Properties and predicted functions of large genes and proteins of apicomplexan parasites
Tiffany Fang, Amir Mohseni, Stefano Lonardi, Choukri Ben Mamoun

TL;DR
This paper explores the functions of unusually large proteins in apicomplexan parasites like Toxoplasma and Plasmodium, which may help them survive and interact with their hosts.
Contribution
The study identifies potential functions of large proteins in apicomplexan parasites, including roles in immune evasion and host interaction.
Findings
Large proteins in apicomplexan parasites contain domains linked to antigenic variation and erythrocyte invasion.
Some large proteins are predicted to participate in metabolic pathways or host interactions.
These large proteins are not strictly conserved and are expressed at low levels.
Abstract
Evolutionary constraints greatly favor compact genomes that efficiently encode proteins. However, several eukaryotic organisms, including apicomplexan parasites such as Toxoplasma gondii, Plasmodium falciparum and Babesia duncani, the causative agents of toxoplasmosis, malaria and babesiosis, respectively, encode very large proteins, exceeding 20 times their average protein size. Although these large proteins represent <1% of the total protein pool and are generally expressed at low levels, their persistence throughout evolution raises important questions about their functions and possible evolutionary pressures to maintain them. In this study, we examined the trends in gene and protein size, function and expression patterns within seven apicomplexan pathogens. Our analysis revealed that certain large proteins in apicomplexan parasites harbor domains potentially important for functions…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
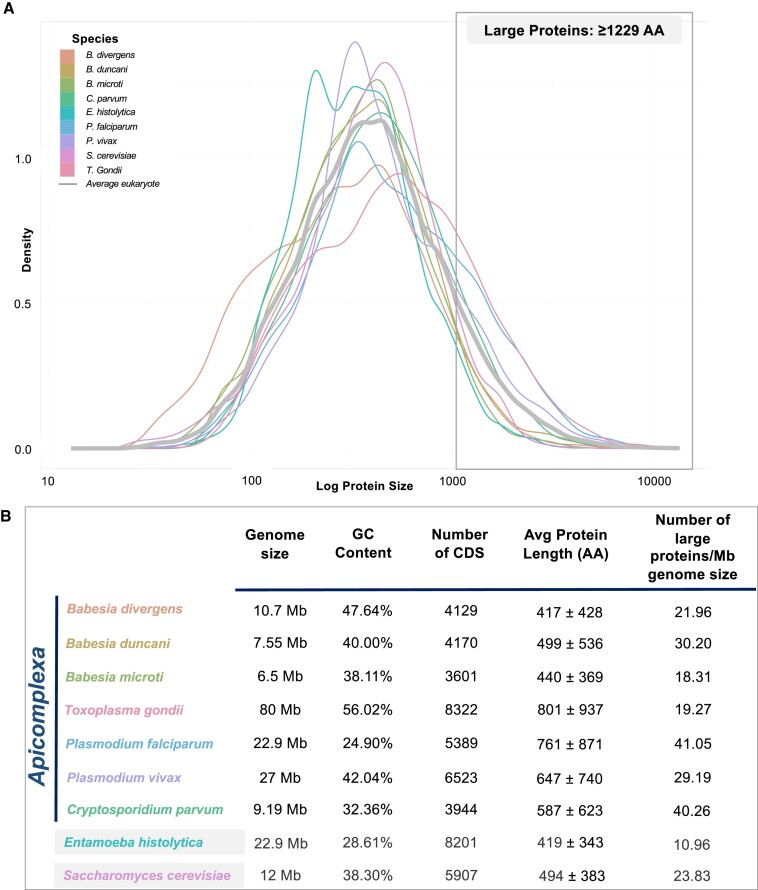 Figure 1
Figure 1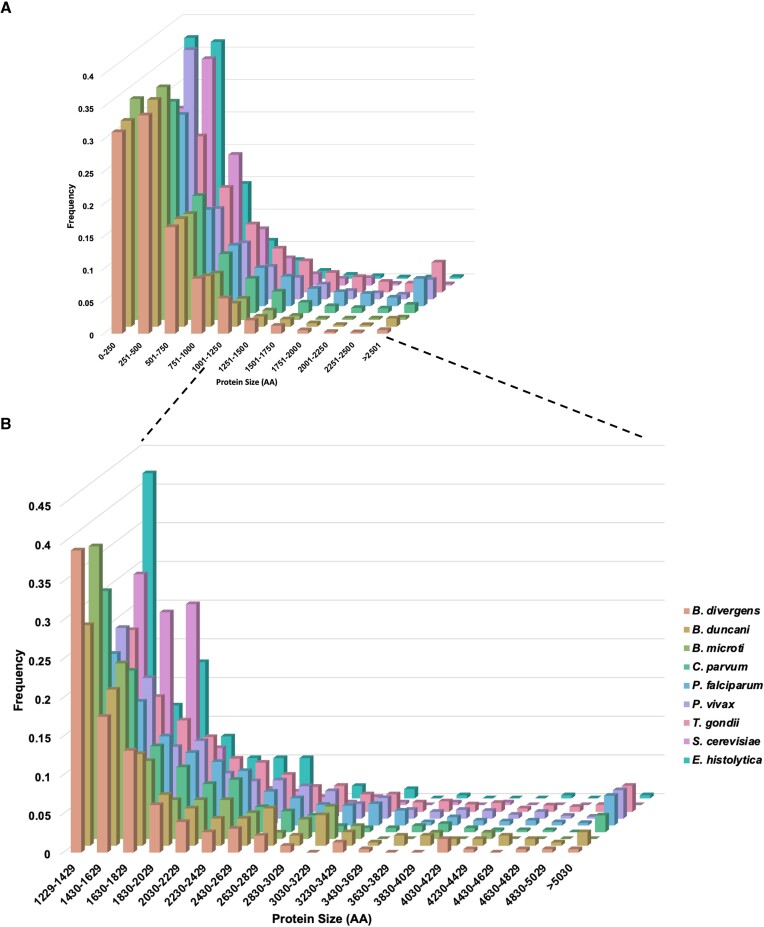 Figure 2
Figure 2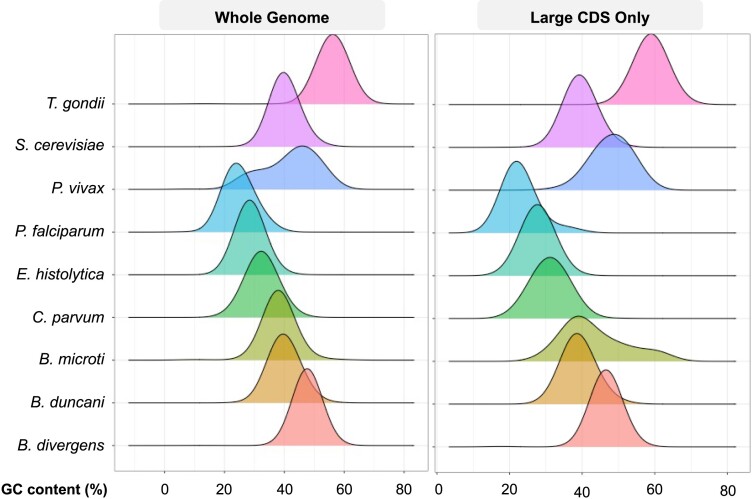 Figure 3
Figure 3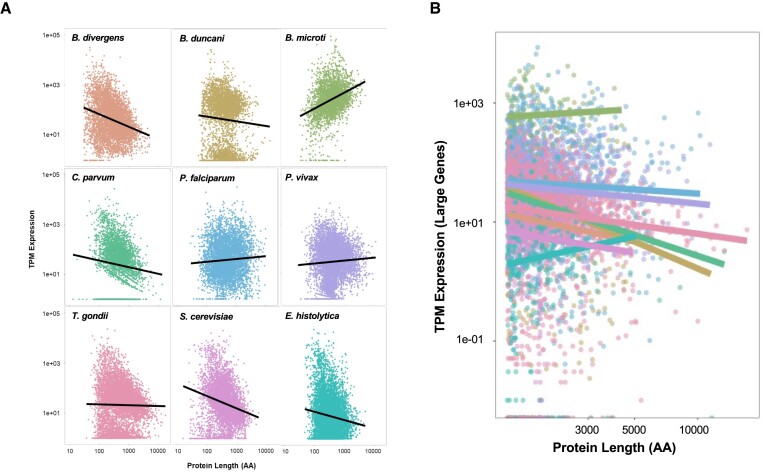 Figure 4
Figure 4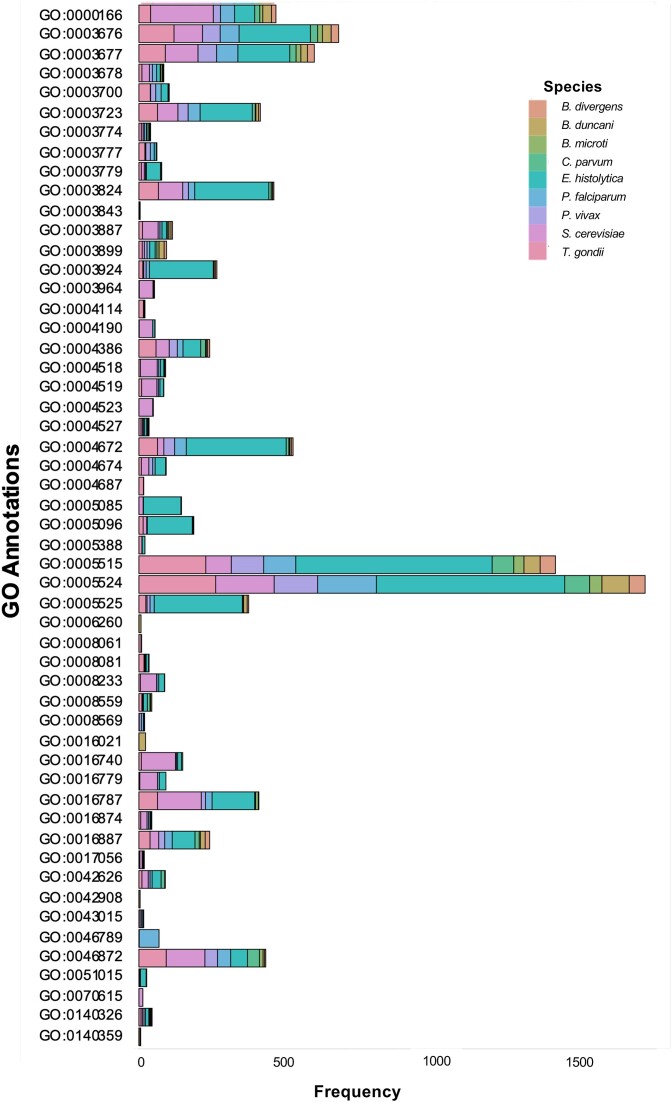 Figure 5
Figure 5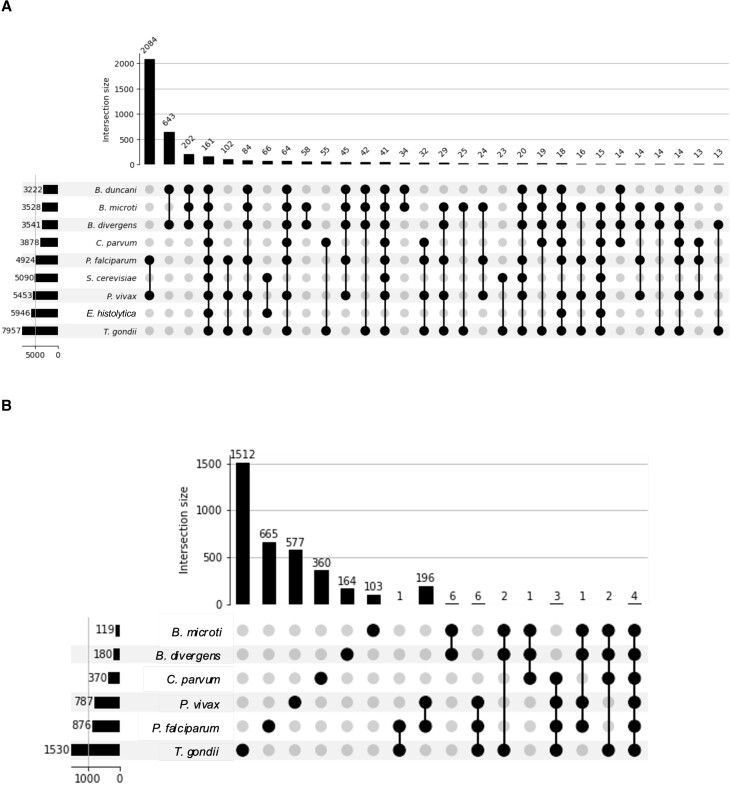 Figure 6
Figure 6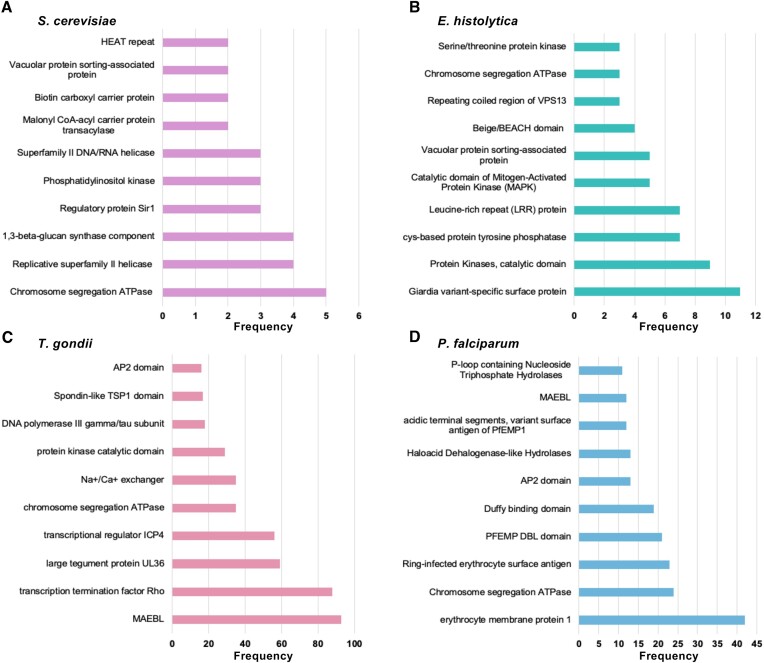 Figure 7
Figure 7- —National Institutes of Health10.13039/100000002
- —Steven and Alexandra Cohen Foundation10.13039/100016230
- —NBIA Disorders Association10.13039/100009582
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsToxoplasma gondii Research Studies · HIV Research and Treatment · Bacteriophages and microbial interactions
Introduction
The phylum Apicomplexa encompasses many species, several of which are responsible for significant diseases affecting humans, livestock and companion animals. Among these vector-borne pathogens are members of the genera Plasmodium and Babesia, which are responsible for human malaria and babesiosis, respectively. The mosquito-transmitted Plasmodium falciparum alone accounts for over 99.7% of fatal human malaria cases with 0.5 million deaths every year (1,2). On the other hand, tick-transmitted Babesia species can induce acute to life-threatening infection in humans and animals (3,4), with hospitalized babesiosis patients exhibiting severe symptoms at a rate exceeding 58% and a 1.6% death rate (5). Human babesiosis is attributed to several species of Babesia, with Babesia microti responsible for the majority of clinical cases reported so far (6). Other species include B. divergens, B. MO1, B. duncani and B. venatorum (4). The clinical presentation of human babesiosis varies widely, ranging from flu-like fever to severe complications such as anemia, disseminated intravascular coagulation, acute respiratory distress syndrome or even fatal outcomes. In Europe, B. divergens, a natural cattle pathogen, is the primary agent responsible for human babesiosis (3). Other members of the Apicomplexa phylum include Cryptosporidium parvum, a human and zoonotic pathogen transmissible through fecal matter that causes acute gastroenteritis (7). Toxoplasma gondii, the etiological agent of toxoplasmosis, affects ∼30% of the global human population and poses the risk of severe complications such as encephalitis, myocarditis and pneumonitis when transmitted through raw meat or contaminated water (8). As obligate intracellular parasites capable of salvaging nutrients from their mammalian host, apicomplexans do not need to support complex metabolic and biochemical processes; as a result, they generally have smaller genomes than free-living eukaryotes, ranging from around 8 to 130 Mb (9).
A recent study by Singh et al. shed light on the genome of B. duncani, revealing a compact 7.55 Mb genome housing 4222 genes with an average length of 1656 bp (9). Approximately 60% of this genome is involved in protein coding. Babesia duncani shares 842 core proteins (comprising 20% of its proteome) with other apicomplexan parasites; out of B. duncani’s 1242 unique proteins, around 70% lack functional annotations (9). Among these hypothetical proteins, BdWA1_000001 stands out as a polypeptide consisting of 11 561 amino acids. In a genome characterized by an average protein length of ∼500 amino acids, the presence of genes encoding such large proteins raises fundamental questions concerning their significance in parasite biology, development and virulence.
The prevalence of such large proteins extends beyond B. duncani, with other apicomplexan species boasting even larger proteins. For instance, the T. gondii proteome encodes seven proteins exceeding 10 000 amino acids, with its largest protein comprising 17 226 amino acids (10,11). Similarly, C. parvum, P. falciparum and Plasmodium vivax all encode proteins exceeding 10 000 amino acids (10,11).
We conducted a comprehensive bioinformatics analysis encompassing the genomes and proteomes of seven apicomplexan parasites, alongside two reference groups: one parasitic protist Entamoeba histolytica and the extensively studied nonparasitic unicellular organism Saccharomyces cerevisiae. Our aim was to uncover discernible patterns in the composition and functionality of large proteins within these organisms. Our analysis of the apicomplexan genomes revealed that the distributions of protein sizes skewed toward the right, revealing a tendency for larger genomes to accommodate larger proteins. Moreover, these larger proteins exhibited lower levels of expression, and their amino acid usage profiles closely mirrored those of their respective genomes. From a functional perspective, we observed that Gene Ontology (GO) functional annotations were not evenly spread across protein sizes. Several functional categories, such as those related to transport, metabolism and nucleic acid regulation, were significantly overrepresented in larger proteins across all seven species. Within these large proteins, we identified conserved domains with similar functions, as well as species-specific domains crucial for various aspects of parasite life cycles. These specialized domains facilitated processes such as host cell invasion, immune evasion and sustained proliferation. Importantly, although these domains were present in different species, they were not restricted to large proteins, suggesting their adaptable roles in diverse biological contexts.
Materials and methods
Genomic data acquisition
Genome assemblies were obtained from Singh et al. (9) (B. duncani) and VEuPathDB (B. divergens, B. microti, C. parvum, P. falciparum, P. vivax, T. gondii, E. histolytica and S. cerevisiae), using the reference genome for each species. The primary data collected included protein length, coding sequence (CDS) length, transcription levels (transcripts per million, TPM), number of transmembrane domains and computed GO function IDs. Protein and transcript sequences were also collected. Codon usage data for whole genomes were taken from organism-specific databases such as AmoebaDB, CryptoDB, FungiDB, PiroplasmaDB, PlasmoDB and ToxoDB.
Figure generation
Heatmaps were generated in R using the ‘heatmap2’ function within the ‘gplots’ package, size histograms of Figure 2 were generated with fixed bins in Excel and all other plots were created with the ‘ggplot’ package.
Protein size thresholding
Using summative data from 485 eukaryotic proteomes (12), the average eukaryotic proteome size and protein length were computed to be 17 215 proteins and 344 amino acids, respectively. Operating under the statistical assumption that eukaryotic protein sizes tend to approximate a log-normal distribution (13), a set of 17 215 normally distributed ‘log-protein size’ random values was generated around a mean of ∼2.53 with a standard deviation of ∼0.33 (calculated using quartile data on the proteomes). Based on this statistical analysis, a large protein is a protein whose length exceeds the 95th percentile of the log-normal distribution, namely 1229 amino acids.
Analysis of genomic properties
The normalized large protein quotient was calculated by dividing the number of large proteins in a specific genome by the genome size in Mb. This value for the ‘average eukaryotic genome’ was calculated by averaging the genome sizes of the previous 485 eukaryotes and dividing them by the average number of large proteins. Since ‘large proteins’ are determined to be the 95th percentile, they would account for 5% of the whole average eukaryotic proteome, thus being 860 proteins. The guanine and cytosine (GC) content was calculated using the ‘GC’ function in the BioPython package of Python.
Analysis of protein properties
Codon frequency data are reported in frequency per thousand; genome-wide amino acid usage was calculated by adding the frequencies of all codons that encode each amino acid. The amino acid frequencies were calculated as the fraction of occurrence across the entire protein.
Statistical GO analyses
A custom script was used to extract the GO annotations of all large proteins; for each annotation, the number of occurrences within the large proteome and that within the whole proteome were recorded (Figure 5A). A P-value was calculated for observing a greater number of occurrences of this annotation according to a hypergeometric distribution, with the threshold of significance set to 0.001; this identified 53 GO classes to be significantly overrepresented.
Motif prediction
Conserved domains for the largest proteins in Supplementary Table S1 were determined using CD-Search (14) based on amino acid sequence. Conserved domains for large proteins in general in Figure 6 were determined using the get_cdd() function within the ‘ragp’ R package (14). PFam domains were identified using OrthoMCL (15). We utilized DIAMOND (16) and configured it with the ‘--sensitive’ option to cluster and identify homologous gene hits of at least 40% identity and 80% coverage across all species mentioned in this study. Additionally, we visualized all overlapping orthologous gene sets using the ‘UpSetPlot’ Python package shown in Figure 7A and B.
Results
Size distribution of apicomplexan proteins
Despite the fact that all the species analyzed in this study belong to the Apicomplexa phylum, their genomes exhibit significant diversity. These parasites possess genomes of varying sizes, ranging from 6.5 Mb in B. microti to 80 Mb in T. gondii (Figure 1B). We calculated a normalized number of large proteins per species (Figure 1B), with a higher value representing a greater number of large proteins per Mb of genome size and more investment of the species into maintaining large proteins. All species in this study show greater large protein density than the average eukaryotic genome (5.157 large proteins per Mb); large protein density is also higher for Apicomplexa than the two outgroups. Notably, all genomes in this study display a right-skewed distribution, with a preponderance of smaller proteins in each size category (Figure 2). However, when we analyze protein size distribution on a logarithmic scale, it conforms to a Gaussian distribution (Figure 1A), a pattern consistent with the average eukaryotic genome. Using this statistical approach, we defined ‘large proteins’ in this study as those exceeding the 95th percentile of the distribution, corresponding to proteins longer than 1229 amino acids. This encompasses 235 proteins in B. divergens, 228 in B. duncani, 119 in B. microti, 1542 in T. gondii, 940 in P. falciparum, 788 in P. vivax, 370 in C. parvum, 251 in E. histolytica and 286 in S. cerevisiae.
(A) Probability density function estimation of log size distribution of proteins in all genomes, smoothed with Gaussian kernel and compared with the ‘average eukaryotic protein size distribution’. (B) Genomic overview including genome size, GC content, number of CDSs, average protein length and number of large proteins normalized over genome size in Mb.
(A) Histogram showing distribution of protein size relative to number of total proteins in all parasite proteomes divided into size bins of 250 amino acids. (B) Distribution of protein size ratio over large proteins in all parasites.
GC content and protein size
By examining the GC content of CDSs, we found that GC content distribution varies among species (Figure 3), with P. falciparum displaying the lowest average GC content at 24.9% and T. gondii the highest at 56.02% (Figure 1B). Interestingly, the GC content distributions in long CDSs (encoding large proteins) often mirror the overall GC content of the species. However, for certain species, large CDSs exhibit either more extreme (e.g. P. falciparum’s large CDS has an even lower average than the overall) or different (e.g. P. vivax and B. microti) GC content distributions compared to the overall genome. While most GC distributions approximate a normal distribution, the large CDSs of B. microti and whole genome of P. vivax appear bimodal. Moreover, GC content does not appear to be strongly correlated with CDS length, with Pearson correlation coefficient below 0.35.
GC content distribution by CDS length for whole genome and large genes of apicomplexan and outgroup species studied.
Expression levels of genes encoding large proteins
We further examined the expression profile of genes encoding from all selected species using transcriptional data available in VEupathDB. We found that larger genes encoding large proteins tend to be expressed at lower levels compared to shorter genes, which display a more diverse expression profile (Figure 4A). This trend persists across various species. Further investigation of the expression levels of the largest genes (Figure 4B) showed a similar trend with shorter genes having a more varied range of possible expressions, whereas large genes were almost exclusively expressed at low levels.
Log10-transformed scatterplots of TPM expression level by protein length across (A) the whole genome and (B) large proteins of each species, with trendlines for each.
Amino acid usage in large proteins
When examining amino acid usage in large proteins, we observed significant variation depending on the species and amino acid type (Supplementary Figures S1 and S2). In the case of B. duncani, lysine fraction held a general negative correlation with length across all proteins, but an opposite positive correlation when focusing on large proteins. In contrast, the arginine fraction displayed a general negative correlation with length across all B. duncani proteins including large proteins, whereas the cysteine fraction displayed a general positive correlation with length across all proteins including large proteins (Supplementary Figures S1 and S2). In the case of P. falciparum, asparagine fraction displayed a strong positive correlation among average-size proteins but was less correlated to length in large proteins (Supplementary Figure S4). In contrast, the arginine fraction displayed a general negative correlation with length across all P. falciparum proteins excluding large proteins (Supplementary Figures S2–S4). These patterns of amino acid usage align with codon usage patterns (Supplementary Figure S3) as well as GC fraction. For example, the high AT content of P. falciparum supports the prevalence of the AAU codon and consequently the abundance of asparagine in its proteins.
GO functional analyses of large proteins across Apicomplexa
To discern the role of large proteins, we investigated whether the predicted functions of large proteins differ from those of average-size proteins. Assuming a hypergeometric distribution, we identified significantly enriched categories within each large proteome, with a P-value <0.001, except for B. divergens. Of all the GO functional classes attributed to large proteins, 53 unique GO functions were found to be overrepresented (Figure 5). Common highly represented annotations from all species examined include functions related to protein binding (GO:0005515), ATP binding (GO:0005524), DNA binding (GO:0003677) and nucleic acid binding (GO:0003676). In addition to the GO annotations generally overrepresented in all large proteomes, five parasites exhibited distinctive and disproportionately overrepresented annotations in their large proteomes. Notably, B. duncani’s large proteins showed an overrepresentation of functions related to nuclear export and aminoacylation, Babesia microti’s large proteins were characterized by a higher representation of functions associated with protein ubiquitination, whereas P. falciparum’s large proteins displayed unique and parasite-specific annotations, with a highly significant presence of host cell binding functions (P = 3.21e−39) compared to the rest of the proteome. In the case of P. vivax, unique functions enriched in its large proteins included cysteine peptidases and gamma-tubulin binding proteins, suggesting potential involvement in processes such as protein degradation, amino acid utilization, protein secretion, cytoskeletal organization (17) or nucleation of microtubule heterodimers (18). Toxoplasma gondii’s large proteome exhibited an overrepresentation of phosphodiesterase, chitin binding and zinc binding annotations. Notably, the chitin-binding-like domains of Toxoplasma possess lectin properties that play a pivotal role in host cell binding (19). While zinc ion binding was specifically enriched in T. gondii’s large proteins, the ‘metal ion binding’ annotation was shared among large proteins across the Apicomplexa, indicating a potential role of large proteins in zinc salvage pathways (20). Furthermore, domains linked to functions such as protein phosphorylation, transcription and xenobiotic transport were overrepresented in large proteins from parasites but not in the model organism S. cerevisiae. Entamoeba histolytica displayed significant representation across most of these GO categories, indicating a commonality among Apicomplexa. Altogether, the functions likely served by large proteins can be categorized into three main groups: transport and signaling, metabolic processes, and nucleic acid or protein synthesis.
Frequency of occurrence of significantly overrepresented (P < 0.001) GO classes in large proteins of all parasitic genomes and outgroups determined via hypergeometric distributions (GO annotation glossary is provided in Supplementary Table S3).
Comparative analysis of large protein orthologs in apicomplexan parasites and outgroup organisms
To investigate the potential functional similarities among large proteins within Apicomplexa, we utilized the DIAMOND software package to identify shared ortholog groups between the complete proteomes and the subsets of large proteins from seven apicomplexan parasites and two outgroup species (Figures 6A and B, respectively). Among the 30 species groupings with the greatest number of orthologs, P. falciparum and P. vivax exhibit a shared set of 2084 gene orthologs, while B. duncani and B. divergens share 643 orthologs. Collectively, all species in the study share 161 orthologs across their entire proteomes. Intriguingly, apicomplexan parasites share a greater number of orthologs with S. cerevisiae (41 orthologs) than with E. histolytica (18 orthologs). However, these orthologs are not predominantly large proteins. Specifically, among the large proteins, only 196 orthologs are shared between P. falciparum and P. vivax, with all other intersections containing <10 orthologs (Figure 6B). Not all species are depicted due to the absence of orthologs in some cases, highlighting the lack of orthologs among all the large proteomes included in this study.
UpSet plots illustrating (A) the top 30 ortholog groupings of species in this study (top: total number of shared proteins among different organisms; bottom left: number of shared genes per organism) and (B) ortholog intersections in large proteins.
Functional domain conservation among largest proteins across diverse species
To investigate the functional conservation of the largest proteins among each of the large protein sets across different apicomplexan species, various motif analyses were conducted, including predictions for transmembrane domains, glycosylphosphatidylinositol (GPI) anchors and conserved motifs (Supplementary Table S1). The search for transmembrane domains revealed notable differences among species. For instance, the largest protein in B. duncani, BdWA1_000001, has 18 potential transmembrane domains, whereas the largest protein in P. falciparum, PF3D7_0628100, has only 2 potential transmembrane domains. No transmembrane domains were identified in the largest proteins in the other species analyzed in this study. Conversely, GPI anchor prediction tools did not yield any hits for any of the largest proteins. Subsequent conserved-domain searches were conducted on the largest proteins from each species, and the identified domains were further classified into functional groups such as transport and signaling, canonical metabolic processes, and nucleic acid or protein synthesis (Supplementary Table S1). The only ‘largest protein’ that returned no results is the B. duncani BdWA1_000001, so an additional conserved-domain search was made on the closest size analog of BdWA1_000001 to possibly predict its function. Unique conserved-domain hits are summarized in Supplementary Figure S1, while ortholog groups and PFam domains can be found in Supplementary Table S2. Overall, our analysis revealed a degree of functional similarity among the conserved domains in different species. Notably, the largest protein in B. divergens contains a RAB escort domain linked to membrane trafficking (21) and a chorein-like domain involved in intracellular protein transport and lipid transfer (22). In B. microti, the largest protein harbors a vWF (von Willebrand factor) domain found in plasma proteins such as complement factors, integrins, collagens and other matrix proteins involved in multistep signal transduction pathways (23). Further analysis using PFam domain search found six domains within this protein commonly found in proteins of the AAA (ATPases associated with various cellular activities) family. More specifically, these domains belong to the dynein-related subfamily, suggesting a potential involvement in transport functions (24). In C. parvum, the largest protein displayed 29 conserved domains linked to metabolic processes, including polyketide synthesis and fatty acid metabolism (25,26). One of the domains found in the largest protein of C. parvum is a Kringle domain found in proteins involved in blood clotting (27). The largest protein in P. falciparum contains a HECT domain involved in ubiquitination, and three copies of ankyrin repeats associated with various functions such as transcriptional regulation, ion transporter and signal transduction (28). Similarly, T. gondii’s largest protein also contains a HECT domain. The most notable domain found in the largest protein of P. vivax is associated with pseudouridylate synthase function, which is important for the modification of uracil bases to pseudouridine (29). One region was also similar to TRAFs, which are proteins that regulate cell survival and stress responses in the immune system. Other domain hits were more parasite-specific, including a malarial adhesin-like domain and a microneme/rhoptry antigen.
To broaden our understanding, conserved-domain searches were extended to outgroup species and select Apicomplexa species. Outgroups displayed fewer conserved-domain hits than the apicomplexan species analyzed. In general, these proteins were found to encode housekeeping functions. For example, some of the most common domains in S. cerevisiae large proteins are involved in mitosis (chromosome segregation and replicative helicases), cell wall synthesis, lipid synthesis and intracellular trafficking (Figure 7A). Most of the common conserved domain hits in E. histolytica large proteins are associated with similar functions, notably transport, cell cycle control and signal transduction. The largest protein of E. histolytica contains a Giardia variant-specific surface protein (VSP) domain (Figure 7B) found in cysteine-rich VSP proteins, involved in antigenic variation and survival of the parasite in the host (30). In T. gondii, common conserved domains include those found in merozoite adhesive erythrocytic binding proteins (MAEBL), herpes tegument protein UL36 (31), regulatory protein ICP4 (32) and AP2 factors (Figure 7C). Five of P. falciparum’s 10 most common hits include various domains of the P. falciparum erythrocyte membrane protein 1 (PfEMP1), which is expressed by malaria parasites during their blood-stage development and serves as both surface antigen and adhesion molecule with a critical role in virulence (33) (Figure 7D). This is consistent with data from VEuPathDB annotations, indicating that 61 of the 940 large proteins of P. falciparum are classified as PfEMP1 proteins. Similarly, of the 235 large proteins of B. divergens, 36 are annotated as members of the variable erythrocyte surface antigen.
Top 10 most common conserved domains among large proteins of controls (A) S. cerevisiae and (B) E. histolytica as well as select Apicomplexa (C) T. gondii and (D) P. falciparum.
Discussion
This study is the first of its nature to investigate the properties, distribution, expression and predicted functions of large proteins of apicomplexan parasites. These proteins have been historically understudied, with their functions remaining poorly elucidated even in model organisms. This limited knowledge stems from the inherent challenges associated with expressing these proteins as full-length polypeptides to assess their biochemical, cell biological or structural properties. Moreover, while some pathogens, such as T. gondii, benefit from well-established genetic tools facilitating gene-specific and large-scale functional analyses through gene deletion, editing or regulation, genetic manipulation remains challenging for most apicomplexan parasites. In some cases, such as B. microti or B. duncani, this field of research is in its infancy (34–37). The use of bioinformatics tools offers a unique opportunity to start exploring the hidden world of large proteins in the biology and pathogenesis of these parasites. Our investigation revealed that very large proteins occur infrequently and are also not highly expressed. Additionally, we found a higher abundance of large proteins, including those considered ‘very large’, within the Apicomplexa group compared to the nonparasitic unicellular outgroup S. cerevisiae and the parasitic protist outgroup E. histolytica.
The two populations with bimodal GC distributions show interesting trends. For B. microti, there does not seem to be a clear functional difference between the pool of genes with a median GC of 38% and the one of 60%; the bimodality could be due to the fact that there are only 53 genes encoding large proteins in B. microti, so a larger sample size may yield a greater degree of normality. For P. vivax, across both the 30% GC median group and the ∼45% GC median group, predominant functional annotations include hypothetical conserved Plasmodium genes, Plasmodium interspersed repeat (PIR) genes and zinc finger genes. PIR genes comprise one of the largest multigene families in Plasmodium, with their expression indicative of blood-stage parasitic infection but not necessarily playing a role in antigenic variation (38). The zinc finger protein is a common DNA-binding domain in several transcription factors, not necessarily indicative of differential expressions of any function in particular.
Based on trends in abundance of amino acids, it may be possible to make inferences on the predicted structures of large proteins. Since most higher level computational forms of structure prediction (e.g. AlphaFold) are unavailable to large proteins, any insight into potential structures or domains present in these proteins would provide information into their functions and reasons for evolutionary conservation. However, the role of cysteine abundance patterns remains unclear.
Through GO annotation, we identified specific functional classes that large proteins are more likely to belong to. These functional classes primarily fall into three categories: transport and signaling, metabolic processes, and nucleic acid or protein synthesis, annotations more likely to be essential at least in the blood stages of Plasmodium parasite. Within the category of large proteins, smaller proteins ranging from ∼1000 to 3000 amino acids tend to be better annotated than those exceeding 10 000 amino acids. This discrepancy emphasizes the limitations of predicting the functions of less annotated very large proteins based solely on the overrepresentation of a particular functional class.
Based on conserved domain hits, it is evident that most large proteins fulfill essential housekeeping functions, with specific roles varying among different species. These roles commonly include the maintenance of cell cycle function, transport processes and metabolic functions. Additionally, each species exhibits its own set of species-specific domains. For example, S. cerevisiae expresses domains related to fungal cell wall synthesis, E. histolytica expresses Giardia variant-specific surface domains and P. falciparum expresses PfEMP1-like domains. Interestingly, E. histolytica likely shares domains similar to Giardia due to their common status as intestinal protozoan parasites (39). However, the origin and specific functions of these proteins in E. histolytica remain to be determined. These variant surface domains play a crucial role in antigenic variation among parasites, contributing to their infectivity. Similarly, PfEMP1, a multigene family of highly polymorphic erythrocyte surface proteins, aids in immune evasion and erythrocyte sequestration to promote intraerythrocytic growth and sustain chronic infections (33). Our analysis showed that several large proteins of T. gondii and P. falciparum contain MAEBL and AP2 domains. MAEBL, originally found in P. gallinaceum, mediates binding of parasites to the erythrocyte and is highly conserved among Plasmodium species (40). AP2 genes encode DNA-binding proteins serving as transcription factors, with the AP2 domain being highly conserved among Apicomplexa, including Babesia and Theileria species (41). These domains, however, were found in the large proteins of Babesia species, but appear to occur primarily in proteins shorter than 1229 amino acids (9,41).
While our knowledge about BdWA1_000001, the 11 561-amino-acid-long protein of B. duncani, remains very limited, conserved-domain searches of the largest proteins in other apicomplexan proteomes may provide insight into their functions. Some of these domains are species-specific, such as malarial adhesin-like domains of P. vivax, which could play a role in parasite attachment and host cell entry (42). However, even the domains that are not specific to apicomplexans could possibly play very specialized roles in parasite development and interaction with the host. The largest protein in C. parvum returned 29 domain hits, most of which belonged to polyketide synthesis domains, NAD binding domains and phosphopantethine prosthetic groups, which together appear to be involved in carbon chain elongation for fatty acid synthesis (43). This pathway is likely maintained as C. parvum salvages free fatty acids and uses its own elongation mechanisms to generate complex lipid structures (44). Babesia divergens and B. microti both have more transport-related domains, with B. divergens’s domains being involved in intracellular transmembrane protein trafficking and Rab-mediated vesicle transport and B. microti’s domains more pertaining to nuclear functions such as transcription and export of ribosomes (45–47). The Rab-based vesicle transport mechanism is crucial to the efficacy of intracellular parasite transport, as it modulates both endocytic uptake and secretion to micronemes and rhoptries (48). Although the conserved domains of these largest proteins vary by genome, they all serve essential functions, particularly in transport and signaling or metabolism.
While conserved domains are vital for the functions of large proteins across species, it is crucial to recognize that these domains are not exclusively present in large proteins. Current data do not indicate the presence of selective pressures promoting highly conserved large proteins essential to Apicomplexa functionality. Large proteins across species tend to share functional annotations related to general macromolecule synthesis, particularly fatty acid synthesis and transport mechanisms. These proteins serve as intracellular modulators of secretion pathways, as well as catalysts for replication and synthesis. Further research, encompassing cell biological and genetic studies, is imperative to unravel the functions of these large proteins and their significance in the pathogenesis and virulence of protozoan parasites.
Supplementary Material
lqae032_Supplemental_Files
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Maier A.G., Matuschewski K., Zhang M., Rug M. Plasmodium falciparum. Trends Parasitol. 2019; 35:481–482.30595467 10.1016/j.pt.2018.11.010 · doi ↗ · pubmed ↗
- 2World Health Organization World Malaria Report 2022. World Health Organization. 2022; 14–16.
- 3Ord R.L., Lobo C.A. Human babesiosis: pathogens, prevalence, diagnosis and treatment. Curr. Clin. Microbiol. Rep. 2015; 2:173–181.26594611 10.1007/s 40588-015-0025-z PMC 4649939 · doi ↗ · pubmed ↗
- 4Renard I., Ben Mamoun C. Treatment of human babesiosis: then and now. Pathogens. 2021; 10:1120.34578153 10.3390/pathogens 10091120 PMC 8469882 · doi ↗ · pubmed ↗
- 5Bloch E.M., Day J.R., Krause P.J., Kjemtrup A., O’Brien S.F., Tobian A.A.R., Goel R. Epidemiology of hospitalized patients with babesiosis, United States, 2010–2016. Emerg. Infect. Dis. 2022; 28:354–362.35076004 10.3201/eid 2802.210213 PMC 8798708 · doi ↗ · pubmed ↗
- 6Swanson M., Pickrel A., Williamson J., Montgomery S. Trends in reported babesiosis cases—United States, 2011–2019. MMWR Morb. Mortal. Wkly Rep. 2023; 72:273–277.36928071 10.15585/mmwr.mm 7211 a 1PMC 10027409 · doi ↗ · pubmed ↗
- 7Gerace E., Lo Presti V.D.M., Biondo C. Cryptosporidium infection: epidemiology, pathogenesis, and differential diagnosis. Eur. J. Microbiol. Immunol. 2019; 9:119–123.10.1556/1886.2019.00019 PMC 694599231934363 · doi ↗ · pubmed ↗
- 8Liu Q., Wang Z.D., Huang S.Y., Zhu X.Q. Diagnosis of toxoplasmosis and typing of Toxoplasma gondii. Parasit. Vectors. 2015; 8:292.26017718 10.1186/s 13071-015-0902-6PMC 4451882 · doi ↗ · pubmed ↗