genome sequence of Vibrio diabolicus NVE-VD1 from Nematostella vectensis.
Findings
The Vibrio diabolicus strain was isolated from the starlet sea anemone Nematostella vectensis.
The full genome sequence is reported, supporting future studies on host-microbe interactions.
Abstract
Nematostella vectensis has grown as a model organism for investigating host–bacteria interactions. Here, we report the full genome of Vibrio diabolicus NVE-VD1, an isolate from N. vectensis from the South Carolina Baruch Estuarine Reserve.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
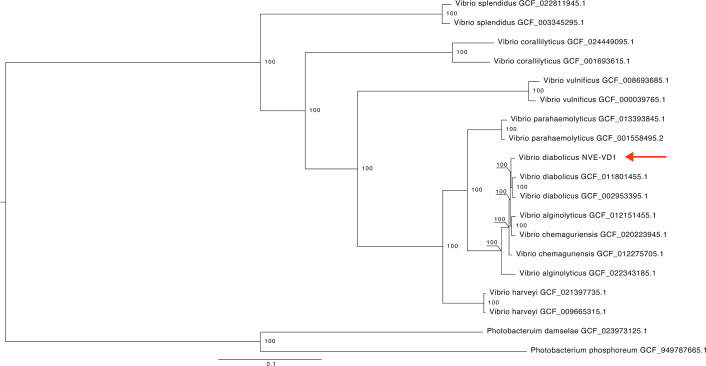 Fig 1
Fig 1|
| |
|---|---|
| Raw reads | 23,441,293 |
| Trimmed reads | 16,975,243 |
| Unicycler | |
| Statistics without reference | Scaffolds |
| # contigs | 93 |
| Largest contig | 1,863,922 |
| Total length | 5,117,331 |
| N50 | 1,302,326 |
| N75 | 849,246 |
| L50 | 2 |
| L75 | 3 |
| GC (%) | 44.75 |
|
| |
| # N’s | 575 |
| # N’s per 100 kbp | 9.77 |
| CDS | 4,541 |
| rRNA | 10 |
| tRNA | 98 |
| tmRNA | 1 |
|
| |
| Completion | 100.00 |
| Contamination | 3.67 |
- —Human Frontier Science Program (HFSP)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVibrio bacteria research studies · Aquaculture disease management and microbiota · Genomics and Phylogenetic Studies
ANNOUNCEMENT
Nematostella vectensis was collected from tidally restricted pools on Goat Island located in the South Carolina Baruch Estuarine Reserve in June 2018 (33°19′50.6″N 79°12′04.2″W). Anemones were then transported to the University of North Carolina at Charlotte (Charlotte, NC, USA). Homogenized anemone was serially diluted and plated on marine broth (24.72 g L^−1^ NaCl, 0.78 g L^−1^ KCl, 1.36 g L^−1^ CaCl_2_, 4.66 g L^−1^ MgCl_2_, 6.30 g L^−1^ MgSO_4_, 0.36 g L^−1^ NaHCO_3_, 10 g L^−1^ peptone, 3 g L^−1^ yeast extract, and 15 g L^−1^ agar) plates. A distinct colony was isolated and struck at minimum three times on fresh media to ensure a pure isolate. The isolate was then grown in marine broth and pelleted by centrifugation for DNA extraction and sequencing at Omega Bioservices. DNA was extracted with the Mag-Bind Universal Pathogen DNA Kit (Omega Bio-tek, M4024-00) and quality-checked by PicoGreen (ThermoFisher) and NanoDrop. The library was prepared by the KAPA HyperPrep for WGS (Roche, KR0961) and sequenced on the Illumina HiSeq X Ten (PE 2 × 150).
Paired-end read files were quality-checked with FastQC v0.11.9 (1). A total of 23,441,293 reads were generated from the sequencing run (Table 1). The raw reads were then adapter-clipped and quality-trimmed using Trimmomatic v0.39, which resulted in 16,975,243 remaining reads (2). Next, the trimmed reads were assembled with Unicycler v0.5.0 for de novo assembly (3). The final assembly metrics were assessed using Quast v5.0.2 (4) and CheckM v1.2.2 (5) to determine genome completeness and identify any potential contamination. The genome was subsequently annotated with PROKKA v1.14.6, using the options --genus Vibrio and –mincontiglen 500 (6). Next, orthogroups containing 1,309 single-copy genes identified with Orthofinder v2.4.0 were aligned using option -A muscle (7). ModelFinder determined that the best-fit model according to the Bayesian Information Criterion was LG+F+R6 (8). This alignment was used to generate a maximum likelihood phylogenetic tree in IQTREE2 v2.1.2, with 1,000 bootstraps (9). This tree was then visualized in FigTree v1.4.4. Lastly, taxonomic ranks were predicted with NCBI’s PGAP build 6771, using the –taxcheck-only option (10). The Vibrio isolate described here was predicted to be Vibrio diabolicus, based on the phylogenetic distance of single-copy orthologs (Fig. 1). The constructed maximum-likelihood tree places this isolate in a monophyletic clade with Vibrio diabolicus. We designate this isolate Vibrio diabolicus strain NVE-VD1. The reference isolate sharing the most genetic similarity with V. diabolicus NVE-VD1 was Vibrio chemaguriensis with an Average Nucleotide Identity of 98.136% (NCBI accession number ASM1227570).
Phylogenetic tree of Vibrio diabolicus NVE-VD1 single-copy genes. The single-copy genes of the clade were identified with Orthofinder v2.4.0 and aligned with MUSCLE. The maximum-likelihood tree was constructed in IQTREE2 v2.1.2 and visualized in FigTree v1.4.4.
The final assembly was 5,117,331 bp, with a GC content of 44.75%. There were 93 contigs, where the L50 was 2 and N50 was 1,302,326 bp. Interestingly, we found that 309 genes were duplicated, including stress/resistance proteins, secretion system genes, transporter genes, and motility/adhesion genes. These duplications may allow for additional flexibility in the interactions the bacteria have with the host.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Andrews S. 2010. Babraham bioinformatics. Babraham Institute, Cambridge, United Kingdom.
- 2Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120. doi:10.1093/bioinformatics/btu 17024695404 PMC 4103590 · doi ↗ · pubmed ↗
- 3Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. P Lo S Comput Biol 13:e 1005595. doi:10.1371/journal.pcbi.100559528594827 PMC 5481147 · doi ↗ · pubmed ↗
- 4Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi:10.1093/bioinformatics/btt 08623422339 PMC 3624806 · doi ↗ · pubmed ↗
- 5Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. Check M: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi:10.1101/gr.186072.11425977477 PMC 4484387 · doi ↗ · pubmed ↗
- 6Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi:10.1093/bioinformatics/btu 15324642063 · doi ↗ · pubmed ↗
- 7Emms DM, Kelly S. 2019. Ortho Finder: phylogenetic orthology inference for comparative genomics. Genome Biol 20:238. doi:10.1186/s 13059-019-1832-y 31727128 PMC 6857279 · doi ↗ · pubmed ↗
- 8Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. Model Finder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. doi:10.1038/nmeth.428528481363 PMC 5453245 · doi ↗ · pubmed ↗