Extraskeletal Ewing Sarcoma: A Case Report
Ryan Denis, Martin Felix, Daniela Mejia, Mikayla Hobbs, Paul Alvarez, Damian Casadesus

TL;DR
A 42-year-old woman was diagnosed with a rare case of Ewing sarcoma in soft tissue, not bone, and showed a positive response to chemotherapy.
Contribution
This case report highlights an unusual presentation of Ewing sarcoma in a middle-aged woman with a soft tissue mass.
Findings
The tumor displayed a positive EWSR1 gene rearrangement via fluorescence in situ hybridization.
The patient showed a favorable response to chemotherapy despite complications.
This case contributes to understanding the atypical manifestations of Ewing sarcoma.
Abstract
Ewing sarcoma is one of the most common primary bone tumors arising from neuroectodermal cells mainly presenting in the younger population. Instances of this highly malignant tumor manifesting outside of the bone and outside of the typical age range create an unfamiliar clinical scenario. In this report, we present a rare extraskeletal Ewing sarcoma in a 42-year-old woman with a subcutaneous soft tissue mass in the posterior chest displaying a positive EWSR1 gene rearrangement via fluorescence in situ hybridization. The patient is currently on a chemotherapy regimen showing favorable response to the tumor size despite additional complications. This overall presentation of Ewing sarcoma allows further understanding of the malignancy and fosters better care for future cases.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
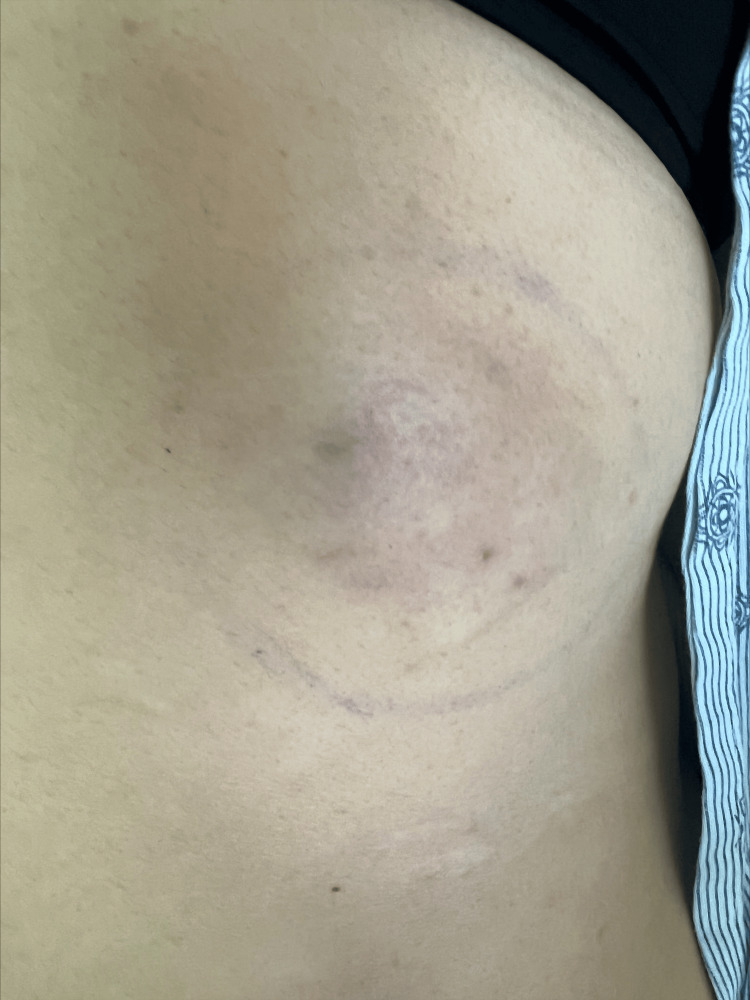 Figure 1
Figure 1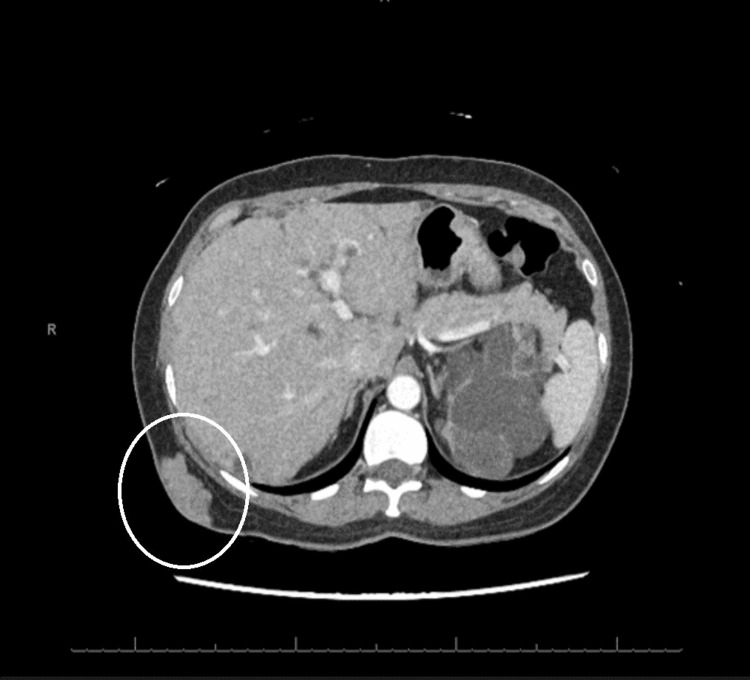 Figure 2
Figure 2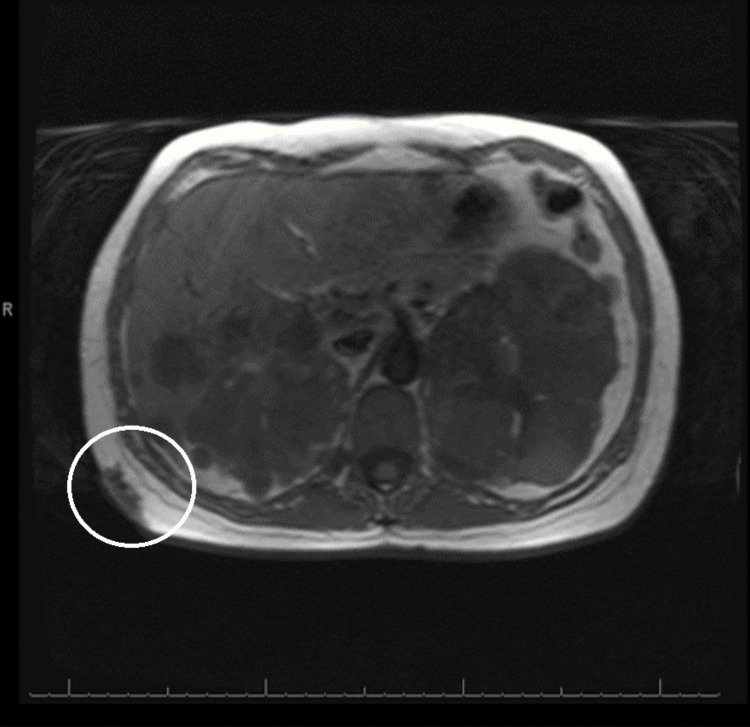 Figure 3
Figure 3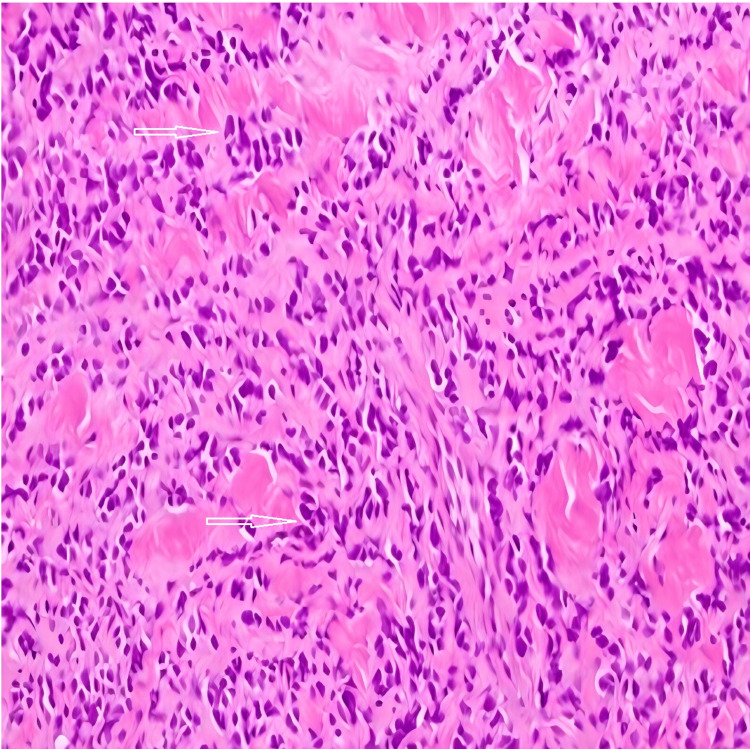 Figure 4
Figure 4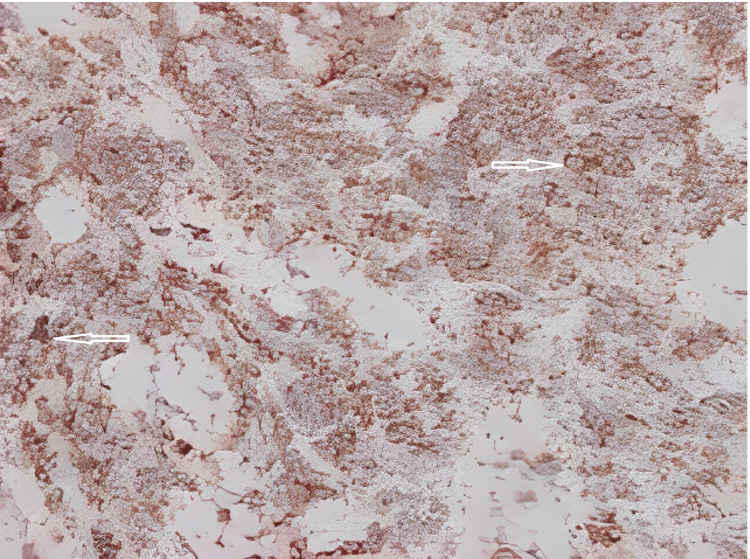 Figure 5
Figure 5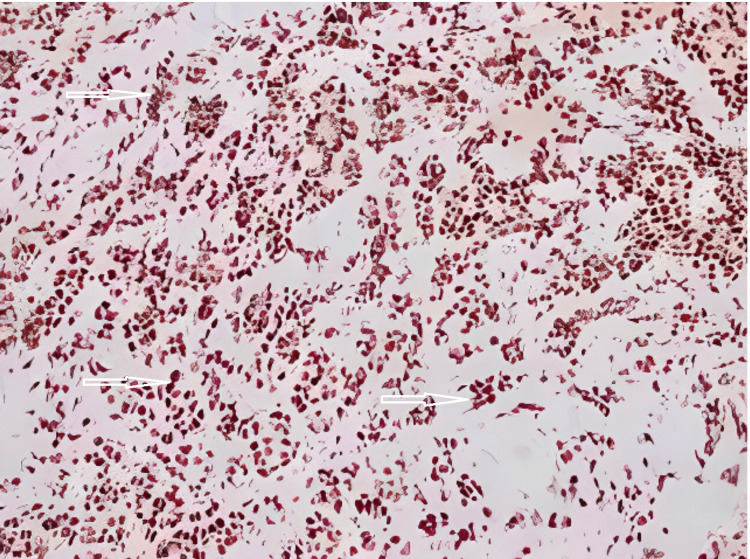 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSarcoma Diagnosis and Treatment · Cardiac tumors and thrombi · Bone Tumor Diagnosis and Treatments
Introduction
Ewing sarcoma (ES), a rare and aggressive form of primary bone cancer, predominantly affects children and young adults. It is characterized by small round cells and comprises 10-15% of all bone sarcomas. This malignancy arises from primitive neuroectodermal cells and commonly manifests within the bone marrow of the long bones, pelvis, and ribs [1]. The peak incidence is typically between 10 and 15 years of age, with around 30% of the cases arising in adults over the age of 20 [1].
However, instances of extraskeletal ES (EES), where the tumor arises outside of the bone, are exceedingly rare and account for 20-30% of all reported cases of ES [2]. EES is characterized by its aggressive nature and tendency to metastasize, posing unique diagnostic and therapeutic challenges when located in soft tissues or other extramedullary sites. For extraskeletal primary tumors, the most common sites of the disease include the trunk (32%), extremity (26%), head and neck (18%), retroperitoneum (16%), and other sites (9%) [3].
The rarity of EES necessitates a meticulous examination of individual cases to enhance our understanding of its clinical behavior, optimal diagnostic approaches, and tailored therapeutic interventions. Our patient’s clinical presentation, diagnostic workup, and treatment course offer valuable insights into the challenges faced by clinicians when confronted with this atypical manifestation of EES. Through the exploration of clinical nuances and therapeutic interventions, this case report seeks to enhance our understanding of EES and ultimately improve the care and prognosis for individuals facing this challenging diagnosis.
Case presentation
A 42-year-old Hispanic woman presented to the emergency department (ED) with a one-day history of worsening right-sided back pain. She had a past medical history of autosomal dominant polycystic kidney disease, hypertension, and meningioma. A month before this visit, she visited the ED with a similar presentation of back pain and complaints of a growing mass in the back. In the ED, vital signs were within normal limits. On physical examination, the patient had a mobile, subcutaneous, right-sided, posterolateral chest mass measuring 4 x 4 cm that was hard on palpation with minimal skin changes (Figure 1).
Subcutaneous mass in the posterolateral right side of the chest.
Imaging with an abdominal and pelvis computed tomography (CT) scan with intravenous (IV) contrast demonstrated a stable soft tissue mass in the subcutaneous fat of the right posterior lateral chest wall (Figure 2). Magnetic resonance imaging (MRI) of the chest with and without IV contrast revealed evidence of polycystic kidney disease affecting the kidneys and liver and a mass in the right-sided posterior chest (Figure 3). The MRI with and without IV contrast did not show any evidence of possible metastatic disease in the thoracic or lumbar spine.
Computed tomography scan with intravenous contrast revealing a soft tissue mass in the subcutaneous fat of the right posterolateral chest wall.
Magnetic resonance imaging of the chest with and without intravenous contrast revealing evidence of polycystic kidney disease affecting the kidneys and liver and a mass in the right-sided posterior chest.
The patient underwent a core needle biopsy. The histopathologic study revealed atypical small round cells consistent with ES (Figures 4-6). The tumor was found to be positive for CD99 and NKX2.2 and negative for AE1/AE3, CK8/CK18, CK7, CK20, desmin, SOX10, BCOR, WT1, CD45, CD31, and merkel antigen, supporting the diagnosis of EES. Further tissue studies detected an* EWSR1* signal in 80% of nuclei, indicating the presence of an *EWSR1 *rearrangement. Other images did not reveal metastatic disease at the time of the diagnosis of the tumor.
Infiltrating tumor in the dermis composed of small round blue cells with minimal cytoplasm and nuclear overlapping (hematoxylin and eosin, 200×).
CD99 immunostaining (a sensitive but not specific marker for Ewing sarcoma) showing positive membranous staining (200×).
NKX2.2 immunostaining (sensitive and specific for Ewing sarcoma) showing positive nuclear staining (200×).
The patient started treatment with a systemic chemotherapy regimen with vincristine, ifosfamide, and doxorubicin. The patient’s course was complicated with neutropenic fever secondary to a vestibular abscess of tooth #19, treated with extraction and amoxicillin-clavulanate. After the first round of chemotherapy, the patient had a discernible response with shrinkage in the size of the tumor.
Discussion
ES of the bone (ESB) is a predominantly pediatric primary malignancy and 10 times more common than EES. The latter type of sarcoma has an incidence of 0.4 per million and is most common in patients below the age of 5 and above the age of 35 and in women [4-6]. On presentation, patients with EES most often report pain in the upper regions of the arms, shoulders, buttocks, and thighs, with 25% of cases involving bone marrow, lung, and bone metastases. Our patient’s presentation is very interesting because the tumor presented at an older age and in the subcutaneous tissue.
The most used imaging modalities for the diagnosis of EES include MRI, CT scan, and ultrasonography. In our patient, the imaging studies revealed the key findings reported in the literature, including a stable, hyperlobulated, distinct borders, and hyperdense lesion located in the subcutaneous fat; however, no areas of necrosis were observed [7-9]. No evidence of metastatic disease in the lungs and bones was observed in our patient. The typical histological findings of clusters of blue small and round with cytoplasmic borders and nucleoli that are difficult to delineate from nearby structures and large and round nuclei were present in our patient [10]. Other studies have revealed positive CD99 and NKX2.2, as well as an abnormal fluorescence in situ hybridization result demonstrating an EWSR1 rearrangement in 80% of cells.
There is currently a very limited amount of information in the literature regarding the best form of treatment for EES; however, surgery with or without radiotherapy, along with chemotherapy, has been described as the most viable option [11,12]. ES of the skin appears to have a more favorable prognosis, with 91% of the patients surviving at 10 years which is higher than ESB [13,14]. Current recommendations endorse a two-week, rather than a three-week, administration interval of vincristine-doxorubicin, ifosfamide-etoposide, and cyclophosphamide cycles, with a maximum event-free survival rate of about 73% [15].
Studies have described fewer than 100 cases of EES in the literature. The tumor is more frequent in white women and the pediatric population and the presence of metastasis is very rare at the time of diagnosis. Our patient, in the fourth decade of life, presented without metastatic disease, and after the treatment, the tumor size improved [13,16-18].
Conclusions
EES is associated with an indolent course, rarely has metastatic disease at the time of diagnosis, and has a favorable prognosis when treated with combined modality therapy. Less invasive chemotherapy should be considered a potential treatment for EES because of the low rate of metastatic disease at diagnosis, local control, good response to treatment, and excellent outcome, as well as decreased side effects and toxicity with a high curative rate. Physicians should consider ES in the differential diagnosis of subcutaneous tumors, even in older age.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ewing Sarcoma Durer S Shaikh H Treasure Island, FL Stat Pearls Publishing 2023 https://www.ncbi.nlm.nih.gov/books/NBK 559183/32644609 · pubmed ↗
- 2Extraskeletal Ewing sarcoma: diagnosis, management and prognosis Oncol Lett Abboud A Masrouha K Saliba M 3542120213374721110.3892/ol.2021.12615 PMC 7967932 · doi ↗ · pubmed ↗
- 3Ewing's sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991 J Clin Oncol Raney RB Asmar L Newton WA Jr 574582151997905347910.1200/JCO.1997.15.2.574 · doi ↗ · pubmed ↗
- 4Extra-osseous Ewing sarcoma Pediatr Hematol Oncol van den Berg H Heinen RC van der Pal HJ Merks JH 1751852620091943732010.1080/08880010902855581 · doi ↗ · pubmed ↗
- 5Clinical features and outcomes in patients with extraskeletal Ewing sarcoma Cancer Applebaum MA Worch J Matthay KK Goldsby R Neuhaus J West DC Dubois SG 3027303211720112169205710.1002/cncr.25840 PMC 3135782 · doi ↗ · pubmed ↗
- 6Extraskeletal versus skeletal Ewing sarcoma in the adult population: controversies in care Surg Oncol Lynch AD Gani F Meyer CF Morris CD Ahuja N Johnston FM 3733792720183021729010.1016/j.suronc.2018.05.016 · doi ↗ · pubmed ↗
- 7A to Z of extraskeletal Ewing sarcoma family of tumors in adults: imaging features of primary disease, metastatic patterns, and treatment responses AJR Am J Roentgenol Javery O Krajewski K O'Regan K Kis B Giardino A Jagannathan J Ramaiya NH 022197201110.2214/AJR.11.666722109315 · doi ↗ · pubmed ↗
- 8Imaging of malignant tumours of the long bones in children: monitoring response to neoadjuvant chemotherapy and preoperative assessment Pediatr Radiol Brisse H Ollivier L Edeline V Pacquement H Michon J Glorion C Neuenschwander S 5956053420041510342810.1007/s 00247-004-1192-x · doi ↗ · pubmed ↗