Fibrodysplasia Ossificans Progressiva: A Case Report
Linzeng Qi, Yongyuan Guo

TL;DR
This paper presents a case of Fibrodysplasia Ossificans Progressiva, a rare genetic disorder causing abnormal bone growth, in a 22-year-old woman.
Contribution
The study contributes a detailed clinical case report with genetic confirmation and treatment outcomes in a young FOP patient.
Findings
The patient exhibited typical FOP features including toe deformities and soft tissue ossification confirmed by imaging and ACVR1 gene mutation.
Treatment with methylprednisolone and alendronate sodium vitamin D3 showed some therapeutic efficacy.
Early diagnosis and comprehensive management can improve quality of life despite the lack of definitive cure.
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare autosomal dominant genetic disorder characterized by congenital great toe malformations and progressive ectopic ossification. We report a typical case of FOP in a 22-year-old female patient presenting with limited movement of the left knee joint, which began following trauma in 2019. Clinical examination revealed a large mass behind the left knee, bilateral great toe deformities, and no palpable superficial lymph nodes, without systemic pain or other discomfort. Imaging and genetic testing further supported the diagnosis of FOP, demonstrating high-density ossification within soft tissues and a mutation in the ACVR1 gene. Treatment involved a combination of methylprednisolone and alendronate sodium vitamin D3 tablets, which yielded some therapeutic efficacy. The discussion emphasizes clinical diagnosis, pathogenesis, and treatment…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
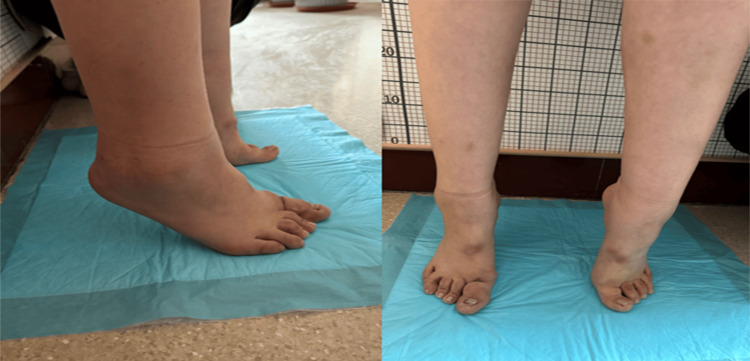 Figure 1
Figure 1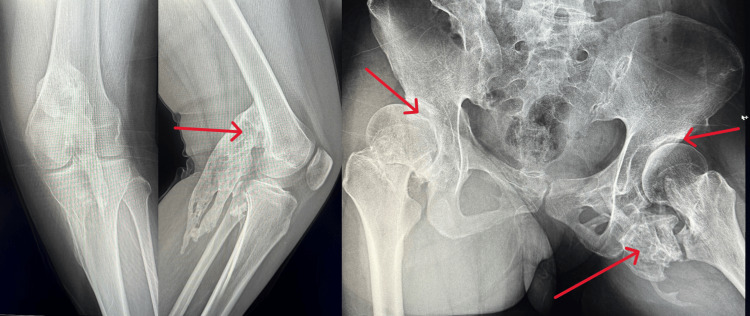 Figure 2
Figure 2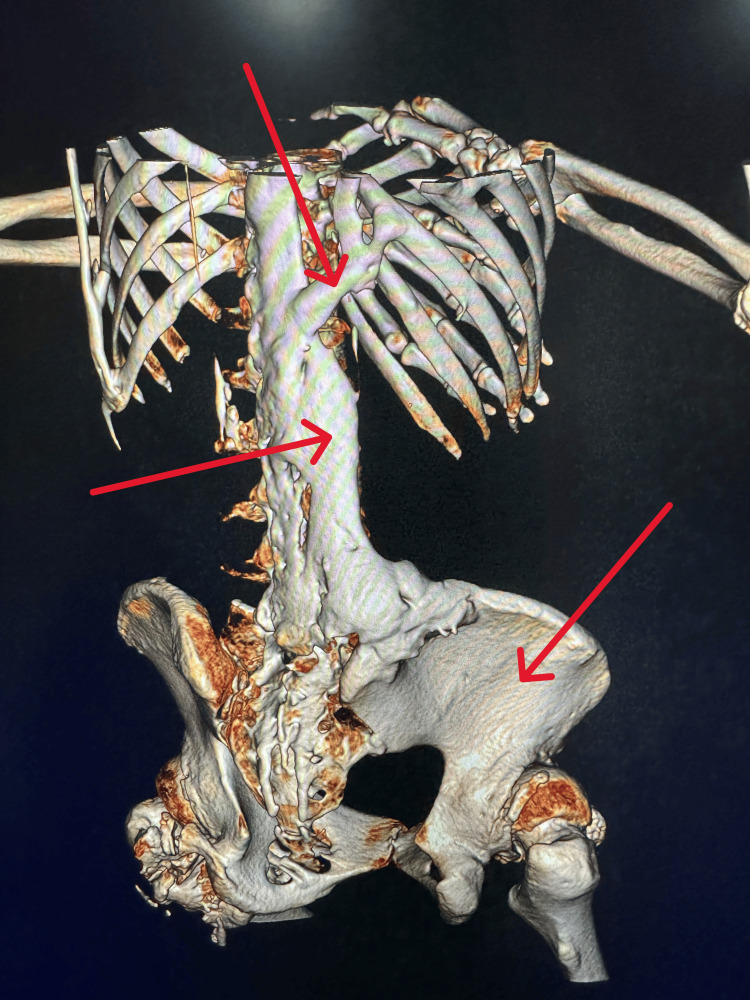 Figure 3
Figure 3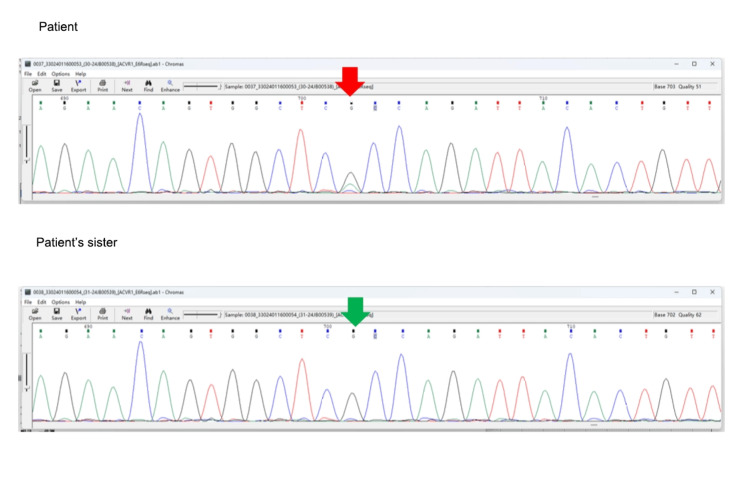 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHeterotopic Ossification and Related Conditions · Medical Imaging and Pathology Studies · Genetic Syndromes and Imprinting
Introduction
Fibrodysplasia ossificans progressiva (FOP), also known as myositis ossificans progressiva (MOP), is a rare autosomal dominant genetic disorder characterized by congenital malformation of the great toes and progressive ectopic ossification, with an estimated incidence of 1/2,000,000 to 1/1,300,000 [1]. Clinically, it is mostly reported as individual cases, with limited research on its pathogenesis and treatment strategies [2]. Due to the challenging clinical course of this patient, we report a typical case of FOP and provide an analysis and summary of its clinical characteristics and treatment.
Case presentation
A 22-year-old female patient was admitted to the Department of Orthopedics at Qilu Hospital of Shandong University on December 3, 2023, due to limited movement of the left knee joint for three years. Since sustaining trauma in May 2019, followed by a 15-day hospitalization at a local hospital, the patient experienced abnormal movement in both feet, left knee, left hip, and right shoulder. Over the subsequent three years, coinciding with the COVID-19 pandemic, the patient's symptoms worsened progressively despite infection with the virus and vaccination. Presently, the patient has lost mobility in the lumbar spine, cervical spine, and left knee, with a large mass behind the left knee originating from the distal femur. Mobility is also compromised in the right shoulder. Muscle strength and sensation in all limbs are normal. The patient exhibits bilateral great toe deformities characterized by outward deviation (Figure 1).
Photographs of the anteroposterior and lateral ankle jointsDouble full arch disappeared, great hallux valgus deformity.
Laboratory investigations including blood routine, inflammatory markers, thyroid function, anti-nuclear antibodies/antibody spectrum, markers of vasculitis, IgG4 levels, and tumor markers were within normal limits. Alkaline phosphatase (ALP) levels showed no significant abnormalities. Whole-body long bone radiography revealed high-density ossification in the soft tissue behind the left knee joint and irregular bone density along the inner edge of the upper segment of the right tibia. The spinal curvature was straightened, with irregular morphology of multiple ribs. Bilateral hip dysplasia and osteoarthritis were noted, along with patchy ossification below the right hip joint (Figure 2).
The X-ray of the left knee and hipThere was a large mass in the left knee joint, and dysplasia and multiple large masses in the hip joint.
Whole-body CT revealed no significant abnormalities in the skull, with partial fusion of cervical spinous processes and posterior edges. Ectopic ossification was observed around the bilateral scapulae, hip joints, knee joints, and lumbar spine with the ilium (Figure 3).
CT imaging Multiple ectopic ossification can be seen throughout the body.
Genetic testing using whole-exome sequencing and copy number analysis revealed a mutation in ACVR1 (NM-001105.4), with a heterozygous pathogenic variant at c.617G>A (p.Arg206His) (Figure 4).
Genetic test results The mutation site of the gene.
The patient's 12-year-old sister had a wild-type ACVR1 genotype. Additionally, mutations in ACAN, LIFR, and HSPG2 genes were observed. Based on the comprehensive clinical history and multidisciplinary discussions between orthopedics and radiology, the patient was diagnosed with FOP. The patient did not report systemic pain or other discomfort, and her condition remained stable. Treatment included intravenous administration of methylprednisolone 40 mg once daily and oral alendronate sodium vitamin D3 tablets 70.14 mg once weekly to suppress ectopic ossification. The patient was discharged with instructions for regular follow-up appointments.
Discussion
FOP is a connective tissue disorder characterized by congenital great toe abnormalities and progressive ectopic ossification. It is a rare, debilitating genetic disease with no specific gender, racial, or geographic predilection [2,3]. Early manifestations are intermittent, gradually progressing from the cervical, thoracic, and dorsal regions to the proximal ends of the limbs. The median lifespan in FOP patients is around 40 years, primarily due to severe infections or multiorgan failure resulting from diaphragmatic involvement [4]. Diagnosis relies mainly on clinical criteria, including congenital great toe malformations, progressive ectopic ossification, and anatomical changes during disease progression [5]. Enhanced CT or MRI can detect ectopic ossification and structural alterations, facilitating diagnosis. In this case, a 22-year-old female patient presented with stable acupuncture stimulation [6,7]. The pathogenesis of FOP primarily involves the bone morphogenetic protein receptor ACVR1, with mutations occurring in approximately 95% of FOP patients, most commonly the exon c.617G>A (p.Arg206His) mutation, leading to classical clinical manifestations. In this case, genetic analysis revealed a de novo heterozygous missense mutation in ACVR1 at c.617G>A (p.Arg206His), with wild-type genotypes observed in the patient's sister. Additionally, the patient exhibited multiple osteochondromas and multiple ectopic ossifications, highlighting the importance of screening for congenital malformations and benign soft tissue tumors in clinical FOP patients. FOP can be misdiagnosed as dermatomyositis, myositis ossificans, osteochondromatosis, or myofibroblastic tumors; therefore, combining imaging and genetic diagnostics can reduce clinical misdiagnosis rates.
Current treatment strategies for FOP emphasize early diagnosis, injury prevention, and rehabilitation exercises [8-10]. This includes avoiding soft tissue and muscle injuries and minimizing local stimulation. Appropriate rehabilitation therapy aims to improve lung function, enhance muscle strength, and increase joint mobility, thereby improving the quality of life in the later stages of the disease. Pharmacological treatments include short-term symptomatic treatments such as non-steroidal anti-inflammatory drugs, mast cell inhibitors, glucocorticoids, and immunosuppressants during early acute inflammatory reactions. However, long-term and high-dose use is not recommended due to potential adverse effects. During the fibroproliferative phase, anti-angiogenic drugs and endothelial growth factor inhibitors may be used. In the ossification formation phase, abnormal expression of bone morphogenetic protein-4 leads to cartilage formation and subsequent ectopic calcification. Therefore, inhibitors of bone morphogenetic protein-4, amino bisphosphonates, and bisphosphonates may have certain therapeutic effects [11].
Emerging therapies targeting ACVR1 include inhibitors of hypoxia-inducible factor-1α for inhibiting ectopic ossification, ACVR1 kinase inhibitors (LDN 212454) for reducing ectopic ossification, and activin A neutralizing antibodies for effectively suppressing ectopic ossification, offering the potential for developing highly selective and specific drugs for FOP treatment. However, it should be emphasized that these measures only delay or alleviate the progression of the disease and do not effectively halt or reverse its progression. Some researchers suggest that low-dose graded radiation therapy for early ectopic ossification may be effective, while others propose that retinoid agonists and fluoroquinolones may effectively suppress ectopic ossification. In terms of gene therapy for FOP, research is progressing in several directions, but it has not yet reached the clinical application stage. There are some advances in gene therapy and the potential for future treatments.
Gene editing technology
Using gene editing techniques such as CRISPR-Cas9, researchers are attempting to correct mutations in the ACVR1 gene in cells of FOP patients to restore normal receptor function. This approach has the potential to directly target the disease-causing genes for repair, but it still faces many challenges, such as ensuring the accuracy and safety of the repair [12,13].
RNA interference (RNAi)
RNAi technology can inhibit the expression of the ACVR1 gene by targeting RNA molecules, thereby reducing abnormal ossification. Researchers have demonstrated the inhibitory effect of RNAi on the pathological process of FOP in animal models, but further research is needed to determine its effectiveness and safety in humans [14].
Gene therapy vectors
Using viruses or other vectors to introduce normal ACVR1 genes into patients' cells to restore receptor function. While this approach has been applied in other gene therapy fields, it is still in the early stages for FOP treatment and requires overcoming safety and efficacy issues associated with gene therapy [15].
Stem cell therapy
Utilizing stem cell technology, researchers are attempting to repair receptor function by implanting normal ACVR1 genes and promoting the regeneration and repair of receptor cells. Although this approach holds promise, it still needs to address safety issues such as transplant rejection and tumor formation [16]. In conclusion, gene therapy holds great potential for the treatment of FOP, but it still faces many challenges. Future research will continue to explore various gene therapy strategies and strive to overcome technical and clinical obstacles to provide more effective treatment options for FOP patients. In summary, there are currently no definitive effective treatment modalities for FOP.
Conclusions
This study presents a typical case of FOP, providing a detailed description and analysis of its clinical presentation, imaging features, and genetic testing results. The patient exhibited typical symptoms of FOP following trauma, including progressive joint restriction, toe malformation, and ectopic ossification within soft tissues. Radiographic examination revealed corresponding ossification images, while genetic testing further confirmed a mutation in the ACVR1 gene, supporting the diagnosis of FOP. Treatment with methylprednisolone and alendronate sodium vitamin D3 tablets showed some therapeutic efficacy. The discussion emphasizes clinical diagnosis and treatment strategies for FOP, including injury prevention, rehabilitation exercises, and pharmacological interventions. Despite the lack of effective treatment modalities currently available, this study provides important clinical insights for the early diagnosis and comprehensive management of FOP, aiming to improve patient quality of life and delay disease progression. Further large-scale clinical research is warranted to explore more effective treatment approaches and provide better medical care and support for patients with FOP.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Fibrodysplasia ossificans progressiva: lessons learned from a rare disease Curr Opin Pediatr Akyuz G Gencer-Atalay K Ata P 7167223120193169357810.1097/MOP.0000000000000802 · doi ↗ · pubmed ↗
- 2Special considerations for clinical trials in fibrodysplasia ossificans progressiva (FOP)Br J Clin Pharmacol Hsiao EC Di Rocco M Cali A 119912078520193028184210.1111/bcp.13777 PMC 6533500 · doi ↗ · pubmed ↗
- 3Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva J Bone Joint Surg Am Kaplan FS Zasloff MA Kitterman JA Shore EM Hong CC Rocke DM 6866919220102019432710.2106/JBJS.I.00705 PMC 2827822 · doi ↗ · pubmed ↗
- 4From mysteries to medicines: drug development for fibrodysplasia ossificans progressive Expert Opin Orphan Drugs Kaplan FS Pignolo RJ Shore EM 637649120132480018010.1517/21678707.2013.825208 PMC 4007356 · doi ↗ · pubmed ↗
- 5Fibrodysplasia ossificans progressiva: a case report J Nepal Health Res Counc Acharya S Joshi S Chaulagain R 245247162018 https://pubmed.ncbi.nlm.nih.gov/29983445/29983445 · pubmed ↗
- 6A recurrent mutation in the BMP type I receptor ACVR 1 causes inherited and sporadic fibrodysplasia ossificans progressiva Nat Genet Shore EM Xu M Feldman GJ 5255273820061664201710.1038/ng 1783 · doi ↗ · pubmed ↗
- 7Acquired and congenital forms of heterotopic ossification: new pathogenic insights and therapeutic opportunities Curr Opin Pharmacol Pacifici M 51584020182961443310.1016/j.coph.2018.03.007PMC 6015534 · doi ↗ · pubmed ↗
- 8Feasibility of bisphosphonate therapy in an Indian pediatric patient of fibrodysplasia ossificans progressiva Int J Appl Basic Med Res Kochar IS Sethi A Ramachandran S 118120920193104117710.4103/ijabmr.IJABMR_52_18PMC 6477960 · doi ↗ · pubmed ↗