GINSA: an accumulator for paired locality and next-generation small ribosomal subunit sequence data
Eric Odle, Samuel Kahng, Siratee Riewluang, Kyoko Kurihara, Kevin C Wakeman

TL;DR
GINSA is a Python tool that automatically links small ribosomal subunit sequences with their geographic locations using global biodiversity data.
Contribution
The novelty lies in automatically linking decentralized genetic data with locality information using global biodiversity infrastructure.
Findings
GINSA demonstrates broad applicability across different taxonomic levels and dataset sizes.
The tool is freely available and can be easily installed via pip from PyPI.
Abstract
Motivated by the challenges of decentralized genetic data spread across multiple international organizations, GINSA leverages the Global Biodiversity Information Facility infrastructure to automatically retrieve and link small ribosomal subunit sequences with locality information. Testing on taxa from major organism groups demonstrates broad applicability across taxonomic levels and dataset sizes. GINSA is a freely accessible Python program under the MIT License and can be installed from PyPI via pip.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
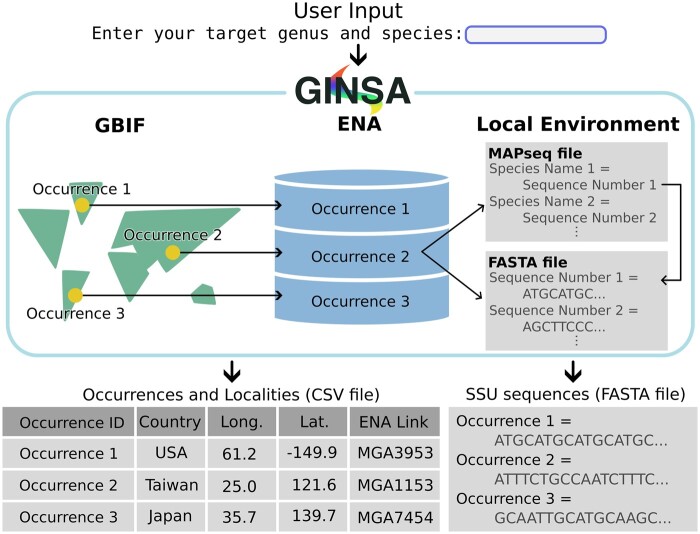 Figure 1
Figure 1| Taxon | Total occurrences | ENA/MGnify |
|---|---|---|
| Animals | 2 097 448 406 | 33 065 |
| Plants | 442 531 533 | 376 547 |
| Fungi | 38 914 204 | 955 943 |
| Bacteria | 22 722 639 | 18 355 383 |
| Protists | 15 895 213 | 3 098 014 |
| Archaea | 442 031 | 335 722 |
| Viruses | 910 025 | 0 |
| Incertae sedis | 8 014 894 | 630 700 |
| Taxon | OC ( | RT (min) | Size (GB) |
|---|---|---|---|
|
| 11 | 3.4 | 0.8 |
|
| 26 | 10.1 | 6.80 |
|
| 190 | 36.0 | 5.26 |
|
| 253 | 152.2 | 58.3 |
|
| 309 | 86.8 | 31.9 |
|
| 549 | 65.3 | 0.118 |
|
| 628 | 336.0 | 79.3 |
|
| 655 | 95.1 | 2.21 |
|
| 1379 | 327.5 | 97.8 |
|
| 2602 | 593.0 | 178.5 |
- —DX Fellowship Code
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Community Ecology and Physiology · Protist diversity and phylogeny · Genomics and Phylogenetic Studies
1 Introduction
Advances in nucleic acid sequencing technologies have led to a rapid increase in the amount of available genetic data (Keen et al. 1996, Warburton and Sebra 2023). To better organize and share this emergent abundance of sequence data between researchers, public databases such as GenBank (Benson et al. 1993, Strasser 2011), the European Nucleotide Archive (ENA) (Burgin et al. 2023), and the DNA Data Bank of Japan (DDBJ) (Tanizawa et al. 2023) were established in the 1980s. For example, evolutionary biologists often rely on small ribosomal subunit rRNA gene (SSU) sequences archived in these databases to study new species. However, sequence databases do not require a complete set of metadata (e.g. site of collection, date of collection, species-level identification, or link to publication) when uploading sequences. Absence of a complete set of metadata can lead to the omission of locality data, forcing biologists to manually seek associated location information elsewhere. To address this disconnect among archived data, we developed GINSA (GbIf Next-gen Sequence Accumulator): a biodiversity research tool that fetches SSU sequences and their associated localities. This tool takes advantage of the Global Biodiversity Information Facility (GBIF) [www.gbif.org], which links taxa (scientific names), localities (sites of occurrence), and SSU sequences.
Manually pairing high-volume SSU sequence and locality data is prohibitively slow. Although GBIF provides links to specific sequences used for identification, researchers must currently follow a convoluted chain of websites to FASTA files stored in off-site repositories (typically ENA for next-generation sequencing). Upon downloading FASTA/FASTQ files, researchers must then manually search massive lists (often hundreds of thousands) of SSU sequences. This time-consuming step is required for each species occurrence on GBIF, for which there can be thousands. Finally, researchers must manually trace sequences back to their occurrence locality from GBIF. We developed GINSA to automate this process.
Previous attempts to address the inaccessibility of sequence metadata include pysrabd (Choudhary 2019), grabseqs (Taylor et al. 2020), and ffq (Gálvez-Merchán et al. 2023). While helpful for specific applications, these tools address use cases that differ from those of GINSA. Python package pysradb provides convenient access to next-generation sequences stored on the National Center for Biotechnology Information (NCBI) Sequence Read Archive but does not focus on data from ENA. Another tool, grabseqs, automates next-generation sequence acquisition for multiple repositories, but requires users to have prior knowledge of the specific accession numbers associated with their organism of interest. Similarly, ffq addresses the difficulty in acquiring sequence metadata from ENA. However, ffq requires database accession or article DOI numbers as input. In contrast, GINSA leverages the structure provided by GBIF to link specific taxa with their known localities and SSU sequences. Users simply enter the name of their target organism (taxon), and then wait for the collection process to finish automatically.
2 Applications and implementation
The GINSA tool provides researchers with large datasets in line with the Big Data (De Mauro et al. 2016, Miralles et al. 2020) nature of current molecular taxonomy. For instance, evolutionary biologists rely on large molecular sequence datasets to study speciation trends (Schlegel 1991, Adl et al. 2019). Large datasets help resolve cryptic diversity, which is a challenge seen across the tree of life—animals (Marchán et al. 2018, Li and Wiens 2023), plants (Vieu et al. 2023, Windham et al. 2023), fungi (Koufopanou et al. 1997, Pringle et al. 2005), bacteria (Meyer et al. 2023), protists (Wakeman and Leander 2013, Krienitz et al. 2015, Martin et al. 2016), archaea (Câmara et al. 2023), and viruses (Roux et al. 2019). Specialists across a range of taxa can therefore use GINSA to collect more data for their phylogenetic (SSU sequence) and biogeographic (locality) analyses.
There are currently over 2.6 billion occurrences on GBIF representing 1.3 million confirmed species. Occurrence coverage is uneven across taxa; animals account for 79.8%, followed by plants at 16.8%, and other taxa at <1.5% each (Table 1). When considering only next-generation SSU sequences archived by ENA (via the publisher MGnify), coverage favors bacteria and protists (Supplementary Fig. S1). This ENA/MGnify subset includes approximately 23.7 million GBIF occurrences, which together comprise the pool of data accessible by GINSA. Moreover, this data pool is expected to grow. Since 2008, 25–50 thousand new species from each major global region have been added to GBIF every two years (Waller 2020).
The GINSA tool offers an efficient method for accessing ENA next-generation sequence repositories linked to GBIF taxon occurrences (Fig. 1). Users enter a search taxon, then GINSA queries GBIF for all recorded occurrences of that taxon. Next, the program extracts respective ENA links from GBIF occurrence records, downloading and processing FASTA/FASTQ files into a curated list of SSU sequences. The following details outline how GINSA automates this task.
Chart visualizing the GINSA workflow. User input is taken as a GBIF search taxon. Occurrences are then linked with their source sequences archived on ENA. Output CSV and FASTA files link GBIF occurrence IDs, localities, and sequences.
2.1 User prompt
Upon running GINSA, users are prompted for two inputs: the project folder path and the target taxon. All subsequent sub-folders and output files are saved inside the project folder. Search taxa are parsed in Python as a string, and users may enter either one-word (e.g. genus name Lecudina) or two-word (e.g. species name Lecudina longissima) queries.
2.2 GBIF taxon search
An API call searches GBIF for all instances of the queried taxon, and a list is generated by the function search_species_occurrences() for all matching GBIF occurrences.
2.3 FASTA and MAPseq download
Sequential API calls are made to ENA/MGnify for each occurrence linked from GBIF. Next, the function ssu_fasta_grab() downloads FASTA/FASTQ files containing SSU sequences belonging to the search taxon. This process is then repeated by mapseq_grab() for the associated MAPseq files. These MAPseq files are necessary because next-generation sequencing read assembly generates long lists of sequences with complex names.
2.4 SSU contig decode
For each occurrence, a text search is run within the MAPseq file to locate all sequences associated with the search taxon. Next, sequence labels are tracked to specific sequences in the corresponding FASTA/FASTQ file.
2.5 Generate FASTA master file
Extracted SSU sequences are gathered into a file named seq_master.FASTA alongside a corresponding metadata table named occurrences.csv. Users may annotate seq_master.FASTA with additional GBIF metadata (e.g. latitude, longitude, or country of origin). A script (misc/suffix_annotator.py) demonstrating annotation with occurrences.csv is provided on the project GitHub repository.
3 Availability and testing
The GINSA project was written using Python (Van Rossum and Drake 1995) version 3.12 and is free to use under the MIT License. Code was structured into two scripts: a command line interface (CLI) implementation named GINSA_cli.py and a graphical user interface (GUI) implementation named GINSA_gui.py. Following installation via pip,pip3 install GINSAthe CLI can be run with a single line of text:GINSA_cli <path/to/project/directory> <"search taxon">Moreover, the GUI can be run by simply entering:GINSA_gui
A broad range of taxonomic groups were represented when testing GINSA (Table 2). These groups include animals (arthropod genus Lambia), plants (Aneura mirabilis, Chrysymenia brownii), fungi (Malassezia globosa), bacteria (Altibacter lentus), protists (Lecudina longissima, Tetraselmis marina, Lecudina tuzetae, and Labyrinthula spp.), and archaea (Nanohaloarchaea). This set of taxa allowed us to evaluate the speed and utility of GINSA across multiple taxa and occurrence sizes. Testing was performed on an Intel Xeon W-2235 CPU 3.80 GHz system with 31.0 GiB of available memory running Linux kernel 5.15.0–87. Network download speed during testing was stable, ranging from 443 to 540 Mbp.
Following testing, GINSA exhibited applicability across a spectrum of taxa and occurrence sizes. Taxa with smaller datasets (Lambia spp., Lecudina longissima) took less time to analyze than taxa with larger datasets (Malassezia globosa, Labyrinthula spp.) (Table 2). All tests completed without interruption, although the larger taxa required significantly more storage (97.8–178.5 GB). Network speed and local storage capacity were the only observed bottlenecks to performance. With sufficient storage and internet connectivity, taxa with an even greater number of GBIF occurrences could theoretically be analyzed using GINSA.
Neither runtime nor output directory size were linearly associated with occurrence count. These observations are attributed to the presence of GBIF occurrences identified through means (e.g. human identification, museum specimens, and Sanger sequencing) other than next-generation sequencing. For example, although Aneura mirabilis had 549 occurrences on GBIF, only two of those occurrences linked back to next-generation SSU sequences. For this reason, GINSA generates an output plot summarizing the proportion of searched occurrences containing next-generation SSU sequence data. Examples of these plots are provided on the project GitHub page (https://github.com/ericodle/GINSA).
4 Conclusion
This article introduced GINSA (GbIf Next-gen Sequence Accumulator), a novel tool designed to bridge the gap between genetic sequence data and locality metadata. Rapid growth in the amount of data from next-generation sequencing technologies has generated increasing demand for more efficient methods to pair sequence information with biogeographic context. The GINSA tool addresses this challenge by automating the collection of SSU sequences and locality metadata for a given taxon through integration with the Global Biodiversity Information Facility (GBIF). By streamlining the process of accessing and pairing these crucial data, GINSA enables researchers to work more efficiently. This tool has a beginner-friendly design, open-source code base, and is applicable across major organism groups. As such, GINSA is offered as a free resource for evolutionary biologists navigating the complexities of cryptic speciation and Big Data research.
Supplementary Material
btae152_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adl SM , Bass D, Lane CE et al Revisions to the classification, nomenclature, and diversity of eukaryotes. J Eukaryot Microbiol 2019;66:4–119.30257078 10.1111/jeu.12691 PMC 6492006 · doi ↗ · pubmed ↗
- 2Benson D , Lipman DJ, Ostell J. Genbank. Nucleic Acids Res 1993;21:2963–5.8332518 10.1093/nar/21.13.2963 PMC 309721 · doi ↗ · pubmed ↗
- 3Burgin J , Ahamed A, Cummins C et al The European nucleotide archive in 2022. Nucleic Acids Res 2023;51:D 121–5.36399492 10.1093/nar/gkac 1051 PMC 9825583 · doi ↗ · pubmed ↗
- 4Câmara PE , Bones FLV, Lopes FAC et al DNA metabarcoding reveals cryptic diversity in Forest soils on the isolated Brazilian Trindade Island, South Atlantic. Microb Ecol 2023;85:1056–71.35484416 10.1007/s 00248-022-02018-4 · doi ↗ · pubmed ↗
- 5Choudhary S. pysradb: a python package to query next-generation sequencing metadata and data from NCBI sequence read archive. F 1000 Res 2019;8:532.31114675 10.12688/f 1000 research.18676.1PMC 6505635 · doi ↗ · pubmed ↗
- 6De Mauro A , Greco M, Grimaldi M. A formal definition of big data based on its essential features. Library Rev 2016;65:122–35.
- 7Gálvez-Merchán Á , Min KH, Pachter L et al Metadata retrieval from sequence databases with FFQ. Bioinformatics 2023;39:btac 667.36610997 10.1093/bioinformatics/btac 667PMC 9883619 · doi ↗ · pubmed ↗
- 8Keen G , Burton J, Crowley D et al The genome sequence database (GSDB): meeting the challenge of genomic sequencing. Nucleic Acids Res 1996;24:13–6.8594561 10.1093/nar/24.1.13PMC 145611 · doi ↗ · pubmed ↗