Decoding the Neurodevelopment and Seizure Puzzle: A Pediatric Case of DYRK1A Gene Mutation and Autosomal Dominant Mental Retardation Type 7
Abdulrahman A Aldoseri, Rashed N Buhaza, Raafat Hammad Seroor Jadah

TL;DR
This paper presents a rare case of a child with a DYRK1A gene mutation causing developmental delay and epilepsy, highlighting the importance of genetic testing for accurate diagnosis.
Contribution
The paper adds to the limited clinical understanding of DYRK1A mutations through a detailed pediatric case report.
Findings
The patient exhibited global developmental delay, epilepsy, and dysmorphic features consistent with DYRK1A mutation.
Whole Exome Sequencing confirmed the diagnosis, emphasizing the role of genetic testing in rare disorders.
The case underscores the clinical variability and diagnostic challenges of Autosomal Dominant Mental Retardation Type 7.
Abstract
Autosomal Dominant Mental Retardation Type 7 is a disorder caused by pathogenic variants in the DYRK1A gene. Clinical features associated with this gene mutation include focal dysmorphism, developmental delay, and epilepsy. In this report, we present a case of an 8-year-old boy with a DYRK1A gene mutation, whose clinical manifestations underscore the rarity and clinical challenges of this genetic condition. The patient is a known case of global developmental delay with intractable epilepsy on multiple anti-epileptic medications. Upon examination, the patient showed delayed developmental milestones, hypotonia with brisk deep tendon reflexes, as well as dysmorphic features in the form of microcephaly, deep-set eyes, prominent ears, and a short nose. MRI was done, and findings were suggestive of a DYRK1A gene mutation. The diagnosis was later confirmed by Whole Exome Sequencing (WES). Our…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
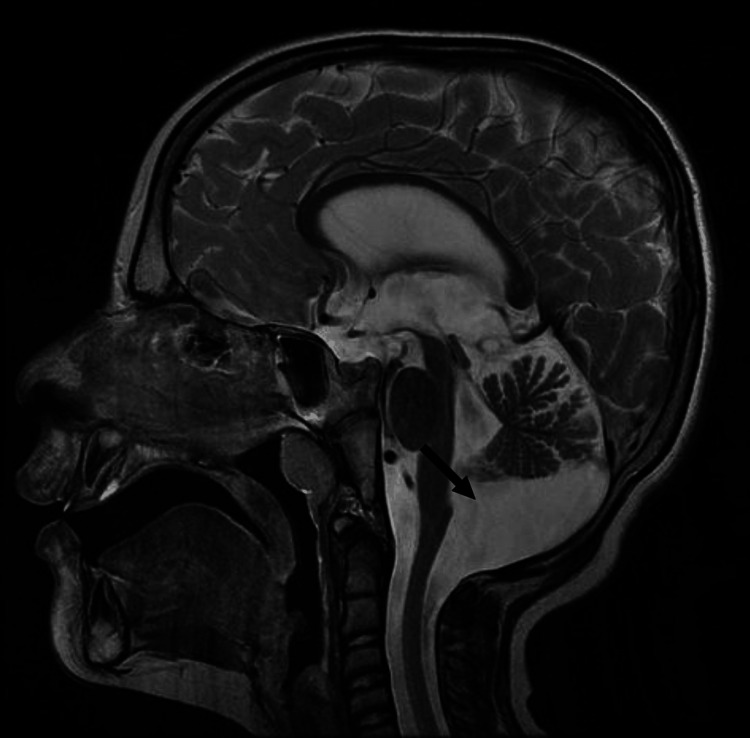 Figure 1
Figure 1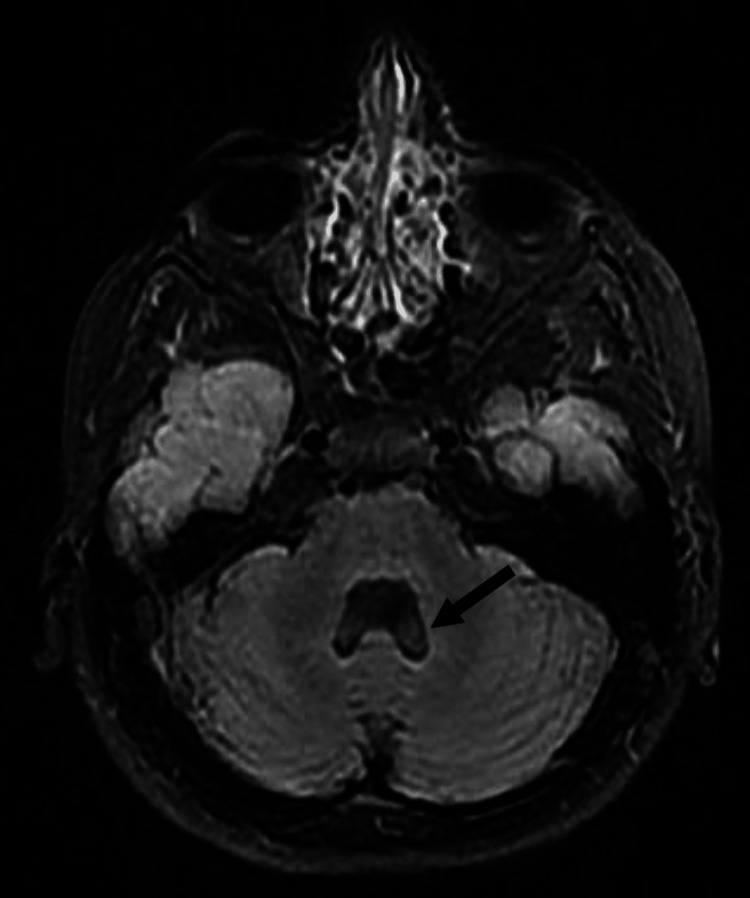 Figure 2
Figure 2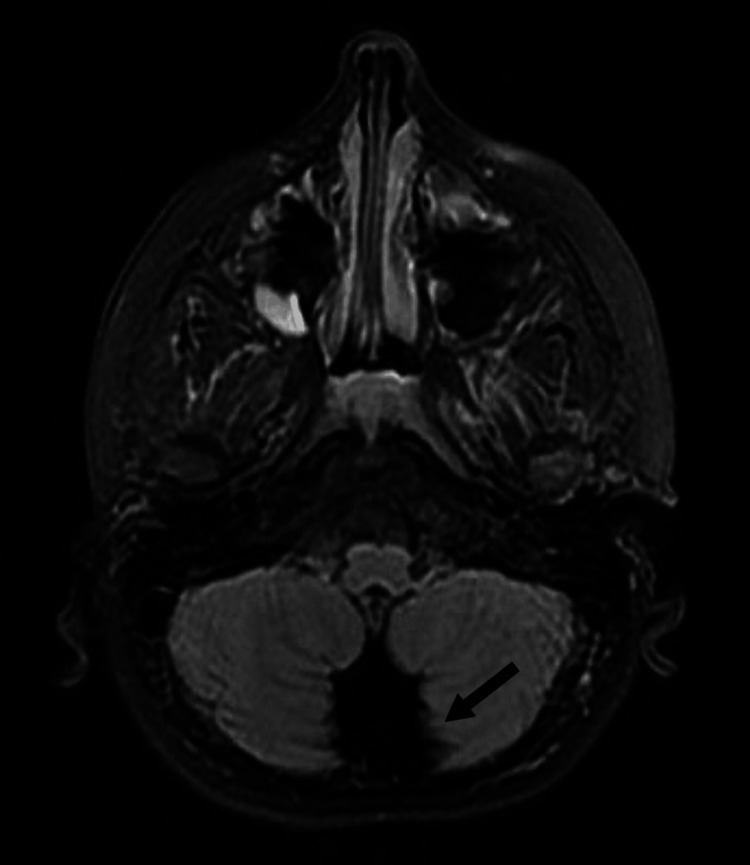 Figure 3
Figure 3| GENE | VARIANT COORDINATES | AMINO ACID CHANGE | SNP IDENTIFIER | ZYGOSITY | NI SILICO PARAMETERS* | ALLELE FREQUENCIES** | TYPE AND CLASSIFICATION*** |
| DYRK1A | p.(Leu197Serfs*12) | N/A | heterozygous | PolyPhen: N/A, Align-GVDG: N/A, SIFT: N/A, MutationTaster: N/A, Conservation_nt: N/A, Conservation_aa: N/A. | gnomAD: -, ESP: - 1000 G: -, CentoMD: -. | Frameshift Likely pathogenic (class 2) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDown syndrome and intellectual disability research · Genetics and Neurodevelopmental Disorders · Congenital heart defects research
Introduction
Within the landscape of rare genetic disorders, the *DYRK1A *gene mutation is a rare genetic disorder with only 68 cases has been reported in the literature leading to Autosomal Dominant Mental Retardation Type 7 with strikingly uncommon occurrence [1]. Manifesting in dysmorphic features, intellectual delay, and invariable form of epilepsy [2], this genetic anomaly has been identified in only a handful of cases worldwide. Insights gained from these cases enable healthcare providers to comprehend the intricate interplay between genetic factors and neurodevelopment disorders. The possibilities of *DYRK1A *gene mutation should be strongly considered in pediatric patients who present with such phenotype associated with this rare genetic disorder.
Following the confirmation of diagnosis by genetic testing, genetic counseling plays a crucial role in guiding families through the complexities of genetic testing and inheritance patterns [2]. By raising awareness of this rare genetic disorder, healthcare professionals can improve diagnostic accuracy and facilitate timely interventions for affected individuals.
Case presentation
This is an eight-year-old boy, with a known case of global developmental delay (GDD) with intractable epilepsy on multiple anti-epileptic medications including levetiracetam, topiramate, and oxcarbazepine. This patient initially presented at the age of eleven months with frequent typical febrile convulsions, which later became an atypical form of febrile convulsion due to the long duration of the seizure, which lasted for more than 30 minutes. The patient also showed a progressive delay in his developmental milestones as he started to sit without support at the age of ten months and started to walk at the age of three years with poor verbal communication in the form of speech delay, saying only two to three words at the age of three and half years. He is a product of term delivery to non-consanguineous parents with no postnatal complications. No family history of epilepsy or seizure disorder.
Neurological examination showed an active child with poor cognitive and verbal communication and dysmorphic features in the form of microcephaly, deep-set eyes, prominent ears, and a short nose. Skin examination was negative for neurocutaneous stigmata. He is hypotonic with brisk deep tendon reflexes, however, no clonus, with normal power. Cranial nerve examinations were normal with no cerebellar signs. The gait examination was normal and the rest of the systemic examinations were unremarkable.
Magnetic resonance imaging (MRI) showed posterior fossa and 4th ventricle enlargement in sagittal T2 sequence images (Figure 1).
Enlargement of posterior fossa and 4th ventricle.
Axial T2 fluid-attenuated inversion recovery (FLAIR) images also showed severe vermis hypoplasia (Figures 2, 3), as well as enlargement of the 4th ventricle (Figure 2), and significant posterior fossa enlargement (Figure 3).
Enlargement of the 4th ventricle with vermis hypoplasia.
Posterior fossa enlargement with vermis hypoplasia.
The patient underwent whole exome sequencing (WES) test, and a heterozygous likely pathogenic variant was identified in the *DYRK1A *gene.
The identified variant interpretation was as follows: The *DYRK1A variant c.588dup p. (Leu197Serfs12) creates a shift in the reading frame starting at codon 197. The new reading frame ends in a stop codon 1 positions downstream. It is classified as likely pathogenic (Table 1).
The result is consistent with a genetic diagnosis of Autosomal Dominant Mental Retardation Type 7. Genetic counseling was done, and parental carrier testing was recommended.
Discussion
DYRK1A, located on chromosome 21q22.13, encodes a dual-specificity tyrosine-phosphorylation-regulated kinase, playing a pivotal role in neurodevelopment. This gene's mutations result in variable clinical presentations, contributing to the limited recognition of this rare condition [3].
Moreover, patients harboring *DYRK1A *mutations consistently exhibit developmental delays, speech impairments, and refractory epilepsy, as observed in our case. These manifestations highlight the neurological impact of *DYRK1A *on cognitive and motor functions, posing diagnostic and therapeutic challenges [4].
Our patient presented with dysmorphic features in the form of a beaked nose and micrognathia, with intractable epilepsy on multiple epileptic medications. Distinctive facial characteristics, to illustrate, a prominent forehead, beaked nose, micrognathia, and hypertelorism have been identified in several cases with *DYRK1A *mutations [5]. All in all, these dysmorphic features offer clinical clues, reinforcing the link between genetic abnormalities and phenotypic expressions.
The conducted neuroimaging, such as MRI, often revealed cerebellar anomalies in *DYRK1A *cases. Hypoplasia of the inferior cerebellar vermis and the presence enlarged of cystic areas within the posterior fossa are recurrent findings [6]. These neuroimaging patterns enhance our understanding of the structural impact of *DYRK1A *mutations.
Our patient’s brain MRI showed features suggestive of vermian hypoplasia. Whole Exome Sequencing emerged as a pivotal tool in diagnosing rare genetic disorders, including DYRK1A mutations. This technique offers a comprehensive view of genetic variants, aiding clinicians in unraveling complex cases [7]. Hence, the patient presented in this case was diagnosed based on Whole Exome Sequencing.
Conclusions
This case underscores the rarity and clinical complexity of *DYRK1A *gene mutations, contributing to the pool of global knowledge on Autosomal Dominant Mental Retardation Type 7. It presented with a typical clinical picture and was diagnosed by Whole Exome Sequencing, a diagnostic test. Through our report, we aim to fortify the medical community's awareness and appreciation of the intricate interplay between genetics and neurodevelopmental disorders.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1DYRK 1A syndrome Gene Reviews [Internet] van Bon BWM Coe BP de Vries BBA Seattle University of Washington 2015 Dec 17 [Updated 2021 Mar 18]https://www.ncbi.nlm.nih.gov/books/NBK 333438/
- 2The DYRK 1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy J Med Genet Courcet JB Faivre L Malzac P 7317364920122309964610.1136/jmedgenet-2012-101251 · doi ↗ · pubmed ↗
- 3Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK 1A Eur J Hum Genet Bronicki LM Redin C Drunat S 148214872320152592055710.1038/ejhg.2015.29PMC 4613470 · doi ↗ · pubmed ↗
- 4Case report of novel DYRK 1A mutations in 2 individuals with syndromic intellectual disability and a review of the literature BMC Med Genet Luco SM Pohl D Sell E Wagner JD Dyment DA Daoud H 151720162692265410.1186/s 12881-016-0276-4PMC 4769499 · doi ↗ · pubmed ↗
- 5DYRK 1A pathogenic variants in two patients with syndromic intellectual disability and a review of the literature Mol Genet Genomic Med Meissner LE Macnamara EF D'Souza P 08202010.1002/mgg 3.1544 PMC 776756933159716 · doi ↗ · pubmed ↗
- 6DYRK 1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies Eur J Hum Genet Ji J Lee H Argiropoulos B 147314812320152594438110.1038/ejhg.2015.71PMC 4613469 · doi ↗ · pubmed ↗
- 7Whole-exome sequencing for identifying genetic causes of intellectual developmental disorders Int J Gen Med Guo YX Ma HX Zhang YX Chen ZH Zhai QX 127512821420213388005910.2147/IJGM.S 300775 PMC 8053495 · doi ↗ · pubmed ↗