Targeting SMAD-Dependent Signaling: Considerations in Epithelial and Mesenchymal Solid Tumors
Farhana Runa, Gabriela Ortiz-Soto, Natan Roberto de Barros, Jonathan A. Kelber

TL;DR
This paper reviews how targeting SMAD signaling, which can both suppress and promote tumors, could lead to better cancer therapies.
Contribution
The paper provides a review of preclinical studies on SMAD-regulating inhibitors in epithelial and mesenchymal tumors.
Findings
TGF-β/SMAD signaling has dual tumor-suppressing and tumor-promoting roles.
Inhibitors targeting SMAD-regulating proteins show potential in preclinical studies.
Challenges remain due to the complex and context-dependent nature of SMAD signaling.
Abstract
SMADs are the canonical intracellular effector proteins of the TGF-β (transforming growth factor-β). SMADs translocate from plasma membrane receptors to the nucleus regulated by many SMAD-interacting proteins through phosphorylation and other post-translational modifications that govern their nucleocytoplasmic shuttling and subsequent transcriptional activity. The signaling pathway of TGF-β/SMAD exhibits both tumor-suppressing and tumor-promoting phenotypes in epithelial-derived solid tumors. Collectively, the pleiotropic nature of TGF-β/SMAD signaling presents significant challenges for the development of effective cancer therapies. Here, we review preclinical studies that evaluate the efficacy of inhibitors targeting major SMAD-regulating and/or -interacting proteins, particularly enzymes that may play important roles in epithelial or mesenchymal compartments within solid tumors.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
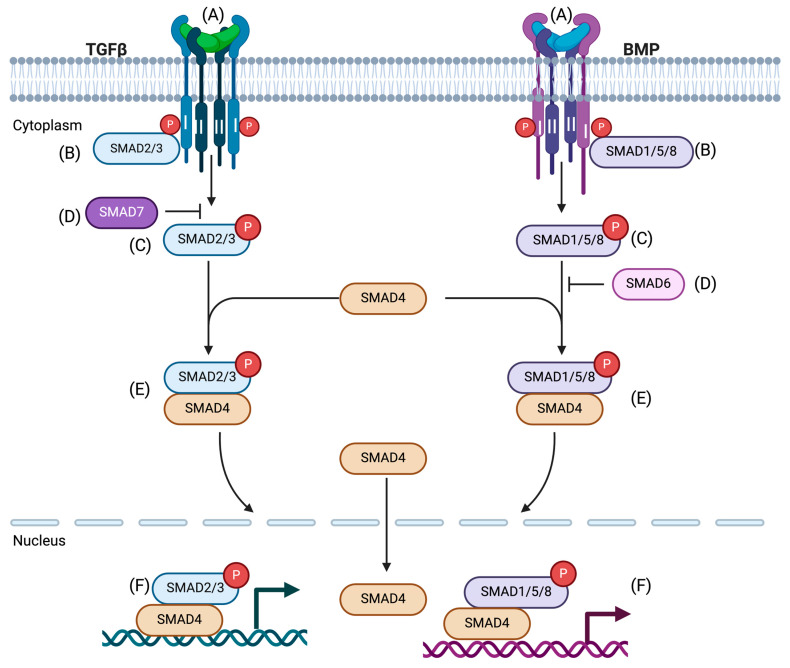 Figure 1
Figure 1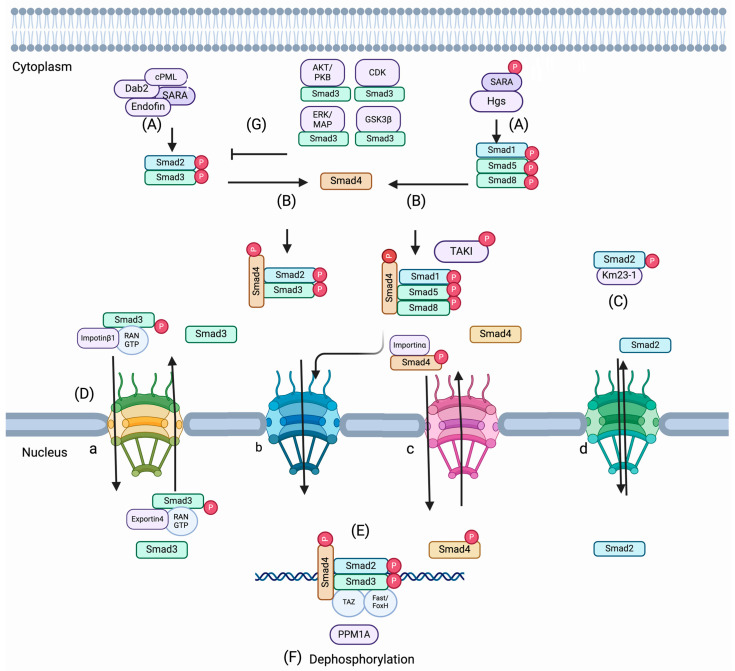 Figure 2
Figure 2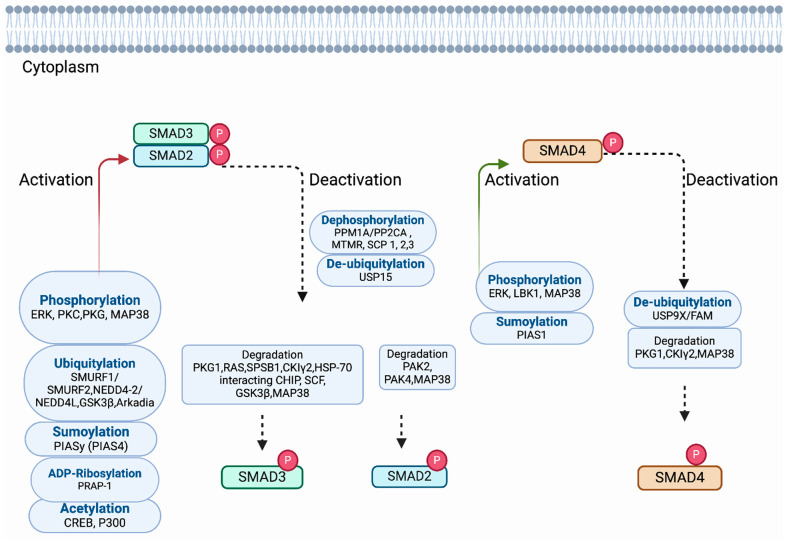 Figure 3
Figure 3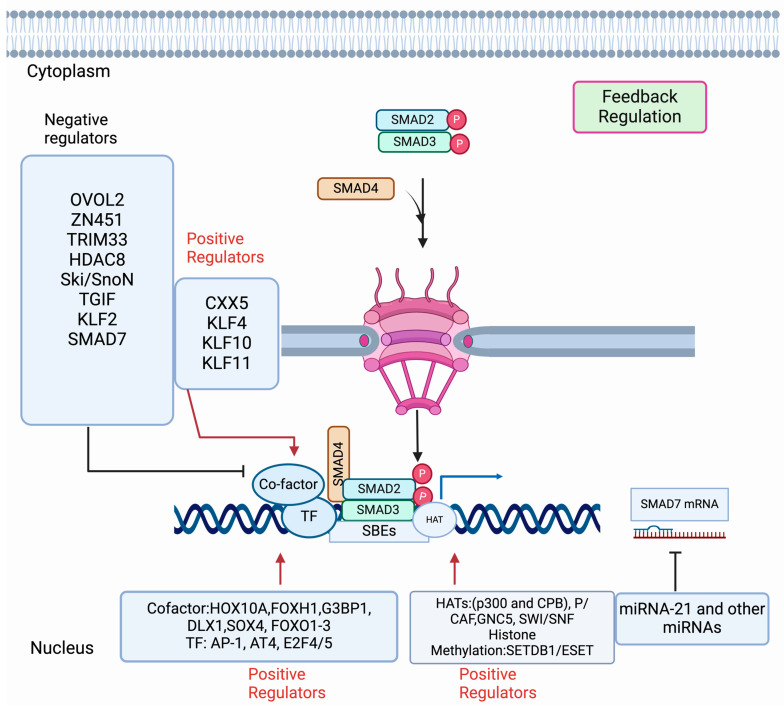 Figure 4
Figure 4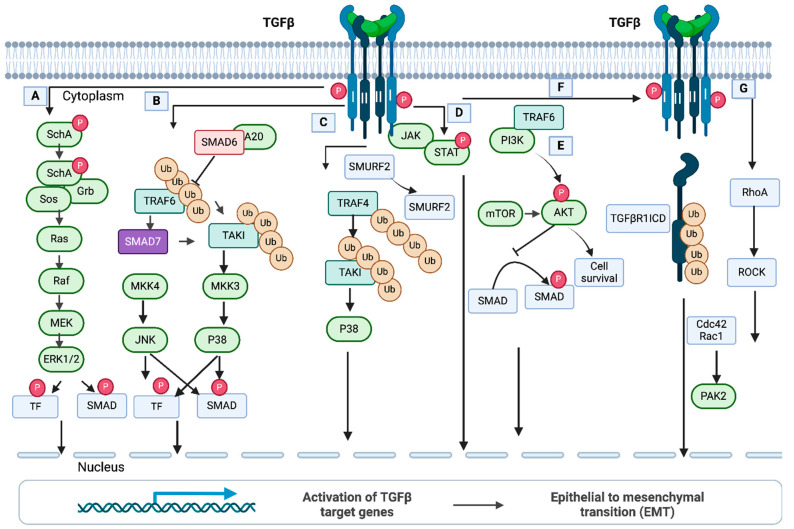 Figure 5
Figure 5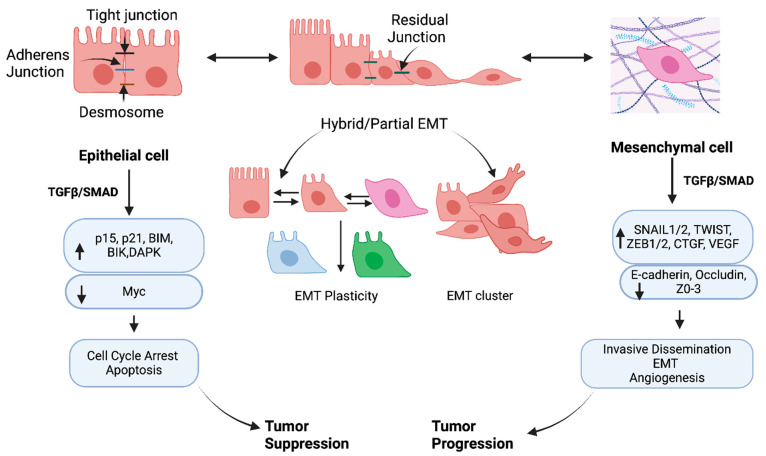 Figure 6
Figure 6- —National Institutes of Health (NIH)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSoybean genetics and cultivation · Legume Nitrogen Fixing Symbiosis · Soil Carbon and Nitrogen Dynamics
1. Introduction
Transforming growth factor-β (TGF-β) superfamily and resulting canonical SMAD signaling have gained significant attention in cancer research since the mid-1980s because of their significance in regulating central functions of the cell, such as proliferation, apoptosis, adhesion, and differentiation. SMADs specifically function to transmit information from extracellular signals received by TGF-β receptors, registering at the plasma membrane to the nucleus downstream of TGF-β. In the nucleus, SMADs cooperate with transcription factors, co-activators, and co-repressor regulators to control gene expression in a context-dependent manner [1,2]. Importantly, many non-SMAD factors are involved in directly controlling how SMAD proteins function in the TGF-β pathway to support, attenuate, or modulate downstream cellular responses.
TGF-β/SMAD plays a biphasic function during tumor progression, where it can suppress or potentiate tumorigenesis in normal and pre-malignant epithelial cells [3]. Signals downstream of TGF-β produced by tumors in the TME (tumor microenvironment) can also activate tumors to undergo an EMT (epithelial–mesenchymal transition) and/or hybrid/partial EMT [4,5,6,7]. EMT is well known to confer invasive, therapy-resistant, and metastatic properties to cancer cells [8,9,10]. Since TGF-β signaling plays a critical role in tumor progression, targeting the downstream SMAD signals presents an enormous challenge in the effort to develop target therapies to eradicate solid tumors effectively. Previous reviews have highlighted novel approaches to targeting signals downstream of TGF-β that focus on microRNAs (miRNAs), long non-coding RNAs (lnRNAs), deubiquitinating enzymes (DUBs), and protein–protein interactions (PPIs) [11,12,13,14,15]. However, the pleiotropic nature of TGF-β signaling contributes to alternate pathway engagement and tumor escape and drug resistance, creating challenges for clinicians and patients. Preclinical findings demonstrate that the TGF-β pathway can be modulated and/or targeted potentially using different alternatives. Although mouse/human chimeric monoclonal antibodies, receptors ligand traps, synthetic DNA antisense oligomers, therapeutic vaccines, and small-molecule inhibitors [16,17,18,19] are worth mentioning, these therapies commonly disrupt normal physiological functions and provide unsatisfactory results in clinical trials [20,21].
Here, we present a consideration of preclinical studies testing the anti-tumor efficacy of pharmacological targeting SMAD-regulating and/or -interacting proteins. We summarize inhibitors targeting the major SMAD-interacting enzymes involved in each pathway in addition to non-SMAD non-canonical pathways downstream of the TGF-β signaling. Finally, we propose how these inhibitors may contribute to abrogating cancer progression induced by TGF-β/SMAD signaling in the epithelial and/or mesenchymal cell types that are commonly found within the solid TME.
2. Canonical TGF-β/SMAD Signaling
The basic principles underlying TGF-β signaling are well-founded. Ligands within the TGF-β superfamily are known as activins/NODALs, bone morphogenetic proteins (BMPs), and growth differentiation factors (GDFs). Ligand-driven signaling activity depends on the formation of pairs with type I and type II receptors. The receptors (serine/threonine kinase) take part in the formation of dimers with disulfide-linked ligand dimers [22,23]. Upon their activation, the type I receptors are trans-phosphorylated by the type II receptors in the juxta membrane region (TTSGSGSG), which drives activation of the type I receptor kinase domain [24,25,26]. These activated receptor complexes subsequently activate a series of downstream signal transduction proteins known as SMAD proteins via phosphorylation that are transported to the nucleus, regulating various transcriptional programs.
Among the eight SMADs in vertebrates, only five are recognized as R-SMADs (Receptor-SMADs). TGF-β- and activins/NODAL-mediated signals activate SMAD2 and SMAD3 predominantly, whereas GDFs and BMPs initiate the activation of SMAD1, SMAD5, and SMAD8 [27]. Two serines are involved to initiate the phosphorylation of receptor and R-SMADs. Primarily, the phosphorylation event of R-SMADs initiates them to form homomeric complexes and subsequently primes them to form complexes with R-SMADs and SMAD4 (Co-SMAD). After activation, the heteromeric complexes (R-SMADs-SMAD4) finally accumulate in the nucleus and bind to DNA (high-affinity sequences) cooperatively with transcription factors and/or co-factors and co-repressors to regulate transcription (Figure 1) [1,2].
3. Activity and Nucleocytoplasmic Trafficking of SMADS
Mechanisms of SMAD Protein Nucleocytoplasmic Trafficking
SMAD proteins exist in dynamic states in the cytoplasm or nucleus with or without the stimulation of TGF-β, respectively. A study by Nicolás et al. [29] showed that both uninduced and induced cells with TGF-β could exhibit variation in SMAD distribution. Accumulating evidence supports that a few proteins are involved in the activation, retention, and trafficking of SMADs from the cytoplasm to the nucleus and vice versa (Figure 2). Activation begins with phosphorylation of the GS region on the type I receptor TGF-β (TGF-βRI) by the type II receptor (TGF-βRII), which leads to the affinity of receptor I for sxsmotif of R-SMADs at the C-terminal [30]. Thereby, the two C-terminal serines of R-SMADs are phosphorylated with substantial interaction between the R-SMADs and the type I receptor. This event is critical to changing the conformation of R-SMADs and allowing them to dissociate consequently from the receptor complex [31]. In this context, the association of the endocytic protein SARA (SMAD Anchor of Receptor Activation) [32,33] and a few SARA-associated proteins (e.g., cPML [34,35], endofin [32], and Dab2 [36] promote R-SMAD activation by recruiting them to the TGF-β receptor. Phosphorylated R-SMADs can interact with SMAD4 and form heteromeric complexes after dissociation from the TGF-β receptor complexes and subsequently transport to the nucleus [35,36,37,38]. It was also found that hepatocyte growth factor-regulated tyrosine kinase substrate (Hgs) is associated with SARA and promotes phosphorylation of SMADs 1, 5, and 8 induced by BMP signals [39,40]. The conformational change driven by the phosphorylation of R-SMAD monomers supports the dissociation from the receptor complex as well as promotes the interaction between the R-SMAD, which is phosphorylated at the C-terminal in addition to SMAD4 and another R-SMAD. Thus, different compositions of SMAD trimeric complexes can exist as a complex of R-SMAD monomers (activated homo or heterotrimer) and/or a complex of phosphorylated one or two R-SMADs with SMAD4 in the cytoplasm [41,42,43,44].
Nuclear translocation of SMAD complexes is sustained by some nuclear pore proteins. Although MH1 domains of SMAD proteins contain NLS-motifs, the regulation of trafficking of SMADs is governed by divergent methods including importin/exportin-mediated, Ran-dependent (Figure 2(Da)); phospho-dependent (Figure 2(Db,Dc)); importin-mediated Ran-independent (Figure 2(Dc)); and importin-exportin-independent and phospho-independent method (Figure 2(Dd)). Many importin/exportin family members and Ran proteins (small Ran GTPase) are involved in the trafficking of R-SMADs. Importin-β1 interacts at the MH1 domain of SMAD3 and is frequently imported into the nucleus through Ran-depended proteins [45], whereas exportin 4, which has a conserved sequence on the MH2 domain of SMAD3, allows the exportation of SMAD3 from the nucleus in a GTPase manner [46]. Importantly, RanBP3 acts as a selective nuclear protein of SMAD2/SMAD3 in the TGF-β pathway. In mammalian cells (human keratinocyte cells, HaCaT, human HEK293T, human HepG2, mouse myoblast cells C2C12) and Xenopus embryos, RanBP3 can directly identify dephosphorylated SMAD2/SMAD3 produced by the phosphatase activity of nuclear SMAD phosphatase. Thereby, the nuclear export of SMAD2/SMAD3 is augmented in a Ran-dependent manner to terminate the TGF-β signal [47]. On the other hand, importin-α mediates the nuclear translocation of SMAD4 [48]. Moreover, nuclear pore proteins (CAN/Nup214 and Nup15) can regulate the cytoplasmic shuttling of SMAD2, SMAD3, and SMAD4 proteins [49,50]. Further, dynein light chain km23-1 and Km23-2 can interact with SMAD2 and SMAD3, which facilitates the nuclear trafficking of the R-SMADS from cytoplasm to the nucleus for target gene transcription [51,52].
Other proteins, like AKT/PKB [53,54], can directly interact with SMAD3 and inhibit its phosphorylation and nuclear accumulation, further inhibiting SMAD3-mediated transcription and apoptosis. On the other hand, ERK/MAP [55,56] CDKs (2/4) [57,58] and GSKB3b [59,60] can inhibit the nuclear accumulation of R-SMADS by phosphorylating the linker region of R-SMADs in both epithelial and fibroblast cells. Nucleocytoplasmic shuttling is mediated by dephosphorylation, which promotes R-SMAD traveling from the nucleus to the cytoplasm. It was found that PPM1A functions as a phosphatase and terminates TGF-β signaling [61]. A study by Dai et al. [62] shows that PPM1A targets the nuclear exporter RanBp3 and thereby controls the transport of SMAD2/SMAD3 from the nucleus to the cytoplasm. RanBP3 is dephosphorylated at Ser 58, which promotes SMAD2/SMAD3 exportation from the nucleus. Thus, PPM1A provides the maximum RanBP3 exporter function for the effective termination of canonical TGF-β signaling.
Intriguingly, it was also found in epithelial, fibroblast, cancer (Hela cells, small lung cancer cell lines, breast cancer cell lines), and embryonic stem cells where SMADs can be reserved in the nucleus through the interaction with high-affinity DNA-binding transcription factors. YAP (Yes-associated protein), co-factors like Fast1/FoxH1, and TAZ (transcriptional co-activator with PDZ-binding motif) can associate with SMADs [63,64,65,66,67]. The nuclear retention inhibits the export of SMADs from the nucleus to the cytoplasm, and it is speculated that the association of YAP and SMAD3 in the nucleus may prevent the interaction of SMAD3 with exportin 4 or RanBP3. Further, it was found that phospho-SMAD-3 in epithelial cells can block the interaction of SMAD-4 with exportin 1 and thereby promote nuclear accumulation of SMAD4 [68].
Inhibitors targeting the SMAD-interacting enzymes such as AKT/PKB (Ser/Thr kinase/protein kinase B), CDK (cyclin-dependent kinase), GSK GSK3b (axin/glycogen synthase kinase 3 beta), and ERK (extracellular signal-related kinase) are cataloged in Table 1 [69,70,71,72,73,74,75,76,77,78,79,80,81,82,83]. These inhibitors act by modulating the phosphorylation activity of the kinases and thereby regulate their interaction, the R-SMADs, and SMAD-mediated signaling in epithelial and mesenchymal cells. Significant advancements have been observed in the area of small-molecule inhibitors of AKT functions through different mechanisms. In addition, pleckstrin homology domain, ATP-competitive, and allosteric inhibitors of AKT are noteworthy in developing clinical trials, and therapeutic benefits in treating different solid tumors have been observed [83]. A preclinical study of MK-2206 in epithelial morphology of pancreatic cancer cells by Wang et al. [72] showed that AKT phosphorylation is inhibited, and cell proliferation is attenuated by the application of the MK-2206 inhibitor. Further, a combination of MK-2206 with gemcitabine enhanced the inhibition of cancer cell proliferation. It suggests that gemcitabine-mediated AKT activation induced increased cell proliferation [72]. In epithelial cells, AKT can act as one of the major mediators of cell survival and apoptosis and regulates the activity of SMAD3 (Figure 2). In epithelioid pancreatic cancer cells, AKT phosphorylation might enhance the activity of SMAD3 and stimulate SMAD-mediated cell proliferation. It is considered that the inhibition of AKT-phosphorylation by MK-2206 may promote the interaction of AKT with SMAD3 and thereby support the inhibition of SMAD3 activation, nuclear accumulation, SMAD3-mediated transcription, and cell proliferation.
Another potential inhibitor is nitazoxanide (NTZ), which targets CDK1 by inhibiting phosphorylation of CDK1 at Thr161, as studied by Huang et al. [74]. It is thought that CDK1 phosphorylation might enhance the activity of SMAD3 and nucleocytoplasmic trafficking and stimulate SMAD-mediated cell proliferation in epithelioid glioblastoma cells. However, NTZ application might modulate SMAD-3 and CDK1 interaction, resulting in inhibition of SMAD3 activation, nucleocytoplasmic trafficking, transcription, and SMAD3-induced cell proliferation. Thus, modulation of the SMAD signaling by inhibiting the phosphorylation of kinase enzymes suggests that NTZ can be a promising CDK1 inhibitor for the development of clinical trials.
4. Regulation of SMAD Activity by Post-Translational Modifications (PTMs)
4.1. Phosphorylation and Dephosphorylation
Activation of R-SMADs is originated by phosphorylation induced by type I receptors of two serines in their carboxy SSXS motif induced by ligands. R-SMAD2 and R-SMAD3 are phosphorylated by the TβRI/ALK-5 (TGF-β-specific type I receptor); R-SMAD 1, 5, and 8 are phosphorylated by ALK-3/BMPRIA (BMP-specific type I receptor, BMP receptor IA). In addition, the structure of R-SMADs with the MHI and MH2 domains and linker region contains substrate sites for other kinases such as Map kinases, cyclin-dependent kinases (CDKs), extracellular signal-regulated kinase 1 (ERK1), and monopolar spindle kinase (MPS1), which regulate the stability of R-SMADs and subsequent transcriptional responses [84,85]. For example, ERK (extracellular signal-regulated kinase 1) phosphorylates SMAD2 on their MH1 domain and enhances SMAD-mediated transcription in epithelial cells [86]. Not all kinases that target R-SMADs potentiate SMAD-mediated transcription. In epithelium and mesenchymal cells, for example, PKC (protein kinase C) directly phosphorylates SMAD3, but this action inhibits SMAD3-dependent transcription [87]. Still, other R-SMAD phosphorylation events generate a platform for R-SMAD degradation via ubiquitin-dependent processes. A study by Saura et al. [88] shows that PKG1 (cGMP-mediated protein kinase 1)-mediated phosphorylation of SMAD3 at the MH1 domain fosters proteasomal degradation of SMAD3 in endothelial cells. Other small GTPase proteins like Ras promote ubiquitin-dependent SMAD2/3 degradation and stabilization of TβRII via the degradation of the SPSB1 protein [89]. Ras interacts and colocalizes with the SPSB (TβRII negative regulator) on the cell membrane, resulting in SPSB1 degradation [89]. Both ubiquitylation and degradation can also occur through phosphorylation of SMAD3 and SMAD2 by casein kinase 1 gamma 2 (CKIg2) and PAK4 [90,91]. And phosphorylation of SMAD2 by PAK2 confers the inactivation of SMAD2 by interfering with TGFβRI-SMAD2 interaction [92] in epithelial cells. SMAD4 is constitutively phosphorylated in epithelial cells (mink lung epithelial cells, Mv1Lu) and cancer cells (human squamous carcinoma cell lines, HSC-4), as shown by a study by Nakao et al. [93]. Although constitutive phosphorylation of SMAD4 has been observed in epithelial and cancer cells, the site of phosphorylation of SMAD4 remains to be identified. ERK promotes SMAD4 nuclear accumulation and enhances SMAD4-mediated transcriptional activity [94]. Other kinases, such as LKB1 (liver kinase B1) and MAP38, phosphorylate SMAD4 on MH1 and MH2 domains, respectively. LBK1 promotes SMAD4 stability by associating with a scaffolding protein LIPI and enhances the SMAD-dependent transcription as a negative regulator in controlling TGFβ gene responses and EMT [95,96]. On the other hand, MAP38-mediated phosphorylation at Ser^343^ of SMAD4 drives positive regulation of transcription in response to TGF-β-mediated apoptosis and cell proliferation in a kinase-dependent manner [96].
The dephosphorylation of C-terminal serine plays a significant role in deactivating the R-SMADs and mitigating the SMAD-mediated signaling. PPM1A (PP2Cα), designated as Mg^+^/Mn^+^-dependent 1A protein phosphatase, was the first phosphatase identified by Lin et al. [61] that dephosphorylates the SMAD2/3 at the C-terminus SXS motif. The phosphate MTMR4 dephosphorylates the activated SMAD2 and SMAD3 in the endosome [97]. Phosphates such as SCP1, SCP2, and SCP3 can remove linker phosphorylation at specific levels without interfering with phosphorylation at the C-terminal of SMAD2/SMAD3 to enhance the TGF-β signal [98]. On the other hand, in mammal keratinocyte cells and Xenopus embryos, dephosphorylation of SMAD1 by SCP1, SCP2, and SCP3 can attenuate the BMP signal [99].
4.2. Ubiquitylation and Deubiquitylation
Ubiquitylation involves the sequential covalent modification of protein substrates with ubiquitin molecules via the action of E1, E2, and E3 ubiquitin ligases, which mark the substrate for further activity or degradation. Regulation of R-SMADs is coordinated by ubiquitylation and ubiquitin-like modifications, which supports SMADs’ stability and subsequent activity and subcellular localization. Several ubiquitin ligases have been involved in the degradation of R-SMADs. HECT (the homologous E6-AP carboxy terminal) family E3 ligases SMURF1 and SMURF2 [100,101,102], NEED4-2/NEDD4L [103], Hsc70-interacting protein (CHIP) [104], and the RING-finger family E3 ligase SCF complexes such as Rbx1, Skp1, Cullins, F-box proteins [105,106] and Arkadia (also known as RNF111) [107,108] are mentioned here. In brief, SMURF1 promotes ubiquitylation of the R-SMADs SMAD1 and SMAD5 and indicates them for degradation [102]. SMURF2 polyubiquitylates SMAD2 and promotes degradation, whereas SMURF2 monoubiquitylates SMAD3, inhibiting the formation of SMAD3 complex [101,102]. NEDD4-2/NEDD4L ubiquitylates both SMAD2 and SMAD3, while CHIP, SCF (Skp1), and GSK3β only trigger SMAD3 ubiquitylation and degradation after phosphorylation [107]. In addition, Akadia triggers both phospho-SMAD2 and SMAD3 ubiquitylation and proteasomal degradation [108].
Monoubiquitylation also regulates the SMAD4-mediated signaling, and it is worth mentioning that SCF^skp2^ E3 ligase ubiquitylates SMAD4 at the MH2 domain [109], enabling SMAD4 mutations in cancer to be degraded. The tripartite motif-containing 33 E3 ligase (TRIM33/TIF1γ/Ectodermin) promotes the disruption of the SMAD complex by SMAD4 monoubiquitylation [110,111]. A deubiquitylating enzyme (DUB) can oppose the action of ubiquitylation on SMADs. For example, USP15 (ubiquitin-specific peptidase) removes the monoubiquitylation of SMAD3 and thereby enhances the access of SMAD complexes to its target promoter domains/complexes [112]. Further, DUB, ubiquitin-specific protease 9x (USP9X/FAM), can reverse the function of monoubiquitylation of SMAD4 at Lys^519^ and inhibit the association of SMAD4 with R-SMADs [113].
4.3. Acetylation, ADP-Ribosylation, and Sumoylation
In addition to phosphorylation and ubiquitylation, acetylation of R-SMADs marks the TGF-β-induced interaction of the transcription co-activators cAMP-response element-binding (CREB) protein (CBP) and p300 by acetyltransferase. SMAD2 is acetylated at the MH1 domain, whereas the MH2 domain of SMAD3 is primarily acetylated, and each acetylation event enhances SMAD-mediated transcription [114,115,116]. ADP-ribosylation is another type of PTM; thereby, one or more ADP-ribose moieties are added to arginine by ADP-ribosyl transferase to control cell signaling and various cellular processes. PRAP1 (ADP-ribose polymerase 1) induces ribosylation of SMAD3 in its MH1 domain and thereby dissociates SMAD3 from the SMAD3/SMAD4 complex. SMAD-mediated TGF-β responses are attenuated by the action PARP-1. Further, overexpression of PARP-1 promotes impaired SMAD3-mediated gene expression and EMT [117]. By SUMOylating, a small ubiquitin-like modifier (SUMO) polypeptide is covalently attached to the target protein. SUMO does not lead to the degradation of the proteins. It was found that SMAD3 was associated with a protein inhibitor of activated STATy (PIAS4, PIASy); SUMO E3 ligase associated with the nuclear matrix could suppress the activity of SMAD3 [118]. Thereby, PIAS4 regulates SMAD-mediated signals by a negative feedback loop. On the other hand, another SUMO E3 ligase, PIAS1 (an inhibitor of activated STAT1), has been identified to promote SMAD4 sumoylation and SMAD-mediated transcription [119].
Table 2 shows the list of inhibitors targeting ERK, PKC (protein kinase C), PKG (protein kinase G), CK1 (casein kinase 1), protein kinase PAK2, PAK4, and LKB1 (liver kinase B1) [120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139]. Others enzyme inhibitors against protein phosphatase, such as (PPM1A/PP2CA, SCP), ubiquitin ligase (NEDD4-2/NEDD4L, HSC70-interacting protein, CHIP, SCFskp1 SCFskp2,) histone acetyltransferase (p300/CREB), and methyltransferase (SET 7/9, SETDB1/ESET, m6 methyltransferase, PRAP1), are listed in Table 2 [140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163]. A study by Vena et al. [131] showed that CK1δ is overexpressed in human pancreatic and bladder epithelial cell lines, and the inhibition of CK1δ by application of SR-309 strongly promotes the antiproliferative effect and sensitizes them to gemcitabine treatments. Casein kinase I (CKI) family consists of different isoforms of alpha (α, β, γ1, γ2, γ3, δ, and ϵ), which have been implicated in critical regulatory roles by interacting with R-SMADs. Figure 3 shows that CK1g2 can promote PTMs of SMAD3 activity by deactivation and degradation of the SMAD3 mediated by ubiquitination and phosphorylation [90]. Considering the negative impact of SMAD-mediated signaling, overexpression of CK1δ might dysregulate R-SMAD activity in epithelial cancer cells and activate further SMAD activity to induce SMAD-mediated protumorigenic signals. Upregulation of deoxycytidine kinase (dCK) using the CK1δ inhibitor, SR-3029, may activate the R-SMADs degradation by phosphorylation and promote an antitumorigenic effect. Another potential inhibitor is FRAX597, a small-molecule ATP-competitive Group I PAK inhibitor. It reduces NF2-deficient schwannoma cell proliferation in culture. In vivo, the inhibitor shows potent anti-tumor activity [133]. In NF2-deficient Schwann cells, originating from mesenchymal stem cells, it might inhibit R-SMAD degradation and promote SMAD-mediated protumorigenic signals. It is considered that the application of FRAX597 further deactivates R-SMADs by inducing PTMs, resulting in anti-proliferative and anti-tumorigenic effects in NF2-deficient Schwann cells. Thus, the FRAX597 inhibitor shows significant potential for the treatment of neurofibromatosis and other cancers.
5. Regulation of SMAD-Mediated Transcription
5.1. Histone Modification
Although SMAD complexes have a low binding affinity, they are guided by many DNA-binding transcription factors, co-factors, repressors, and chromatin modifiers to proficiently control the expression of target genes in a context-dependent manner [164,165,166,167,168,169,170,171]. Here, we discuss and illustrate some SMAD-interacting proteins that regulate SMAD-mediated transcription (Figure 4). It is reported that histone acetyltransferases (HATs) regulate the SMAD-dependent transcription by modifying histones and/or controlling the SMAD activity with chromatin modifiers. Evidence from different studies shows that histone acetyltransferase p300/CBP-associated factor (P/CAF) [172], general control of non-repressed protein 5 (GNC5) [173], switch/sucrose non-fermentable (SWI/SNF) remodeling complex [174], and histone lysine methyltransferase 1 (SETDB1/ESET) [175] are important in executing the function of HATS for SMAD-mediated transcription. The histone acetyltransferases CREB-binding protein (CBP) and p300 govern the SMAD3 transcriptional activity, whereas SMAD4 plays a role as a transcriptional co-activator to stabilize the SMAD complex [176]. P/CAF enhances SMAD3-mediated transactivation driven by the interaction of SMAD3 independently or in association with p300 and [172]. Another HAT, GCN5, plays a role as a co-activator of both TGF-β and BMP-induced SMADs mediated transcription induced by both TGF-β and BMP [173]. SMAD2-mediated transcription is regulated by histone 3 (H3) acetylation with the recruitment of p300 and SWI/SNF and, thereby, confers that chromatin remodeling is necessary for SMAD-mediated transcription [174]. On the other hand, a study by Du et al. [175] shows SMAD-3 mediated recruitment of a histone methaytransferase1, SETDB1/ESET, regulates snail gene expression, EMT, and cancer dissemination.
SMAD-driven transcription acts as positive and negative regulators, thereby maintaining the feedback loop of TGF-β/SMAD signaling. The proteins that act as positive regulators are predominantly FOXA10 (Homeobox A10), FOXH1 (Forkhead pioneer factor), G3BO1 (Ras GTPase-activating protein SH3 domain-binding protein 1), AT4 (activating transcription factor 4), SOX4, DLX1, FOXO1-3, E2F4/5, AP-1, CXXC5, and KLF family member proteins (Figure 4).
HOXA10 binds to the SMAD complex and modulates the expression of snail and slug through m^6^A modification of mRNA and METTL3 expression by enhancing the SMAD signaling [177]. FOX1 can recruit SMAD2 to SMAD4 and assist in binding the SMAD complex with SMAD3:FOXH1 [178]. G3BP1 is another positive co-factor that acts as a novel binding partner to the SMAD complex by activating SMAD signaling and recruiting the SMAD2/SMAD4 complex [179]. The co-factor SOX4 can occupy many genomic loci with SMAD3 in a cell type-specific manner [180]. Forkhead-binding element (FHBE) resides within the SMAD binding region of the p21Cip1 promoter and associates with SMAD2/SMAD3/SMAD4 complex coprecipitated with FOXO1, FOXO2, and FOXO3 [181]. AP-1 and AT4 are downstream regulators of the SMAD complex and mTORC2 and control TGF-β/SMAD and mTOR/RAC1-RHOA pathways independently [182]. DLX1 can positively modulate the TGF-β/SMAD signal by interaction with SMAD4 localized in the nucleus upon TGF-β1 induction [183]. The transcription factor E2F4/5 controls SMAD-mediated transcription in a promoter-specific manner [184,185]. The complex of SMAD3 and the transcription factors E2F4/5, DP1, and corepressor p107 translocate to the nucleus and interact with SMAD4, distinguishing a combined SMAD-E2F site for arresting the cell cycle [185]. Another novel regulator and coordinator of TGF-β, BMP, and Wint signaling is CXXC5, which associates HDAC1 and competes for binding with SMAD complex in hepatocarcinoma cells. CXXC5 reduces the inhibitory effect of HDAC1, resulting in cycle arrest and apoptosis [186,187]. In this context, some Küppel-like factors (KFLs) are well known and can associate with SMADs and act as DNA-binding transcriptional regulators. There are 17 well-known KLFs that have highly conserved C-terminal DNA-binding domains with three C2H2 zinc finger motifs. Some of them play significant roles in feedback regulation in SMAD-mediated transcription. Among them, KLF4, which is up-regulated in vascular smooth muscle cells, recruits p300 and forms an active complex with SMAD2. Thus, KL4 induces the expression of SM22α and α-SMA [188,189]. KLF10 can facilitate multiple TGF-β-induced functions through the expression of SMAD2, inhibition of SMAD7 expression to control the proliferation of epithelial cells, and development of immune and bone cells [190]. Further, KLF11 associates with SMAD3 and enhances TGF-β-induced growth inhibition by repressing SMAD7 and Myc expression in epithelial and pancreatic cancer cells [191].
Among the negative regulators, zinc finger protein OVOL2, 451, histone-binding protein TRIM33 (also E3 ubiquitin ligase activity), HDAC8 (histone deacetylate 8), Ski/SnoN (Sloan Kettering Institute) and SnoN (Ski novel), TGIF, Sp1/KLF-like zinc-finger protein KLF1, and PAX2 are significant to control SMAD-mediated transcription in a cell and context-dependent manner (Figure 4). The zinc finger protein 451 inhibits the recruitment of p300 to the SMAD complex and represses SMAD-mediated transcription, resulting in the reduced H3 Lys9 acetylation of the promoters of target genes [192]. Further, the recruitment of TRIM33 to chromatin is mediated by SMAD4, which promotes histone modification upon binding of TRIM33 and SMAD2/SMAD3 complexes on the regulatory sequences of the target genes. This modification by histone allows the switching of the chromatin state from the poised to the active state and supports the negative feedback mechanism [193].
HDAC8 is class I histone deacetylase, a novel co-factor of the SMAD3/4 complex. A study by Tang et al. [194] shows that chromatin remodeling represses SIRT7 transcription by making a complex with HDAC8 and SMAD2/SMAD3. In contrast, the reduction of SIRT7 activates TGF-β by a feedback loop, which regulates the TGF-β/SMAD signal. The negative regulator OVOL2 induces the expression of SMAD7, thereby reducing the expression of SMAD4 and interrupting the complex formation by interfering with the complex formation between SMAD2/SMAD3 and SMAD4 [195]. Also, the negative regulators Ski and SnoN interact simultaneously with the R-SMADs and SMAD4 and disrupt the ability of the SMAD complexes to turn on the target genes [196,197]. Ski/SnoN actively recruits a transcriptional co-repressor complex containing N-CoR/HDAC to the targeted promoter by preventing the recruitment and binding of R-SMADs to p300/CBP. It has been shown that 5′TG3′-interacting factor (TGIF) represses TGF-β signaling [198] by binding with R-SMAD/SMAD4 complexes. Further, Guca et al. [199] showed that TGIFI-HD (homeodomain) binds to SMADs in a mutually exclusive manner. Thereby, it provides a transcriptional repression system in a context-dependent manner. Among the KLF family, KLF2 employs a negative feedback loop on TGF-β/SMAD signaling by inducing SMAD7 transcription [200]. A negative feedback loop is maintained to regulate TGF-β/SMAD signaling by the action of I-SMADs. It is found that in the absence of a ligand, TGF-β promotes nuclear accumulation of I-SMADs from the nucleus to cytoplasm and promotes SMAD7 mRNA and, thereby, creates a negative feedback loop that firmly controls TGF-β SMAD signaling [201,202]. Enhanced SMAD7 methylation by SET9-mediated TGF-β/SMAD signaling promotes its association with the E3 ubiquitin ligase Arkadia and enhances its ubiquitylation and degradation in lung fibroblast. Pharmacological inhibition or depletion of Set9 showed that high SMAD7 protein levels inhibited the expression of TGF-β/SMAD-mediated genes encoding extracellular matrix components [203].
5.2. Regulation of SMAD-Mediated Transcriptional Activity Post-Transcriptionally
At the post-transcriptional level, SMAD-mediated transcription is regulated by RNA-binding proteins (RBPs), microRNA (miRNA), and non-coding RNAs (ncRNAs). A study by Tripathi et al. [204] shows that the interaction of phosphorylated T179 of SMAD3a with PCBP1 (RNA-binding protein) promotes alternative cancer stem cell marker CD44 splicing. SMAD3 and PCBP1 can cooperate in the variable regions of exons for CD44 pre-mRNA and modulate spliceosome assembly. As a result, mesenchymal isoform CD44s is expressed over epithelial isoform CD44s. Thus, alternative splicing by SMAD3 plays an important role in tumorigenesis. Interestingly, R-SMADs are found to influence miRNA expression, processing, and maturation of 44 types of miRNAs. R-SMADs can bind to the stem region of miRNA at a consensus sequence and recruit p68 (DDX5), an RNA helicase component, DROSHA (DROSHA/DGCR8/RNA) in a ligand-dependent manner. Thus, the post-transcriptional modification induced by TGF-β and BMP signaling can drive increased expression of mature miRNA-21 after processing of primary transcripts of miRNA-21 (pri-miRNA-21) into precursor miRNA-21 (pre-miRNA-21) [205,206]. In addition to miRNA-21, other miRNAs are directly involved in the feedback regulation of SMAD-mediated signaling, as reported by Yan et al. [207]. Additionally, long-coding RNAs (lncRNAs) are recognized as intermediaries of the TGF-β response. A comprehensive analysis by Adylova et al. [208] shows that the different lncRNAs can play a role in positive and negative regulation of TGF-β/SMAD signaling in different cancer cells. Another post-transcriptional negative feedback has been described by Bertero et al. [209] in human pluripotent stem cells in which R-SMADs can associate with the m^6^A methyltransferase complex METTL3-METTL4-WTAP. In the nascent transcripts, N^6^-adenosine methylation on the RNA occurs by the action of the m^6^A methyltransferase, resulting in destabilizing and degrading the transcripts [209].
Table 3 shows the list of inhibitors targeting histone acetyltransferases (GNC5/PCAF, SWI/SNF, histone lysine methyltransferase (SETDB1/ESET), small GTPases (RAS), histone deacetylases (HDAC1, HDAC8), and methyltransferase (m^6^A methyltransferase, SET (7/9) methyltransferase) [123,160,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234]. Some of the potential inhibitors are highlighted here. The application of microRNAs (miRNAs) for cancer therapy and radiosensitivity in tumors has achieved significant consideration. A study by Shao et al. [217] showed that miRNA-621 could enhance the radiosensitivity of hepatocellular carcinoma (HCC) through direct inhibition of SETDB1 and targeting the 3′UTR of SETDB1. It was reported that miRNA-621 and/or the SETDB1 axis activated the p53-signaling pathway and advanced the radiosensitivity of HCC cells. SETDB1/ESET regulates SMAD-mediated transcription to control snail gene expression in epithelial or mesenchymal cells in a context-dependent manner. It is supported that snail may bind directly to the DNA-binding domain of p53 and reduce the p53 tumor-suppressive function. Since the expression of miRNA-621 and SETDB1 are negatively correlated in HCC tissues, miRNA-621 might enhance the radiosensitivity and active p53 signaling pathway in HCC cells by inhibiting SETDB1 expression. Thus, mi-RNA-621 and/or SETDB1 in epithelial cells of HCC can be potentially used as a novel therapeutic target [217].
STM2457 is the first bioavailable small-molecule inhibitor of METTL3 (a regulator of m^6^A methyltransferase) that affects the inhibition of catalytic activity and upregulation of METTL3 increasing PD-L1 and reduction of tumor progression in NSCLC (non-small-cell lung cancer) [233]. Upregulation of METTL3 induced by STM2457 enhances the interaction of METTL3 with the translation initiation machinery to make more circularized mRNA of PDL-1. In this case, STM2457 may inhibit R-SMADs from associating with the m^6^A methyltransferase complex METTL3 to destabilize and degrade the nascent transcripts of PDL-1. Thus, STM2457 is considered a potential suppressor that provides an inhibitory effect in epithelial cells of NSCL.
The SET domain containing lysine methyltransferase (SETD7/9) is involved in various disease-related signaling pathways with a broad group of substrates. Methyltransferase activity of human SETD7 is inhibited by a selective inhibitor, (R)-PFI-2. It was found that the Hippo pathway was modulated by (R)-PFI-2 with increasing nuclear YAP and YAP-mediated gene expression in epithelial cancer cells (MCF7) and murine embryonic fibroblasts [234]. How (R)-PFI-2 works is not well defined, but a crosstalk between TGF-β/SMAD and Hippo signaling suggests that the binding of YAP to SMAD7 may modulate TGF-β signaling. SMAD7 methylation by SET (7/9)-mediated TGF-β/SMAD signaling might promote its association with E3 ubiquitin ligase, thereby enhancing its ubiquitylation and degradation. YAP can associate with the complexes of R-SMADs-SMAD4 and drive their sub-cellular localization and transcription in a context-dependent manner. Thus, (R)-PFI-2 and related compounds can be valuable inhibitors to target methyltransferases.
6. Non-SMAD, Non-Canonical TGF-β Pathway Control
6.1. ERK/MAP Kinase Signaling
The Ras/Raf/MAPK (MEK)/ERK pathway is one of the important signaling cascades and contributes an important role in tumor cell survival and progression. TGF-β activates the ERK-MAP kinase pathway by direct phosphorylation of ShcA, which subsequently activates downstream signals (Figure 5A). A study in both Mv1Lu mink lung epithelial cells and mouse fibroblasts by Lee et al. [235] showed that phosphorylation of ShcA on tyrosine results in the initiation of a docking site for the downstream mediators Grb2 and Sos, which further activate Ras GTPase, Raf, MEK (MAP kinase/ERK kinase), and EK1/2 kinase. In conjugation with SMADs, Erk regulates the transcription of target genes through the downstream transcription factors and regulates EMT. ERK activation in human keratinocytes (HaCaT) is initiated by the TGF-β receptor complexes, which are localized in lipid rafts. Clathrin-dependent endocytosis of TGF-β receptor complexes initiates SMAD activation, SMAD-mediated transcription, and TGF-β/SMAD-directed epithelial cell plasticity [236].
6.2. JNK and p38 MAP Kinase Signaling
Jun N-terminal kinases (JNKs) and p38 mitogen-activated protein kinases (MAPKs) significantly coordinate with many signaling mechanisms associated with stresses. In addition, different cellular functions are regulated by the JNK and p38 pathways. In brief, TRAF6 (Tumor Necrosis Factor Associated Receptor-Associated Factor 6) interacts with the TGF-β receptor complex once receptors are activated by ligand binding. An autoubiquitylation modification induces TRAF6, which activates TAKI by polyubiquitylation at Lys^63^TAK1 and initiates the p38 MAPK pathway (Figure 5B) [237,238]. Importantly, TAK1 is a negative regulator of canonical TGF-β signaling and promotes R-SMAD phosphorylation at the linker region in neural crest-derived mesenchymal cells [239]. In this pathway, I-SMADs play significant roles in regulating p38 MAP kinase signaling. It was found that SMAD7 in prostate cancer cells can act as a scaffolding protein for p38 and its upstream kinases [240]. Also, in AML-12 mouse liver cells and primary hepatocytes, SMAD6 acts as a negative regulator and eliminates K63-linked polyubiquitylation of TRAF6 by recruiting the A20 DUB enzyme induced by TGF-β1 [241].
Another ligase, E3, and TRAF4 activate the MAP kinase pathway (Figure 5C). In this pathway, TRAF4 is auto-ubiquitylated upon ligand binding and is recruited to the TGF-β receptor complex. In breast cancer cells, TRAF4 activates TAKI via polyubiquitylation, which results in the activation of the p38 pathway. Subsequently, SMURF2 is degraded through polyubiquitylation by TRAF4, thereby maintaining the stability of the TGF-β1 receptor [242].
6.3. JAK-STAT Signaling
JAK/STAT (the Janus Kinase Signal Transducer and Activator of Transcription) pathway is a prime regulator of cell function. (Figure 5D). Jak/STAT-mediated downstream effects may vary and drive hematopoiesis, tissue repair, inflammation, immune surveillance, apoptosis, and adipogenesis [243]. A study by Tang et al. [244] shows that JAK1 is a constitutive TGF-βRI that is essentially involved in STAT phosphorylation within a short period after TGF-β stimulation. Once SMADs are activated, another phosphorylation of STAT is started for de novo protein synthesis. Thus, the non-SMAD JAK1/STAT pathway in hepatic stellate cells is essential for the expression of TGF-β subset genes.
6.4. PI3/AKT/mTOR Signaling
The two pathways, PI3K (Phosphatidylinositol-3-kinsae)/Akt and Mammalian Target of Rapamycin (mTOR), play roles in many different cellular functions (Figure 5E). In epithelial cells, the TGF-β type I receptor (TGF-βRI) can bind PI3K and modulate the kinase activity [245]. Further, a study by Hamidi et al. [246] demonstrated that in prostate cancer cells, TRAF6 can polyubiquitylate p85α, a regulatory subunit of PI3K, and make a complex between p85α and TGF-βRI to activate PI3K and AKT. Moreover, Lamouille et al. [247,248] showed that TGF-β can induce mTORC2 (Mammalian Target of Rapamycin Complex 2), which further phosphorylates and activates AKT, resulting in cell size changes and epithelial cell progression through EMT.
6.5. TGF-β Type I Receptor (TGF-βRI) Intracellular Domain Signaling
TGF-βRI intracellular domain signaling (Figure 5F) promotes TGF-β-mediated tumor invasion. It is reported by Mu et al. [249] that in prostate cancer cells, TGF-β uses TRAF6, resulting in Lys63-linked polyubiquitination of TGF-βRI and promoting cleavage of the extracellular domain of TGF-βRI by TACE (TNF-alpha converting enzyme). The newly formed intracellular domain (ICD) of TGF-βRI can bind with p300 for transcription of genes, thereby driving tumor invasion by induction of SNAIL and MM2 [250]. TRAF6 can polyubiquitylate a membrane-bound protein, PS-1 (membrane-embedded protease presenilin-1), to initiate a proteolytic cleavage on TGF-βRI. Thereby, the ICD (the complete intracellular domain) of the receptor is formed to assist the translocation of TGF-βRI-ICD to the nucleus [251]. In the nucleus, TGF-βRI-ICD binds to the promoter and turns on the transcription of the genes encoding TGF-βRI. In prostate cancer, TRAF-6-mediated Lys6-linked ubiquitination of the TGF-βRI intracellular domain is important for the modulation of TGF-β and regulation of other genes controlling the cell cycle, proliferation, differentiation, and migration [251].
6.6. Rho-(like) GTPase Signaling
Rho GTPases encompass a branch of the Ras superfamily with 22 genes in humans, and among them, RhoA, Rac1, and Cdc42 are the best exemplified (Figure 5G). They have a significant role in many cellular processes, mainly in cell morphology regulation, cell adhesion, and cytokinesis production. In many studies of mammalian cells, the constitutive and dominant negative mutants were used to examine the function of Rho GTPases. Bhowmik et al. [252] showed that in epithelial cells, the RhoA-dependent signaling pathway stimulates the formation of stress fibers induced by TGF-β. As a result, epithelial cells can transform into cells with mesenchymal characteristics such as increased N-cadherin expression and motility with the loss of E-cadherin (markers of TGF-β-induced EMT). TGF-β induction can be reduced by blocking RhoA or its downstream target, p160^Rock^, expressed by the dominant-negative mutants. Another study by Edlund et al. [253] showed that TGF-β1-treated human prostate carcinoma cells (PC-3U) can promptly form lamellipodia by rearranging the actin filament system. This response was independent of SMAD for a short time, but it requires Rho GTPases Cdc42 signaling for the long term.
A regulator of epithelial cell polarity, Par-6, can negatively control Rho-GTPase signaling by cooperating with TGF-β [254]. Par6 is associated with the TGF-βRI, phosphorylated by TGF-βRII, and thereby it recruits SMURF1 ubiquitin ligase (Figure 5G). Rapid degradation of RhoA GTPase is initiated through polyubiquitylation by SMURF1. This introduces the loss of a tight junction of epithelial cells. Further, Wilkes et al. [255] showed that signals from the TGF-β receptor activate PAK2 (STE20 homolog) in mammalian cells. PAK2 activation was observed in fibroblasts (not in epithelial cell cultures) mediated by signals from SMAD2 and/or SMAD3. In fibroblasts, Rac1 and Cdc42 might regulate PAK2 activity induced by TGF-β. However, targeting PAK2 by morpholino antisense oligonucleotides and dominant negative PAK2 can prevent the morphological features. Thus, PAK2 is considered a novel SMAD-independent pathway that distinguishes TGF-β signaling in fibroblasts and epithelial cells.
6.7. Crosstalk between SMAD and Other Signaling Pathway Molecules
The intracellular signaling network, the SMAD pathway, and the other pathways are connected to develop crosstalk. The crosstalk within the pathways plays an important role in the regulation of biological processes. The crosstalk can occur at different levels by altering signaling components, transcriptional modification, and chromatin modification or by direct interactions between intracellular signaling components with SMADs. Here, we briefly mention the crosstalk of SMADS with Yes-associated proteins with a PDZ-binding motif (YAP/TAZ) and interactions with other proteins, such as TRAP-1, km23-1, and PKA. The binding of YAP to SMAD7 by TGF-β and Hippo signaling was the first reported crosstalk that resulted in the increased inhibition of TGF-β signaling [256]. YAP can bind to the PpxY motif and phosphorylate the SMAD1 linker region by CDK9 in mammalian cells [257]. Neural cell (mESC) differentiation by BMP signal is suppressed by YAP-SMAD1 binding and further SMAD-1-dependent transcription. TAZ and YAP can associate with a heteromeric complex of R-SMAD-SMAD4 mediated by TGF-β signaling [258]. In contrast, another study showed that SMAD nuclear localization in response to TGF-β is not dependent on the levels of YAP or TAZ [259]. Also, it was found that the localization of YAP/TAZ can overcome cell cycle arrest stimulated by TGF-β and promote a pro-tumorigenic transcriptional program [260]. It is reported that TGF-β/SMAD/YAP/TAZ crosstalk also plays an important role in cell differentiation, proliferation, and fibrogenesis in a context-dependent manner [259,260].
The protein kinase A (PKA) signaling pathway is described by Wang et al. [261] in mesangial cells. PKA activates TGF-β stimulated cAMP response element-binding protein phosphorylation and expression of fibronectin. A study by Yang et al. [262] reported that PKA is independent of cAMP by aiding an interaction of PKA holoenzyme subunits with activated SMAD2/SMAD3. The interaction domains of SMAD4 and PKA-R and their functional roles are defined by the study. It was shown that amino acids 290–300 of the SMAD4 linker region are critical for the specific interaction of SMAD4 and PKA-R for the regulation of TGF-β-mediated cellular functions (e.g., PKA activity, CREB phosphorylation, induction of p21, and inhibition of growth). In pancreatic cancer cells, SMAD-PKA plays a role in TGF-β-induced EMT and tumor growth.
TLP is a TRAP-1-like protein that acts as an adaptor protein that mediates the interaction with the TGF-β type II receptor and the downstream effector SMADs. Felici et al. [263] proposed through a study that SMAD2 and SMAD3 might be balanced by TLP signaling through the localization of SMAD4 intracellularly. Thus, the specificity of TGF-β transcriptional responses can occur in a context-dependent manner.
Table 4 shows the inhibitors of SMAD-interacting enzymes (Jun N-terminal kinase (JNK), p38 MAP kinase, AKT, TRAF6, RhoA, PKA) that control non-SMAD noncanonical TGF-β pathways [264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288]. A potential inhibitor, SP0016125, targets c-Jun N-terminal kinase (JNK) and stimulates TGF-β-induced apoptosis of the RBE human cholangiocarcinoma cell line [264]. The result showed that SP0016125 increased the TGF-β-induced SMAD2 and SMAD3 phosphorylation, which promoted TGF-β1-induced transcriptional response and apoptosis in RBE cells. Depletion of SMAD4 reduced the effect of SP600125 on the transcriptional response and apoptosis. Further, TGF-β-induced apoptosis was abolished using the pan-caspase inhibitor Z-VAD-fmk. These findings indicate that SP600125 enhances TGF-β-induced apoptosis of RBE cells by promoting SMAD-dependent caspase activation through a SMAD-dependent pathway. Further, a study by Lu et al. [265] showed a novel pro-apoptotic role in combination with dihydroartemisinin (DHA). In human lung adenocarcinoma cells (ASTC-a-1) induced by DHA, SP600125 synergistically enhances apoptosis through Bax translocation and other intrinsic apoptotic pathways like caspase activation and the mitochondrial pathway. These findings suggest that SP600125 may enhance TGF-β-induced apoptosis in lung adenocarcinoma through a SMAD-dependent caspase pathway. Therefore, SP0016125 is considered a novel therapy for the treatment of cholangiocarcinoma and lung adenocarcinoma epithelial or mesenchymal cells that express JNK.
A potential inhibitor named LY2228820 (an imidazole derivative) is a selective ATP-competitive inhibitor of the α-and β-isoforms of P38α mitogen-activated protein kinase (p38 MAPK) [274]. In vivo cancer models of breast, ovarian, lung, glioma, and myeloma showed that tumor growth was potentially delayed with LY2228820 treatment. Since p38α regulates cytokine (TNF, IL-Iβ, IL-6, IL-8, etc.) production in TME, it is considered that TRAF6 expression in multiple cancer cell lines (MDA-MB-468, OPM-2, A549, A2780) may also control cytokine production. Further, it is hypothesized that auto-ubiquitylation of TRAF6 upon ligand binding might result in recruiting the TGF-β receptor complex, which in turn actives TGF-β/SMAD-mediated target genes to stimulate EMT. Therefore, LY2228820, a p38 MAPK inhibitor, has been optimized for various purposes such as potency, selectivity, bioavailability, and efficacy in animal models of human cancer.
Another important inhibitor of the non-SMAD, noncanonical TGF-β pathway is PKI protein targeting PKA, a well-known regulator of physiological and oncogenic functions. A study by Hoy et al. [289] found that PKI can modulate tumor growth by a molecular switch to drive GPCR-Gαs-CAMP signaling toward EPAC-RAP1 and MAPK. Amplification of PKIA is common in prostate cancer cells, and depletion of PIKA showed reduced migration and tumor growth. To understand the mechanism of PKIA, it is hypothesized that the expression of PIKA and EPAC may modulate MAPK or ERK/MAPK signaling. In conjugation of SMADs, Erk may regulate downstream gene expression for EMT in prostate cancer cells.
7. TGF-β/SMAD Mediated Progression in Solid Tumors
The TGF-β/SMAD signaling pathway plays a dual role in cancer progression. In addition to promoting apoptosis and cell cycle arrest in epithelial cells, TGF-β/SMAD and non-SMAD signaling enhances EMT, cell migration, invasion, angiogenesis, and stemness. TGF-β/SMAD exhibits a tumor suppressor phenotype in epithelial cells at the early stages of tumorigenesis. In contrast, at the later stages, TGF-β/SMAD signals drive oncogenesis and metastasis in mesenchymal properties of cancer cells (Figure 6). Here, we show SMAD-dependent canonical and non-canonical signals mediated by cancer cells with epithelial, EMT, hybrid/partial EMT, and mesenchymal properties associated with cancer progression and metastasis.
7.1. Epithelial Cells
The typical characteristics of epithelial cells are described and summarized by Huang et al. [289]. Briefly, epithelial cells comprise strong apical and basal polarity with plasma membranes placed toward and away from the lumen, respectively. Different proteins are found in each membrane. Proteins in the membrane assist in the transportation and localization of targeted molecules to specific cellular regions with various activities (Figure 6). Both SMAD-dependent canonical and non-canonical pathways predominantly regulate non-cancerous epithelial cells at the pre-malignant stage by cell cycle progression through CDK (G1-Arrest Activating Cyclin-Dependent Kinase) inhibitors p15 and p21. Further, the downregulation of an important oncogene, c-Myc, drives the proliferation and inhibits the transcriptional activity of p15 and p21 [3]. Importantly, apoptosis is initiated by the signals modulating the expression of B-cell lymphoma-2 (BCL-2) family members, BIM (BCL2L11), FAS (death receptor fibroblast-associated antigen (FAS)), DAPK (death-associated kinase), BH3-protein BIK, and caspases, which induce both intrinsic and extrinsic apoptosis [290,291,292]. Further, tumor suppression mediated by TGF-β/SMAD signaling has been demonstrated in some solid tumors like breast cancer, prostate cancer, melanoma, and colon cancer [293]. The non-canonical TGF-β/SMAD is linked to p38 MAPK and caspase-8-dependent programmed cell death, which exhibits a tumor suppressor role by inducing apoptosis [294]. Further, to drive tumor cell death by activating programmed cell death, TGF-β/SMAD promotes the regulation of immune cell function [295,296].
7.2. Cancer Cells with EMT, and Hybrid/Partial EMT
The transition of cancer cells is dynamic, allowing cells to go from epithelial to mesenchymal states and vice versa. A change from epithelium to the mesenchymal state is referred to as EMT (epithelial–mesenchymal transition), which allows cells to gain migratory properties by modifying the adhesion molecules expressed by the cells. The reverse process is MET (mesenchymal–epithelial transition), which is associated with loss of migratory properties rather than adopting an apicobasal polarization [4]. Thus, beyond epithelial phenotype, various EMTs can impact cell behaviors, physiology, and ecology, allowing the transition of different phenotypes (Figure 6). The EMT is implemented for pleiotropic signaling through canonical and non-canonical SMAD pathways. Thereby, specific transcription factors (TFs) named EMT-TFs (e.g., SNAIL, ZEB, TWIST) are expressed to regulate EMT and MET.
Along with transcription, miRNA, post-translational and epigenetic regulators mediate EMT in cancer progression [289]. EMT has been established as a spectrum rather than a linear process supported by recent studies [5,6,7] (Figure 6). A study by Pastushenko et al. [6] used different markers, CD106, CD51, and CD61, showing that the different states of the EMT spectrum can form a “hybrid” (Figure 6). A single-cell analysis found that partial/hybrid EMT expresses SNAIL1/2, ZEB1/2, and TWIST1/2 during mouse organogenesis in a manner similar to epithelial gene expression [297]. Further, using the single-RNA sequencing technique with primary and metastatic head and neck squamous cells, it was shown that that cell contained a gene signature of partial EMT [298]. Partial EMT is also highly aggressive and invasive since the cells present several classical features of EMT. Expression of vimentin (VIM), TGF-β1, and extracellular matrix genes are worth mentioning. Although the EMT-TF expression was low, transcription of epithelial genes was still maintained within the partial EMT. The EMT phenotype can be variable, such as pancreatic ductal adenocarcinoma resulting from adjusting epithelial junction proteins that showed no change in expression, contrasting the common repression of epithelial characteristics of cells [299]. Moreover, many carcinoma cells, such as breast and colorectal cancer cells, can utilize this partial/hybrid EMT program to make a cluster of cells contrasting the single-cell pattern, which is connected to traditionally defined EMT mechanisms [299].
In addition, EndMT (endothelial–mesenchymal transition) shows similarity to EMT in the context of molecular processes and phenotypes. The loss of endothelial junctions, EMT-TF activation, and upregulation of mesenchymal markers push EndMT formation. Endothelial cells downregulate the VE (vascular endothelial cadherin), where cell-type-specific changes distinguish EndMT from EMT [300].
7.3. Cancer Cells with Mesenchymal Characteristics
Upon EMT, cells repress their epithelial phenotypes and gain mesenchymal and invasive properties during cancer progression. The hallmarks of EMT are increased N-cadherin and VIM expression with decreased expression of E-cadherin, ZO1, and desmoplakin [301,302,303]. In cancer cells with mesenchyme morphology, the major EMT-TF are zinc finger binding transcription factors, including SNAIL1/2, E-box binding homology frame factors (ZEB1/2), and BHLH (basic Helix–Loop–Helix) factors (TWIST1/2) [304]. Both SNAIL1 and SNAIL2 drive tumorigenesis induced by EMT. During the upregulation of mesenchymal phenotypic markers, including VIM and fibronectin, SNAIL1 directly inhibits TJ formation by decreasing epithelial markers like E-cadherin and claudin expression. E-cadherin positively correlates to patient survival, whereas the overexpression of MMPs provides aggressiveness of tumors [305]. On the other hand, SNAIL2 promotes the loss of cell adhesion and polarity by decreasing e-calmodulin levels, thereby influencing migration and metastasis in breast and ovarian cancer [306,307,308].
In tumor stem cells, ZEB1/2 expression in epithelial cells causes EMT and mesenchymal phenotypes with invasiveness, metastatic, and dedifferentiation potential [309]. In vivo study of pancreatic cancer supports that ZEB1 is essential for tumor invasion and metastasis [310]. Collective data show that ZEB1/2 expression in breast, colorectal, and pancreatic cancers affects poor patient outcomes [311,312,313,314].
Like SNAIL, TWIST1 can suppress the expression of E-cadherin and promote N-cadherin expression, which affects cell adhesion and cell motility [315,316]. Tumor invasion and metastasis are driven by the overexpression of TWIST [317]. TWIST can be activated by various signals, such as HIF-1α (hypoxia-inducible factor-1α), during the progression of EMT. In a hypoxic condition, activation of TWIST by HIF-1α allows the cell to disseminate with metastatic potential [315]. Further, in a mouse model of squamous cell carcinoma, it was found that TWIST can facilitate the cancer cells to undergo EMT and dissemination in the circulation [318].
7.4. Cancer Cells with Stemness Characteristics
Cancer cells have a group of tumor-initiating cells known as cancer stem cells (CSCs), which can differentiate, renew, and generate tumor heterogeneity within the populations of cells [319]. CSCs studied in breast cancer models revealed that EMT correlates with CSC generation by repressing epithelial and mesenchymal properties [320]. It was reported that the expression of CSC and EMT markers were observed in mammary epithelial and carcinoma cells by the direct induction of TGF-β/SMAD signaling, which promotes mammosphere, soft agar colony, and tumor formation [321]. Also, it was shown that autocrine TGF-β/SMAD signaling is crucial within the subpopulation of immortalized breast epithelial cells to maintain its mesenchymal phenotype and tumorigenicity [321]. It is suggested that in this system, BMP signaling may or may not antagonize TGF-β-induced EMT and CSC generation [322]. Thus, enhanced TGF-β/SMAD signaling can promote EMT and CSC properties in cancer cells, further allowing CSC in cancer cell invasion and dissemination.
7.5. Cancer Dissemination and Metastasis
Tumor cells interact with ECM components (collagen, fibronectin, laminin) and cells in the tumor stroma in vivo. In TME, TGF-β/SMAD plays significant roles in cancer cells driven by autonomous tumor cell signaling. Cancer metastasis is promoted in stromal fibroblast and mesenchymal stem cells stimulated by TGF-β/SMAD signaling [8,9,10,323]. Therefore, ECM and stroma are the primary sites for cancer dissemination and metastasis, which are initiated through invasion. Further, intravasation into the blood circulation, extravasation at distant sites, and adaptation at a secondary site within a new microenvironment proceed [324].
Epithelial plasticity plays a vital role in invasion by carcinoma cells and further promotes their dissemination and metastasis [325,326]. Mesenchymal gene expression is often increased in circulating tumor cells, which disseminate through individual as well as collective cell migration [327]. However, many carcinoma cells, such as breast and colorectal cancer cells, can utilize partial/hybrid EMT programs that favor migration as clusters over signal cell migration defined by traditional EMT migration [301]. Interestingly, leading cells in a cluster with mesenchymal characteristics boost invasion, whereas most stalking cells are hybrid/partial EMT or remain epithelial [328]. At the invasive edges, cells with EMT plasticity respond to TGF-β/SMAD signaling to facilitate dissemination through blood vessels or lymphatics [326]. In squamous cell carcinoma, SNAIL induces the expression of claudin-11, a tight junction protein, and advocates cell migration as a tumor cell cluster [278].
Reversible EMT is supported by a model in which transient expression of EMT transcription factors affects the reversion of EMT and metastatic colonization [11,12,13]. TGF-β/SMAD signaling, which stimulates EMT and dissemination, needs to be repressed for metastatic reversal to epithelial morphology [323]. Breast cancer cells can switch from cohesive to single-cell motility mediated by localized and reversible TGF-β/SMAD. To examine SMAD localization in breast cancer progression, Giampieri et al. [323] demonstrated that TGF-β is activated locally and transiently in a motile cell. A transcriptional program involving several factors (SMAD4, NEED9, EGFR, RhoC) can actively modulate cell motility from cohesive to a single cell. Inhibition of signaling can prevent single-cell motility, but collective cell migration was not inhibited. However, TGF-β/SMAD signals can stabilize mesenchymal phenotypes within the tumor cells and are not shown as supportive for leading MET.
A study by Katsuno et al. [329] showed that the prolonged TGF-β/SMAD signals could promote EMT in epithelial cancer cells. Short exposure to TGF-β/SMAD induces the opposite effect and drives MET for colonization in the lung. Moreover, it was shown that the prolonged TGF-β/SMAD signals further modulate mTOR signaling, which contributes to cancer cell stemness and drug resistance. Therefore, it is important to either short exposure of TGF-β/SMAD signals or other signals to balance between TGF-β signaling and switching of EMT to MET. Another study showed that EMT reversed during migration, not in circulation. This study observed MET that was formed after enormous cycles of cell division to reach a secondary site during colonization [11]. Furthermore, it was found that the hybrid EMT has high tumor-initiating and self-renewal capacity in primary cancer cells, especially in breast and prostate cancer [330,331]. Therefore, it is suggested that EMT plasticity and stemness can play a role in the metastatic colonization of tumor cells. Future studies are required to understand the mechanism of EMT plasticity in cancer cell progression, dissemination, and metastasis.
Dormancy is another stage of the tumor cells that allows the cell to be dormant in an arrest phase at primary or secondary sites [332]. A recent review by Fares et al. [331] described that a delayed adaption of disseminating cancer cells (either single invading or cluster cells in circulation) to their secondary at a new microenvironment causes dormancy. TGF-β/SMAD signals induce the tumor suppressor gene DEC2 (chondrocyte 2), which assists cells in entering a quiescence state by inhibiting CDK4 and activating p27.
Tumor dormancy is maintained by the intracellular signals from the two important SMAD-independent pathways, RAS-MEK-ERK/MAK and PI3K-AKT [333].
Some studies [334,335] show that EMT and cancer cell dissemination can occur at the pre-malignant or epithelial stage of tumorigenesis, contrasting with the concept of whether mesenchymal cells aid metastasis in the later stage of tumorigenesis. Before primary tumors are detected, invasive cells with EMT characteristics are detected in models of various solid tumors, such as in pancreas, lung, and breast cancer cells. The evidence supports that such cells can disseminate and then undergo dormancy. Further reactivation of dormancy allows them to form metastatic tumors
8. Conclusions
This review highlights the comprehensive understanding of the functions of SMADs and SMAD-interacting and/or SMAD-signal-modulating proteins in tumorigenesis. The phenotypes of solid tumors are flexible and can switch through epithelial, EMT, partial/hybrid EMT, mesenchymal, and MET states. This phenotypic plasticity creates challenges to developing new therapies for the treatment of cancer. Therefore, we summarize the important enzymes (involving both TGF-β/SMAD and non-SMAD-mediated TGF-β signaling pathways) and the inhibitors targeting the enzymes (Table 1, Table 2, Table 3 and Table 4). The preclinical findings of the inhibitors on solid tumor models have the potential to understand their inhibitory mechanisms and future perspective. The inhibitors can act by different mechanisms that induce cell cycle arrest, apoptosis, modulation of EMT, and dissemination, thereby inhibiting cancer cell proliferation and tumor growth. Although the findings are preliminary, some considerations are required for prospects. Detection of novel therapies is essential to increase efficacy. Further well-designed clinical trials are important to validate safety, effectiveness, and tolerability with the patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shi Y. MassaguéJ. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus Cell 200311368570010.1016/S 0092-8674(03)00432-X 12809600 · doi ↗ · pubmed ↗
- 2MassaguéJ. TGFβ signalling in context Nat. Rev. Mol. Cell Biol.20121361663010.1038/nrm 343422992590 PMC 4027049 · doi ↗ · pubmed ↗
- 3Mukherjee P. Winter S.L. Alexandrow M.G. Cell cycle arrest by transforming growth factor beta 1 near G 1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction Mol. Cell Biol.20103084585610.1128/MCB.01152-0919948884 PMC 2812244 · doi ↗ · pubmed ↗
- 4Thiery J.P. Acloque H. Huang R.Y.J. Nieto M.A. Epithelial-Mesenchymal Transitions in Development and Disease Cell 200913987189010.1016/j.cell.2009.11.00719945376 · doi ↗ · pubmed ↗
- 5Nieto M.A. Huang R.Y.-J. Jackson R.A. Thiery J.P. EMT: 2016 Cell 2016166214510.1016/j.cell.2016.06.02827368099 · doi ↗ · pubmed ↗
- 6Pastushenko I. Brisebarre A. Sifrim A. Fioramonti M. Revenco T. Boumahdi S. Van Keymeulen A. Brown D. Moers V. Lemaire S. Identification of the tumor transition states occurring during EMT Nature 201855646346810.1038/s 41586-018-0040-329670281 · doi ↗ · pubmed ↗
- 7Varga J. Greten F.R. Cell plasticity in epithelial homeostasis and tumorigenesis Nat. Cell Biol.2017191133114110.1038/ncb 361128945230 · doi ↗ · pubmed ↗
- 8MassaguéJ. TG Fbeta in Cancer Cell 200813421523010.1016/j.cell.2008.07.00118662538 PMC 3512574 · doi ↗ · pubmed ↗