Synthesis under high pressure: crystal structure and properties of cubic Dy36O11F50[AsO3]12 ∙ H2O
Felix Christian Goerigk, Ralf Jules Christian Locke, Thomas Schleid

TL;DR
This paper reports the synthesis and crystal structure of a new compound, Dy36O11F50[AsO3]12 ∙ H2O, under high pressure.
Contribution
The study presents a novel multi-anionic compound with a unique cubic crystal structure and unusual coordination geometries.
Findings
The compound has a non-centrosymmetric cubic structure with a large unit cell.
Dy3+ cations exhibit diverse coordination geometries, including capped trigonal prisms and square antiprisms.
The structure contains interstitial water molecules confirmed by Raman spectroscopy.
Abstract
The multi-anionic compound with the composition Dy36O11F50[AsO3]12 ∙ H2O, which can be described in the non-centrosymmetric cubic space group F 4¯3 c, already shows an unusually large unit cell with an axis of a = 2587.59(14) pm. Its crystal structure exhibits isolated ψ1-tetrahedral [AsO3]3– anions, but both the coordination numbers and the linking schemes of the Dy3+-centered polyhedra differ significantly from the mostly layered structures described so far in literature. (Dy1)3+ is sevenfold coordinated by oxygen atoms and F− anions, forming a capped trigonal prism [(Dy1)O4.333F2.667]8.333–, and the remaining two cations (Dy2)3+ and (Dy3)3+ both reside in an eightfold coordination of anions. In both cases they form slightly distorted square antiprisms, which have the compositions of [(Dy2)O3.667F4.333]8.667– and [(Dy3)O4.667F3.333]9.667–, respectively. Some of the oxygen atoms are…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
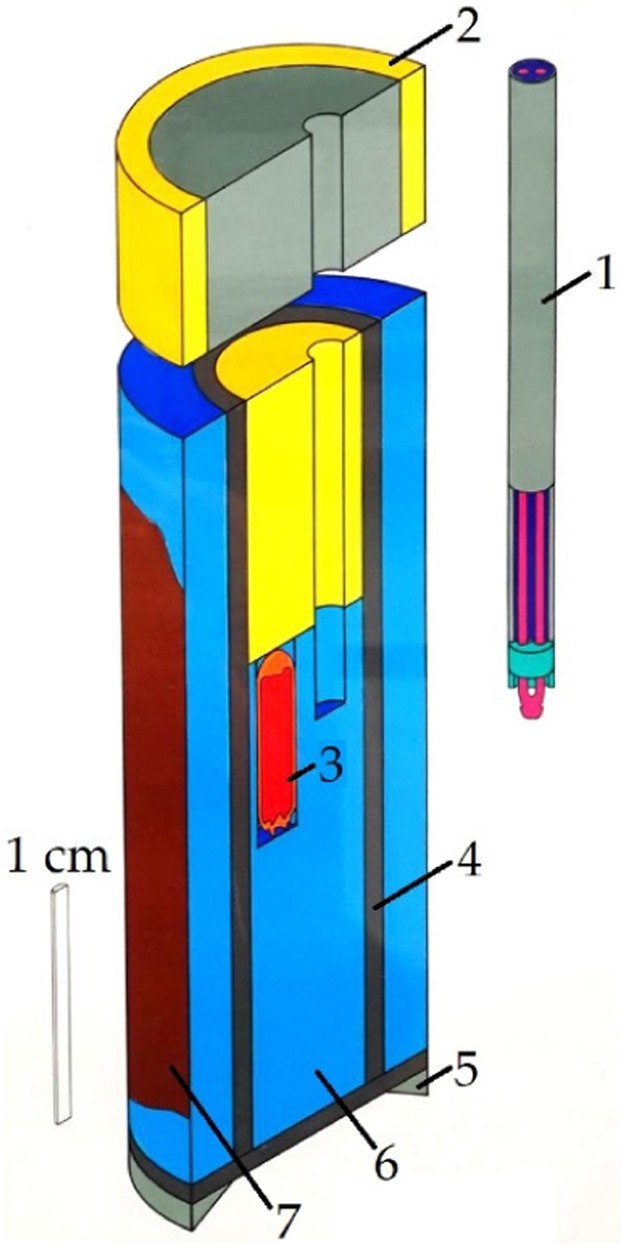 FIGURE 1
FIGURE 1 FIGURE 2
FIGURE 2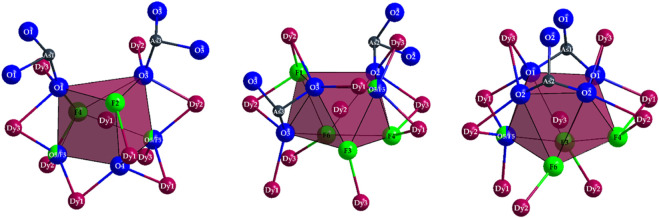 FIGURE 3
FIGURE 3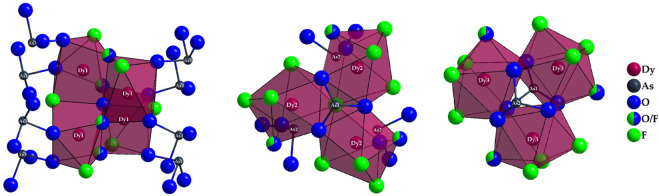 FIGURE 4
FIGURE 4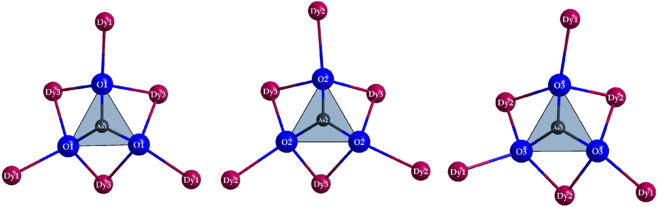 FIGURE 5
FIGURE 5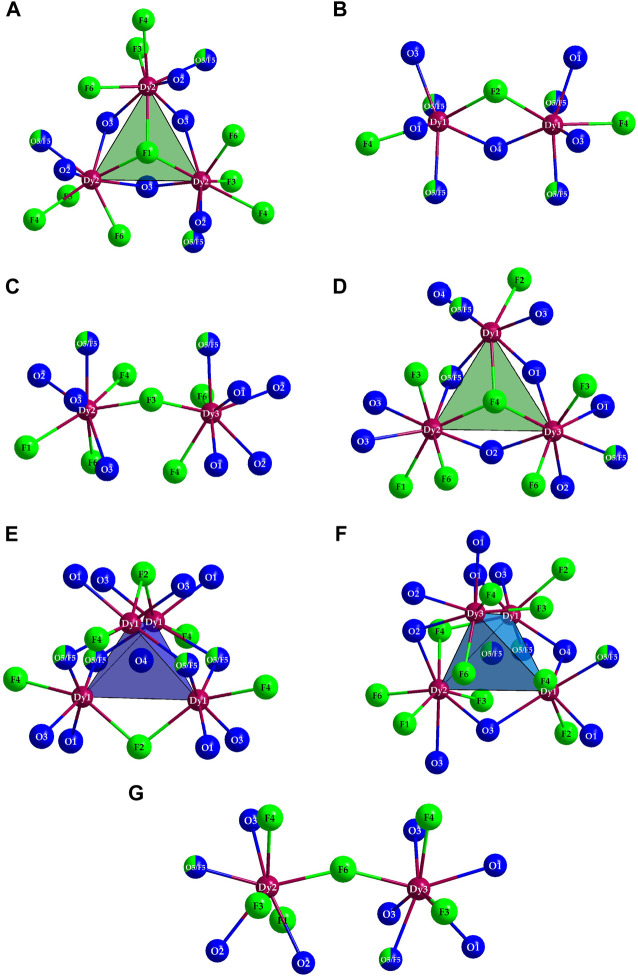 FIGURE 6
FIGURE 6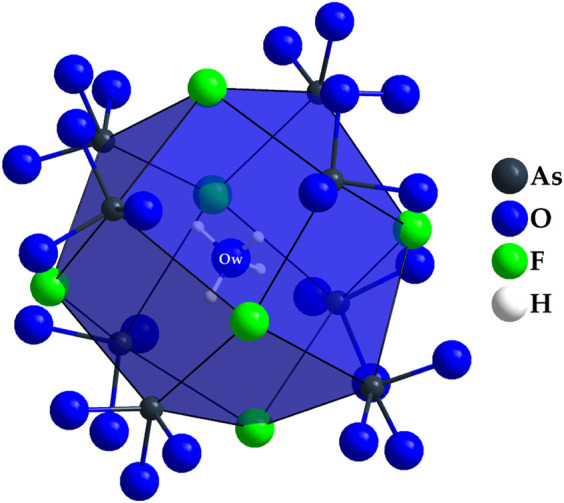 FIGURE 7
FIGURE 7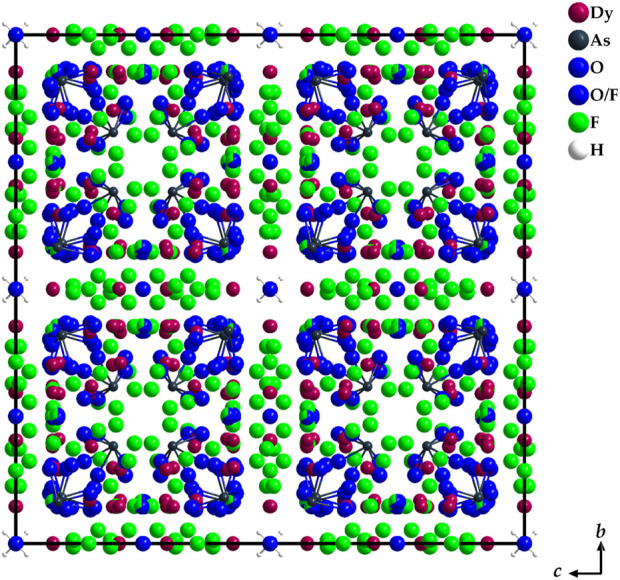 FIGURE 8
FIGURE 8 FIGURE 9
FIGURE 9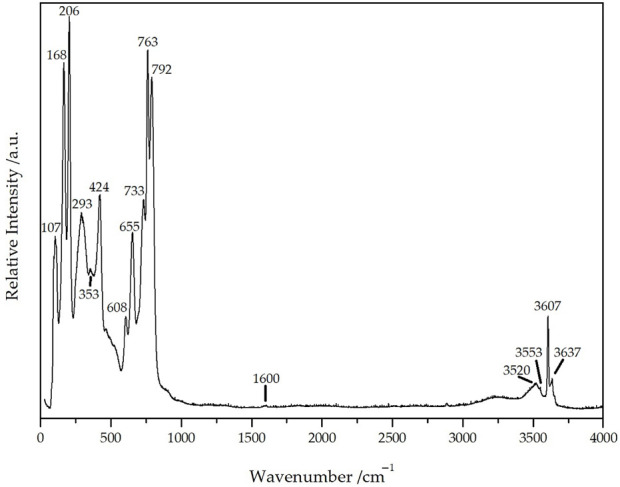 FIGURE 10
FIGURE 10 FIGURE 11
FIGURE 11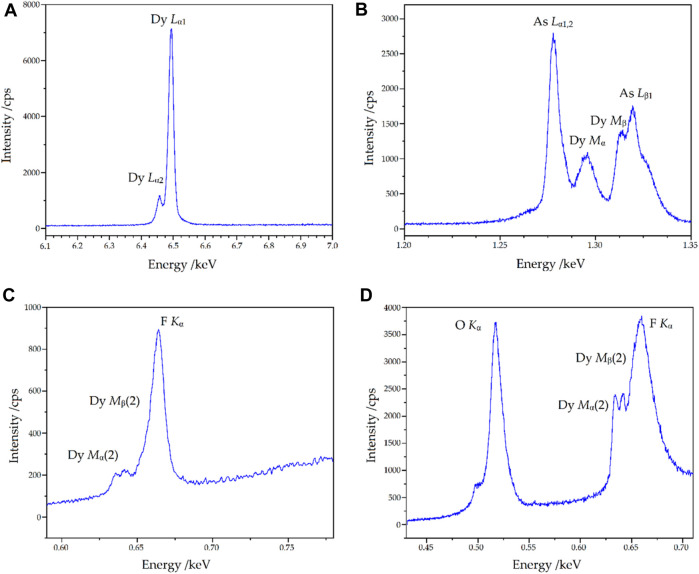 FIGURE 12
FIGURE 12| Formula | Dy36O11F50[AsO3]12 ∙ H2O |
|---|---|
| Crystal system | cubic |
| Space group |
|
| Lattice parameter, | 2587.59(14) |
| Number of formula units, | 8 |
| Calculated density, | 6.492 |
| Molar volume, | 1304.523 |
| Measurement limit, 2 | 60.96 |
| Measurement range, ± | 36 |
| Electron sum, | 28848 |
| Absorption coefficient, | 35.332 |
| Diffractometer | Stoe Stadi-Vari |
| Analytical radiation | Mo- |
| Measured reflections | 200828 |
| Symmetrically independent ones | 2221 |
|
| 0.092/0.034 |
|
| 0.044/0.029 |
| w | 0.061/1.017 |
| Flack X parameter | –0,01(3) |
| Residual electron density, | 1.58/–1.71 |
| CSD number | 2310780 |
| Atom | Site |
|
|
|
|
|---|---|---|---|---|---|
| Dy1 | 96 | 0.203007(16) | 0.001157(16) | 0.072966(16) | 183.4(9) |
| Dy2 | 96 | 0.294678(16) | 0.084582(15) | 0.145213(16) | 158.3(9) |
| Dy3 | 96 | 0.190631(16) | 0.148864(16) | 0.072279(15) | 155.0(9) |
| As1 | 32 | 0.08211(3) | 0.08211(3) | 0.08211(3) | 158(3) |
| As2 | 32 | 0.19160(3) | 0.19160(3) | 0.19160(3) | 153(3) |
| As3 | 32 | 0.40950(4) | 0.40950(4) | 0.40950(4) | 174(3) |
| O1 | 96 | 0.0666(2) | 0.0984(2) | 0.1467(2) | 174(12) |
| O2 | 96 | 0.2152(2) | 0.1345(2) | 0.1612(2) | 153(12) |
| O3 | 96 | 0.3458(3) | 0.1094(3) | 0.0710(3) | 208(13) |
| O4 | 24 | 1/4 | 0 | 0 | 193(25) |
| O5* | 96 | 0.2476(2) | 0.0822(2) | 0.0725(2) | 169(11) |
| F1 | 32 | 0.3341(2) | 0.3341(2) | 0.3341(2) | 324(23) |
| F2 | 48 | 0.1432(4) | 0 | 0 | 475(26) |
| F3 | 96 | 0.3137(3) | 0.0065(3) | 0.1130(3) | 348(15) |
| F4 | 96 | 0.2220(2) | 0.0261(2) | 0.1618(2) | 301(14) |
| F5** | 96 | 0.2476(2) | 0.0822(2) | 0.0725(2) | 169(11) |
| F6 | 96 | 0.2668(2) | 0.1969(2) | 0.0734(2) | 306(14) |
| Ow | 8 | 0 | 0 | 0 | 1557(243) |
| H*** | 32 | 0.0215 | 0.0215 | 0.0215 | – |
| Contact | Multiplicity | Distance/pm | Contact | Multiplicity | Distance/pm |
|---|---|---|---|---|---|
|
|
| ||||
| Dy1–O4 | 1× | 224.6(4) | Dy3–F3 | 1× | 224.3(6) |
| Dy1–O5/F5 | 1× | 230.7(6) | Dy3–O5/F5 | 1× | 226.9(5) |
| Dy1–O1 | 1× | 232.8(6) | Dy3–F6 | 1× | 232.9(5) |
| Dy1–O5'/F5′ | 1× | 239.2(6) | Dy3–F4 | 1× | 235.9(6) |
| Dy1–F4 | 1× | 243.8(6) | Dy3–O1 | 1× | 239.2(6) |
| Dy1–F2 | 1× | 244.2(7) | Dy3–O2 | 1× | 241.6(6) |
| Dy1–O3 | 1× | 244.3(7) | Dy3–O2′ | 1× | 247.2(6) |
|
| Dy3–O1′ | 1× | 250.6(7) | ||
| Dy2–F3 | 1× | 224.0(6) |
| ||
| Dy2–O5/F5 | 1× | 224.4(6) | As1–O1 | 3× | 177.0(6) |
| Dy2–F6 | 1× | 230.6(6) | As2–O2 | 3× | 178.2(6) |
| Dy2–F1 | 1× | 239.8(4) | As3–O3 | 3× | 179.2(7) |
| Dy2–O3 | 1× | 241.9(7) |
| ||
| Dy2–F4 | 1× | 245.3(7) | Ow–H | 2× | 96.4 |
| Dy2–O2 | 1× | 246.4(6) | |||
| Dy2–O3′ | 1× | 251.7(6) | |||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInorganic Fluorides and Related Compounds · Crystal Structures and Properties · Metal-Organic Frameworks: Synthesis and Applications
Introduction
Regardless of the oxidation state at the involved arsenic atoms, rare-earth metal oxoarsenates can serve well as either host materials or concentrated phosphors for lumogenious applications. This holds for the monazite-, xenotime-, and scheelite-type oxoarsenates(V) RE[AsO_4_] (Schäfer and Will, 1971; Lohmüller et al., 1973; Long and Stager, 1977; Schäfer et al., 1979; Brahim et al., 2002; Kang et al., 2005a; Kang, 2009; Kang and Schleid, 2005; Schmidt et al., 2005; Hartenbach et al., 2006; Golbs et al., 2009; Metzger, 2012; Metzger et al., 2016; Ledderboge et al., 2018; Goerigk, 2021; Adala et al., 2022) (RE = rare-earth metal: Sc, Y, La, Ce–Lu), where O^2−^-to-As^5+^ ligand-to-metal charge-transfer processes (LMCT) within the tetrahedral [AsO_4_]^3−^ anions have a beneficial impact on the necessary energy transfer as well as for the oxoarsenates(III) RE[AsO_3_] and RE 4[As_2_O_5_]2[As_4_O_8_] (Ben Hamida et al., 2005; Kang, 2009; Kang and Schleid, 2006; Ben Hamida, 2007; Metzger, 2012; Metzger et al., 2012; Ledderboge et al., 2014; Ledderboge, 2016; Locke et al., 2023), where the O^2−^-to-As^3+^ LMCT is supported by the lone-pair antenna at the trivalent arsenic centers. The latter occur as ψ^1^-tetrahedral [AsO_3_]^3−^ groups either isolated in the first cases (Pb[SeO_3_]- or K[ClO_3_]-type RE[AsO_3_]) or vertex-condensed to di- and tetranuclear anions (pyro-[As_2_O_5_]^4−^ and cyclo-[As_4_O_8_]^4−^) for the latter ones (RE 4[As_2_O_5_]2[As_4_O_8_] ≡ 2 × RE 2_As_4_O_9). Driven by the influence of fluxing halides during the corresponding preparation efforts, halide-derivatized rare-earth metal(III) oxoarsenates(III) were obtained for the first time, exhibiting the empirical formula RE 5 X 3[AsO_3_]4 (X = F (Ledderboge and Schleid, 2014; Ledderboge, 2016; Goerigk, 2021), Cl (Kang, 2009; Hamida and Wickleder, 2006; Ben Hamida, 2007; Schander, 2009; Ledderboge, 2016; Goerigk et al., 2019), and Br (Ledderboge, 2016; Goerigk, 2021)). With X = Cl and Br as soft halide anions, they occur as layered structures, while the fluoride-derivatives represent three-dimensionally hard materials according to the Pearson HSAB concept of “hard and soft acids and bases“ (Pearson, 1963). For this reason, they are well-suited as host substrates, which secure energy transfer with minimal losses from the rigid lattice and its hard components (RE ^3+^, F^−^ and [AsO_3_]^3−^) to the Ln ^3+^ activator cations, such as Eu^3+^ and Tb^3+^ as most prominent ones. The crystallization and single-phase preparation of most fluoride oxoarsenates(III) RE 5_F_3[AsO_3_]4 was not an easy task in the past, so we tried different synthetic pathways to tackle this challenge. In the case of Dy_5_F_3_[AsO_3_]4, one of these attempts involved a droplet of water added to the appropriate mixture of the well-ground solid starting materials (Dy_2_O_3_, DyF_3_, and As_2_O_3_ in a 2 : 1: 2 molar ratio) in order to heat it in a sealed gold ampoule within the set-up of a Boyd and England-type piston-cylinder high-pressure apparatus. As a surprising result, big well-faceted single crystals of what turned out to be Dy_36_O_11_F_50_[AsO_3_]12 with one molecule of interstitial water per formula unit were obtained. We report here on its fascinating unique crystal structure and several analytic methods to confirm its true nature as dysprosium(III) oxide fluoride oxoarsenate(III) hydrate according to Dy_36_O_11_F_50_[AsO_3_]12 · H_2_O.
Experimental
Since phase-pure and single-crystalline RE 5_F_3[AsO_3_]4 representatives are relatively difficult to access and preparative attempts to obtain them with conventional techniques from fused silica ampoules often led to oxosilicates, a reaction under high-pressure conditions in an inert gold capsule was considered as an alternative. Dysprosium sesquioxide (Dy_2_O_3_), dysprosium trifluoride (DyF_3_), and arsenic sesquioxide (As_2_O_3_) served as reactants in a molar ratio of 2 (164 mg): 1 (48 mg): 1 (87 mg). A fine blend of the reactants was prepared and filled into a gold ampoule (4 mm diameter and 10 mm length), which already contained some demineralized water (30 µL). To prevent water loss while sealing the ampoule, the upper fold was closed by cold welding with a pressure of almost 10 tons at the fold. The ampoule produced this way was placed in a rock-salt pressure cell (Figure 1) and then inserted into the pressure tube of the end-loaded high-pressure piston-cylinder reactor (Boyd and England-type) (Boyd and England, 1960; Massonne and Schreyer, 1986). The operating pressure was set to just 8.5 kbar at a temperature of 500°C for 4 days, then lowered to 400°C and dwelled for a further 3 days. After the end of the experiment and opening of the ampoule, a few very large cube-shaped colorless transparent single crystals, some with an edge length of several 100 µm (Figure 2), were found. These did not cause any rotation of the linearly polarized light in transmitted light under crossed polarizers, which is why the presence of a crystal in the cubic crystal system was assumed (Nickel, 1971). Due to the size, the habit, and the apparently cubic symmetry, it was initially wrongly assumed that the crystals should be sodium chloride (NaCl) deriving from the pressure cell. However, as the crystals did not dissolve in water, one of them was isolated and examined using single-crystal X-ray diffraction. The measurement was carried out with a STADI-VARI single-crystal diffractometer (Stoe & Cie, Darmstadt, Germany). A face-centered cubic metric with a ≈ 2590 pm was found, which could not be assigned to any known class of rare-earth metal(III) compounds. The composition Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O in space group F 3c was determined using direct methods and the structure was refined with the SHELX-97 (Sheldrick, 1997; Herrendorf et al., 1999; Sheldrick, 2008) program package. Based on the selected reactants and their initial weights, however, a synthesis without secondary phases was not possible, as the stoichiometric coefficients for the used weights of the reactants did not allow this. The powder X-ray diffraction pattern of Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O was recorded using a STADI-P diffractometer (Stoe & Cie, Darmstadt, Germany) with Ge(222)-monochromatized copper radiation (λ = 154.06 pm). The sample material was measured in transmission geometry in order to minimize the influence of texture effects and to receive an acceptable signal-to-noise-ratio. About 50 mg of the product was slightly ground and transferred on an amorphous adhesive film (scotch magic tape). For investigations using the Bragg–Brentano geometry, there was not enough sample material available (only about 150 mg product). In the measurement setup, a Stoe position-sensitive detector (PSD) with an angular resolution of 0.073° was employed. As step size, 0.02° in the area from 5° to 70° (2θ) was chosen. A Raman spectrum for the single crystal (excitation wavelength: λ = 638 nm) measured with a Raman microscope (XploRa, Horiba, Kyoto) can be found as well as electron-beam microprobe measurements (SX-100, Cameca, Gennevilliers) in the corresponding subsections.
Schematic structure of the rock salt-type pressure cell of the Boyd and England piston-cylinder apparatus 1) sheathed thermocouple (NiCr-Ni), 2) upper seal (fired pryophyllite ring and steel plug), 3) gold capsule with sample material (4 mm outer diameter), 4) steel furnace, 5) lower piston ring, 6) rock-salt cell, and 7) copper paste (Boyd and England, 1960; Massonne and Schreyer, 1986).
A colorless transparent single crystal of Dy36O11F50[AsO3]12 ∙ H2O.
Results and discussion
Crystal-structure description
The cubic compound Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O crystallizes in the non-centrosymmetric space group F4 c with a = 2587.59(14) pm and eight formula units in the unit cell (Table 1). Three crystallographically unique sites are present for the Dy^3+^ cations, all of which reside at general Wyckoff sites 96h (Table 2). (Dy1)^3+^ is sevenfold coordinated by oxygen atoms and F^−^ anions, forming a capped trigonal prism [(Dy1)O_4.333_F_2.667_]^8.333–^ (Figure 3, left). The “odd" stoichiometric coefficients are due to the common O5/F5 position, which has to be statistically occupied to ^2^/3 by oxygen and to ^1^/3 by fluorine for the overall stoichiometry to remain charge neutral, unless further mixed occupancies are to be introduced on other anion sites. Here, dysprosium-oxygen bonds with lengths of 225–244 pm and distances of the (Dy1)^3+^ cation to the F^−^anions of 231–244 pm occur (Table 3). The two cations (Dy2)^3+^ and (Dy3)^3+^ have eightfold coordination spheres by anions, in both cases forming slightly distorted square antiprisms, which show compositions of [(Dy2)O_3.667_F_4.333_]^8.667–^ and [(Dy3)O_4.667_F_3.333_]^9.667–^ (Figure 3, mid and right). The interatomic distances from the Dy^3+^ cations to the anions occurring here are in quite similar ranges with values of d(Dy2–O) = 224–252 pm, d(Dy2–F) = 224–245 pm and d(Dy3–O) = 227–250 pm, d(Dy3–F) = 224–235 pm, although by increasing the coordination number from seven to eight, a slight increase for the maximum distance can be observed.
Coordination polyhedra of the compositions [(Dy1)O4.333F2.667]8.333– (left), [(Dy2)O3.667F4.333]8.667– (mid), and [(Dy3)O4.667F3.333]9.667– (right) as well as the As3+ cations covalently bonded to most oxygen atoms and their remaining oxygen atoms in ψ1-tetrahedral [AsO3]3– anions in the crystal structure of Dy36O11F50[AsO3]12 ∙ H2O.
In the binary dysprosium sesquioxide (Dy_2_O_3_, bixbyite type), the dysprosium-oxygen distances are in the range of 215–254 pm, while in dysprosium oxide fluoride (DyOF, YOF type), they occur in a narrower interval of 227–234 pm. Thus, the refined bond lengths in Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O are in good agreement with those of the simpler representatives Dy_2_O_3_ and DyOF (Rudenko and Boganov, 1970; Dutton et al., 2012). For the distances to the F^−^ anions, a similar situation is observed, as they are 242–249 pm in DyOF and in binary DyF_3_ (YF_3_ type) in between 234 and 274 pm (Garashina and Sobolev, 1971; Dutton et al., 2012). Here, F^−^ anions always show slightly longer bonds to the Dy^3+^ cations as compared to the O^2–^ anions, while the O^2–^ and F^−^ anions as well as arsenic-bonded oxygen atoms in Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O have quite similar distances to the Dy^3+^ cations (Table 3).
The polyhedra around the Dy^3+^ cations are linked by both corners and edges with different patterns. For a better understanding, in the following, first the linkages of Dy^3+^-centered polyhedra to each other with crystallographically identical Dy^3+^ cations are described. The [(Dy1)O_4.333_F_2.667_]^8.333–^ polyhedra are connected to each other by common edges, and always four of these polyhedra form a tetrahedral body in which the central oxide anion (O4)^2–^ is tetrahedrally surrounded exclusively by four (Dy1)^3+^ cations (Figure 4, left). Two oxoarsenate(III) units [AsO_3_]^3–^ are attached to each polyhedron, with only one common oxygen atom being present. In the case of the [(Dy2)O_3.667_F_4.333_]^8.667–^ antiprisms, the situation is different: here, only three polyhedra are linked to each other exclusively by common edges. It is interesting to note that all three polyhedra share the (F1)- anion and the three oxygen atoms of an oxoarsenate(III) unit of the (As2)^3+^ cation provide the remaining edge links.
Linkage pattern of Dy3+-centered coordination polyhedra in the crystal structure of Dy36O11F50[AsO3]12 ∙ H2O with drawing of the linkage to polyhedra with the crystallographically identical central cations (Dy1)3+ (left), (Dy2)3+ (mid), and (Dy3)3+ (right).
The [(As2)O_3_]^3–^ anion thus bridges all three (Dy2)^3+^-centered polyhedra. This creates a distorted tetrahedral gap at the center of an imaginary triangular surface through the three (Dy2)^3+^ cations (Figure 4, mid). In the case of the [(Dy3)O_4.667_F_3.333_]^9.667–^ polyhedra, the situation is quite similar. Here, three antiprisms are connected by common edges, but the main difference to the case of the (Dy2)^3+^ cation lies in the bridging atoms. Here, the linked [(Dy3)O_4.667_F_3.333_]^9.667–^ polyhedra are surrounded on both sides by ψ^1^-tetrahedra [(As3)O_3_]^3–^, forming an empty trigonal prism from the oxygen atoms of these oxoarsenate(III) units (Figure 4, right).
The other linkage patterns are quite similar and are described here, but not further shown graphically. The polyhedra around (Dy1)^3+^ are connected with three (Dy2)^3+^-centered polyhedra each, with one edge and two corner links present. There are also three contacts to the (Dy3)^3+^-centered polyhedra, with two edge and only one corner linkage occurring here. For the [(Dy2)O_3.667_F_4.333_]^8.667–^ antiprism there are, in addition to the already mentioned, three contacts to the capped [(Dy1)O_4.333_F_2.667_]^8.333–^ prisms (twice via edge and once via corner); there are also contacts to four (Dy3)^3+^-centered polyhedra, with two edge and two corner-connections. The [(Dy3)O_4.667_F_3.333_]^9.667–^ antiprism is linked to three (Dy1)^3+^- and four (Dy2)^3+^- centered polyhedra, where it is linked to the [(Dy1)O_4.333_F_2.667_]^8.333–^ polyhedra twice via edge and once over corner and to the [(Dy2)O_3.667_F_4.333_]^8.667–^ antiprisms twice via edge and twice via corner.
The crystal structure exhibits three crystallographically different sites for the As^3+^ cations as well (Table 2). The first and second coordination spheres of these arsenic centers are identical in all three cases, only the bond lengths show a slight variance. In all cases, isolated ψ^1^-tetrahedra [AsO_3_]^3–^ are formed, whose oxygen atoms each have contact with a terminal and two bridging Dy^3+^ cations (Figure 5). Thereby, the arsenic-oxygen distances with values of 177–179 pm are in a rather narrow range, but very close to the typical arsenic(III)-oxygen bonds in claudetite-I (172–181 pm) (Pertlik, 1978a), claudetite-II (177–182 pm) (Pertlik, 1975), and arsenolite (179 pm, 3×; Table 3) (Pertlik, 1978b), to name just those of the crystalline As_2_O_3_ modifications.
The first and second coordination sphere of the As3+ cations occurring in the crystal structure of Dy36O11F50[AsO3]12 ∙ H2O as ψ1-tetrahedral [AsO3]3– anions with their Dy3+ decoration.
In the crystal structure, there are also eight anion positions, which do not maintain any contacts to the As^3+^ cations (Figure 6). The corresponding elements were assigned to the sites in such a way that all oxygen atoms are tetrahedrally surrounded by Dy^3+^ cations (C.N. = 4), while angled and trigonal coordination spheres also occur for the F^−^ anions (C.N. = 2 and 3). Two peculiarities stand out here: on the one hand, there is a mixed-occupied position with the (O5)^2–^ and (F5)^–^ anions, which is necessary for the charge neutrality of the compound; on the other hand, it is not possible to say with certainty whether only this site is actually mixed and all the other anion positions of Table 2 are occupied by only one kind of non-metallic element.
The anion sites occurring in the crystal structure of Dy36O11F50[AsO3]12 ∙ H2O without binding contacts to As3+ cations and their extended environments for (F1)− (A), (F2)− (B), (F3)− (C), (F4)− (D), (O4)2− (E), (O5/F5)1.667− (F) and (F6)− (G).
The (O4)^2–^-centered (Dy^3+^)4 tetrahedron could also be mixed with fluoride, since the coordination spheres hardly differ. However, since the counts (= multiplicity of Wyckoff positions) of the (O4)^2–^ and (O5)^2–^ sites and the possible variance of the negative charges due to the different Wyckoff positions (24d versus 96h) differ, charge neutrality would not be achievable by a pure mixed occupation of the (O4)^2–^ site. From a merely mathematical point of view, it would also be possible for both positions to be mixed, but for the sake of simplicity, only a mixed occupation of the (O5)^2–^ site was assumed as a structural model. Furthermore, oxygen as an O^2–^ anion also strives for higher coordination numbers than F^−^ anions, which is why a mixed occupation of the only two- and three-coordinated anion sites can be regarded as rather unlikely. The tendency of oxygen atoms, which are not covalently bonded to As^3+^ cations, to be coordinated by Ln ^3+^ cations in quaternary lanthanoid(III) oxide halide oxoarsenates(III) in the form of [OLn 4]^10+^ tetrahedra is already well-known from literature. In the compounds with the structured formula Ln 3_OX[AsO_3]2 (Ln = La–Nd, Sm–Dy for X = Cl, Ln = La–Nd, Sm, Gd–Dy for X = Br, and Ln = Pr for X = I) such [OLn 4]^10+^ tetrahedra are present, which share common edges according to {[OLn ]^4+^} (e = edge-connecting) to infinite chains along their tetragonal c-axis (Kang et al., 2005b; Kang, 2009; Kang and Schleid, 2007; Ben Yahia et al., 2009; BenYahia et al., 2010; Ledderboge, 2016).
The point of highest residual electron density is located at the origin of the unit cell (8a: 0, 0, 0). In terms of the scattering power, the intensity corresponds to a position occupied by an oxygen atom. However, this hypothetical oxygen atom has no binding contacts with other particles present in the unit cell. The closest contact is at 368 pm to As^3+^ cations and at 371 pm to F^−^ anions, well outside the range of plausible chemical bonding. However, based on the synthesis parameters, one explanation would be that this could be the oxygen atom of a crystal water molecule. Upon further search for a suitable hydrogen atom, residual electron density can also be found at a distance of about 96.4 pm (32e: x, x, x, with x = 0.0215) from this oxygen atom. However, due to the applicable symmetry operations, this hydrogen atom would be arranged tetrahedral around the central oxygen atom, a circumstance that does not seem to make sense from a structural-chemical point of view. An under-occupation of this hydrogen position by one half to achieve a neutral water molecule (H_2_O) would be possible and very likely with a H–O–H angle of 109.5°, but hardly be detected by X-ray diffraction. The hypothetical empirical formula of the title compound would then be Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O. The coordination environment of this interstitial crystal-water molecule is shown in Figure 7 with minimal H∙∙∙As distances of 272 pm and H∙∙∙F distances of 325 pm, both far too long for significant bridging hydrogen bonds. Figure 8 presents a section of the whole crystal structure including the cell edges.
Tetrahedral coordination environment of the oxygen atom Ow in the crystal structure of Dy36O11F50[AsO3]12 ∙ H2O, which can be explained by an intercalated crystal-water molecule in a rhomboid-dodecahedral cavity formed by (As1)3+, (As2)3+, and (F2)–.
Section of the crystal structure of Dy36O11F50[AsO3]12 ∙ H2O as viewed along the a-axis. The covalent bonds of the As3+ cations to their oxygen atoms within the ψ1-tetrahedral [AsO3]3– anions and the ones in the crystal-water molecules are emphasized.
Powder X-Ray Diffraction
In order to investigate the composition of the obtained material, powder X-ray diffraction techniques were applied (Figure 9). Since the available amount of the sample was rather low, the signal-to-noise-ratio of the data is challenging.
Measured (blue) and calculated (green) powder X-ray diffractogram of Dy36O11F50[AsO3]12 ∙ H2O. Unindexed reflections are marked with asterisks.
The target compound Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O could be identified as the main phase in the mixture. However, multiple reflections that do not belong to the target compound are also present (marked with “asterisks" *). Regarding the reaction conditions, the theoretical patterns of the starting materials and possible by-products were used to check if one or several of these phases are present in the reaction mixture. Nevertheless, none of the tested compounds (As_2_O_3_ in its claudetite- and arsenolite-type, respectively, Dy_2_O_3_ (A-, B- and C-type), DyOF, DyF_3_, Dy[AsO_4_] (xenotime- and scheelite-type), and even Dy_5_F_3_[AsO_3_]4) could be identified as possible side-phases.
Since no As_2_O_3_ and Dy_2_O_3_ seem to be residual, a complete reaction of the starting materials should have taken place that lead to multiple products of which the target compound Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O can be seen as the main phase.
Raman spectroscopy
To further investigate the mysterious crystal and to verify the interstitial crystal-water molecules, a single-crystal Raman spectrum of Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O was recorded with an excitation wavelength of λ = 638 nm (Figure 10). The peaks in the range from 100 to 500 cm^–1^ (107, 168, 206, 293, 353, 424 cm^–1^) can be assigned to the stretching vibrations ν(Dy(O,F)) and the deformation vibrations δ(AsO_3_) with the three strongest ones probably belonging to ν(Dy(O,F)) and lattice vibrations. The two peaks at 608 and 655 cm^–1^ with the shoulder at 700 cm^–1^ are caused by the antisymmetric stretching vibrations ν as(AsO_3_) of the three crystallographically different [AsO_3_]^3–^ anions, whereas the significantly stronger three peaks at 733, 763, and 792 cm^–1^ undoubtedly belong to the symmetric stretching vibrations ν s(AsO_3_) of these units. The slight elevation at 1600 cm^–1^ can be assigned to the deformation vibrations δ(H_2_O) of the crystal-water molecules, while the very sharp peak at 3607 cm^1^ belongs to the symmetrical valence vibration ν s(H_2_O) of them and the somewhat smaller one at 3637 cm^–1^ stems from the antisymmetrical one ν as(H_2_O). The two weaker and broader peaks at 3520 and 3553 cm^–1^ can be interpreted as results from the stretching vibrations ν(H_2_O)_ n _ of other very few water species that are bound to the detected crystal-water molecule, but may also well be surface-bonded water of the investigated crystal. The very broad band from 3000 to 3400 cm^–1^ can be attributed to air humidity and is always observable in spectra from this kind of instrument (Weidlein et al., 1981; Weidlein et al., 1986).
Single-crystal Raman spectrum of Dy36O11F50[AsO3]12 ∙ H2O recorded at an excitation wavelength of λ = 638 nm.
Microprobe analysis
Electron microscopy and X-ray spectroscopy methods were used to further characterize Dy_36_O_11_F_50_[AsO_3_]12 ∙ H_2_O. Figure 11 shows a backscattered electron image of a single crystal, which demonstrates the above-average crystal size and the almost isotropic crystal growth. Before the single-crystal X-ray diffraction, the likewise recognizable covering particles were rinsed off by washing them in paraffin oil.
Backscattered electron image of a single crystal of Dy36O11F50[AsO3]12 ∙ H2O recorded at an accelerating voltage of 15 kV and a current of 4 nA. The formed edges of the crystal are clearly visible.
Qualitative wavelength-dispersive X-ray spectra (WDXS) were recorded to verify the elements assumed in the single-crystal structure refinement. For the lighter elements, pseudo-crystal multilayer elements were used as diffraction crystals in the spectrometers in order to reach the low-energy regions. The relevant spectra of the measurements carried out on the single crystal are shown in Figure 12. The energy range for the heavy atoms dysprosium and arsenic corresponds to the expectations. In the energy ranges not shown here outside these characteristic lines, there are no extraneous bands from other types of atoms. It can also be seen that it would not be possible to detect arsenic using the (energy-dispersive) EDXS method, as interferences of Dy-M α and Dy-M β with the As-L α and As-L β lines occur, which can be resolved in the wavelength-dispersive system. The following findings can be obtained in the low-energy range of Figures 12C, D: it can be seen that both fluorine and oxygen are present and, furthermore, higher orders of the Dy-M α and Dy-M β lines are also detectable in the fluorine region, but with sufficiently low interference, at least for qualitative detection.
Wavelength-dispersive X-ray spectra (WDXS) of Dy36O11F50[AsO3]12 ∙ H2O in different energy ranges with the relevant emission lines depicted. Given are the regions of Dy-L α [LiF crystal, (A)], As-L α and As-L β [TAP crystal, (B)], as well as O-K α and F-K α (on two different spectrometers with multilayer elements, (C, D), respectively).
Conclusion
An unexpected cubic dysprosium(III) oxide fluoride oxoarsenate(III) hydrate with the composition Dy_36_O_11_F_50_[AsO_3_]12 · H_2_O could be obtained by water-assisted high-pressure synthesis from cold-welded gold ampoules in an attempt to synthesize Dy_5_F_3_[AsO_3_]4. Its crystal structure features Dy^3+^ cations with coordination numbers of seven and eight with respect to the non-metal elements (O and F) along with discrete ψ^1^-tetrahedral [AsO_3_]^3−^ anions. Interstitial crystal-water molecules are trapped within a large cavity confined by eight arsenic atoms and six fluoride anions. Upon heating some crystals up to 500 °C for several days, they were destroyed owing to decrepitation under water-release. From a distance, the “odd” composition Dy_36_O_11_F_50_[AsO_3_]12 · H_2_O with a = 2587.59(14) pm for Z = 8 could well be misinterpreted as “Dy_3_OF_4_[AsO_3_] · ^1^/12 H_2_O” with Z = 96 and even the low water-content might have been overlooked. Under these circumstances, the resulting formula “Dy_36_O_12_F_48_[AsO_3_]12 · H_2_O” for Z = 8 would have only 12 + 48 = 60 non-metal elements without bonds to arsenic instead of 11 + 50 = 61 as in the true composition. But after all, we have found no evidence for an under-occupation concerning any of these seven non-metal positions in Table 2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adala N.Marzougui B.Ben Smida Y.Marzouki R.Ferhi M.Onwudiwe D. C., (2022). An experimental and theoretical study of the structural, optical, electrical, and dielectric properties of Pr As O 4 . J. Alloys Compd. 910, 164894. 10.1016/j.jallcom.2022.164894 · doi ↗
- 2Ben Hamida M. (2007). Oxo-Selenate(IV) und Oxo-Arsenate(III) der Selten-Erd-Metalle und ihre Derivate. Dissertation. Oldenburg: Carl von Ossietzky Universität Oldenburg.
- 3Ben Hamida M.Warns C.Wickleder M. S. (2005). Syntheses and Crystal Structures of RE 2As 4O 9 (RE = Nd, Sm): Oxo-Arsenates(III) according to RE 4(As 2O 5)2(As 4O 8) Exhibiting the Cyclic As 4O 8 4– Anion, Z. Naturforsch. 60b, 1219–1223.
- 4Ben Yahia H.Pöttgen R.Rodewald U. C. (2009). Crystal Structure of the New Phosphate Ag Mn PO 4 , Z. Naturforsch 64b, 896–900.
- 5Ben Yahia H.Pöttgen R.Rodewald U. C. (2010). Condensed [O Pr 4]10+ and Discrete [As O 3]3−-Ψ1-Tetrahedra in Pr 5O 4Cl[As O 3]2 , Z. Naturforsch 65b, 1289–1292.
- 6Boyd F.England J. (1960). Apparatus for phase-equilibrium measurements at pressures up to 50 kilobars and temperatures up to 1750°C. J. Geophys. Res. 65, 741–748. 10.1029/jz 065i 002p 00741 · doi ↗
- 7Brahim A.Ftini M. M.Amor H. (2002). Cerium arsenate, Ce As O 4 , Acta Crystallogr. E 58, i 98–i 99.
- 8Dutton S.Hirai D.Cava R. (2012). Low temperature synthesis of Ln OF rare-earth oxyfluorides through reaction of the oxides with PTFE. J. Mater. Res. Bull. 47, 714–718. 10.1016/j.materresbull.2011.12.014 · doi ↗