Transfer freier Aminogruppen via α‐Aminierung von Carbonylen
Minghao Feng, Anthony J. Fernandes, Ana Sirvent, Eleonora Spinozzi, Saad Shaaban, Nuno Maulide

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
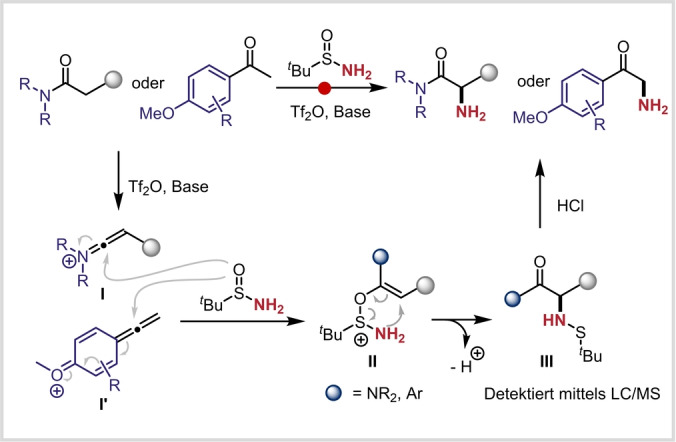 Figure 1
Figure 1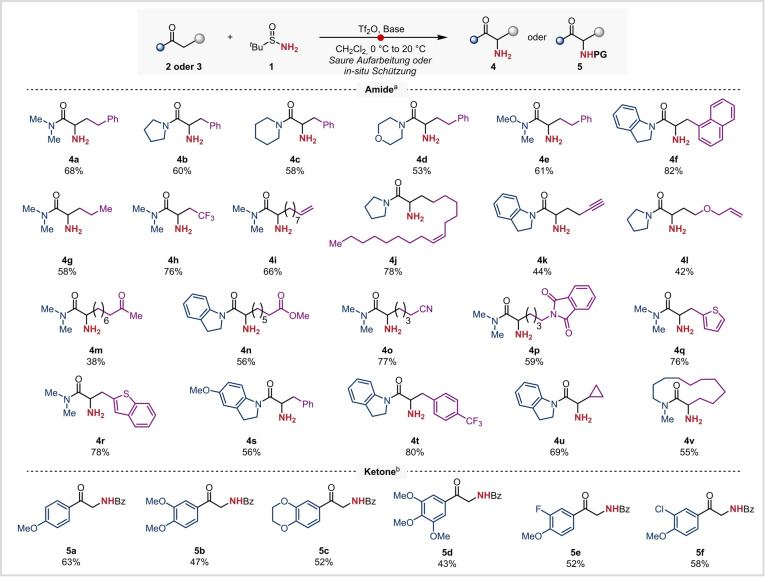 Figure 2
Figure 2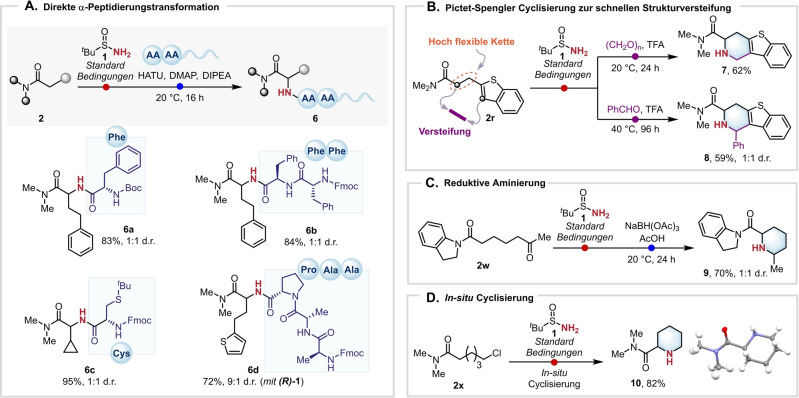 Figure 3
Figure 3 Figure 4
Figure 4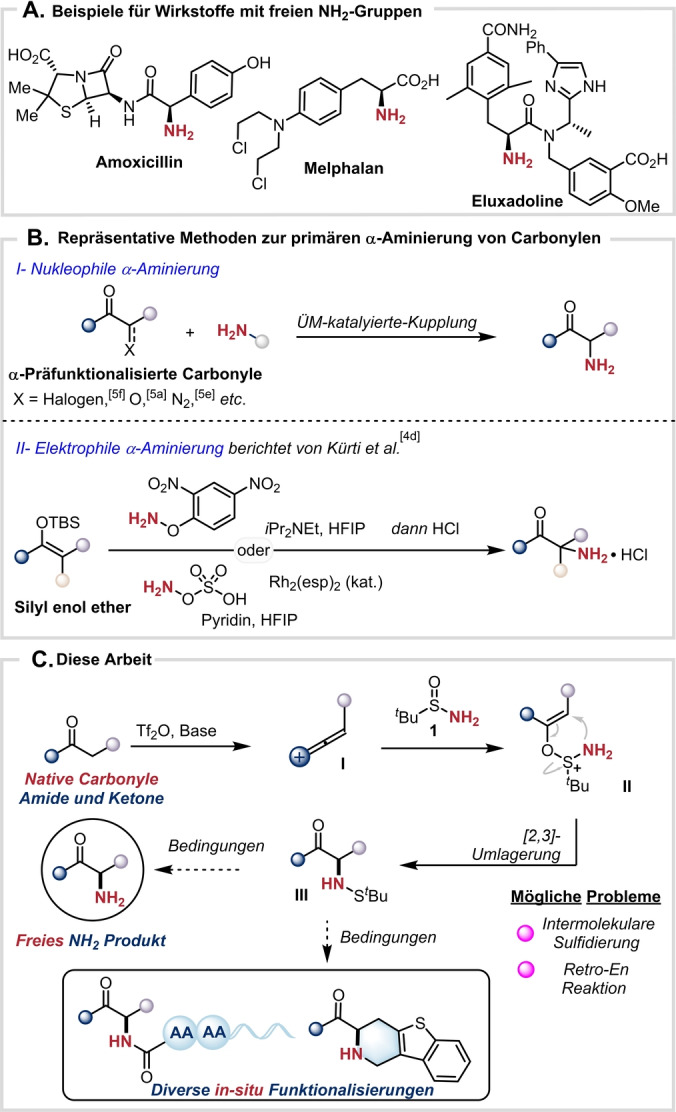 Figure 5
Figure 5- —H2020 European Research Council 10.13039/100010663
- —Austrian Science Fund 10.13039/501100002428
- —Bundesministerium für Digitalisierung und Wirtschaftsstandort 10.13039/501100012416
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Synthetic Organic Chemistry · Asymmetric Hydrogenation and Catalysis · Chemical Synthesis and Analysis
Stickstoff ist das vierthäufigste Element in pharmazeutisch aktiven Wirkstoffen (Schema 1A),[1, 2] wodurch die Entwicklung neuer Verfahren zur Installation von Aminogruppen an einer vorab genau definierten Position in einem Molekül von hoher Bedeutung ist.[3] Die direkte Einführung einer freien, primären Aminogruppe (NH_2_) ist hierbei synthetisch besonders attraktiv, da so die Notwendigkeit zusätzlicher Transformationen zur deren Freisetzung umgangen wird. Bemerkenswerterweise sind in der Fachliteratur jedoch nur wenige Verfahren zur Herstellung primärer α‐Aminocarbonylverbindungen bekannt.[4] Von besonderer Problematik sind hierbei die nicht zusammenpassenden elektronischen Eigenschaften des α‐Kohlenstoffatoms der Carbonylverbindung sowie des Aminostickstoffatoms, wodurch in vielen Fällen eine Präfunktionalisierung des Carbonylpartners in Form einer α‐Halogenierung notwendig wird (Schema 1b).[5] Während entsprechende Aminierungen mit elektrophilen Aminierungsreagenzien eine wichtige Alternative darstellen,[6] führen entsprechende Methoden typischerweise jedoch zunächst zu α‐Hydrazinyl‐ oder α‐Aminoxyprodukten, die mittels weiterer Folgereaktionen erst in die gewünschten primären Aminoderivate überführt werden müssen. 2019 berichteten Kürti und Mitarbeiter über eine einfache Synthese von primären α‐Aminoketonen ausgehend von Silylenolethern und O‐(2,4‐Dinitrophenyl)hydroxylamin bzw. Hydroxylamin‐O‐sulfonsäure (Schema 1B).[4d] Methoden für eine direkte oxidative Aminierung, die die dargestellten Probleme umgeht, sind in der Literatur beschrieben, jedoch ist für diese die Verwendung von nukleophilen (meist cyclischen, sekundären) Aminen als Aminierungsreagenzien von entscheidender Bedeutung.[7] Die direkte Installation einer primären NH_2_‐Gruppe in α‐Position einer Carbonylfunktion ist bisher nicht bekannt und verbleibt ein gleichermaßen herausforderndes wie attraktives Ziel.
Ladungsbeschleunigte Sulfoniumumlagerungen haben sich als nützliche Strategie für die Knüpfung von C−C und C−N Bindungen erwiesen.[8] Kürzlich beschrieb unsere Gruppe erfolgreich eine direkte Kupplung von Amiden und Sulfiniminen, die die Synthese von β‐Aminoamiden via einer [3,3]‐sigmatropen Sulfoniumumlagerung ermöglicht.[8i] Basierend auf diesen Ergebnisse stellten wir die Hypothese auf, dass die Umsetzung von tert‐Butylsulfinamid[9] (1) mit einem geeigneten Vinylkation (I)[10, 11] in der Bildung einer neuen C−N Bindung resultieren und so einen direkten Zugang zur Einführung einer primären Aminogruppe in einem Schritt ermöglichen sollte. Als mögliche Unwägbarkeiten der beschriebenen Strategie wurden etwaige Nebenreaktionen von (II) wie z. B. intermolekulare Sulfidierung[12a] bzw. Retro‐En‐Reaktion[12b] evaluiert. Diese würden schlussendlich zu unerwünschten α‐Sulfanylcarbonylen bzw. den eingesetzten Startmaterialien führen. Abschließend müssten noch geeignete Bedingungen zur direkten Spaltung der N−S Bindung von Sulfenamid III identifiziert werden (Schema 1C).
Wir begannen unsere Studien zur α‐Aminierung von Amiden mit N,N‐Dimethyl‐4‐phenylbutanamid (2 a) als Modellsubstrat. Unter Verwendung von Bedingungen die sich in vorhergehenden Studien als optimal für elektrophile Amidaktivierung (Tf_2_O, 2‐Iodpyridin) erwiesen haben, führte die Addition von tert‐Butylsulfinamid 1 an das aus 2 a resultierende Keteniminiumion nach anschließender saurer Aufarbeitung (HCl in Dioxan) zur Bildung des erwünschten α‐NH_2_ Amids 4 a (68 % Ausbeute).[8i] Zusätzlich konnten 15 % Startmaterial nach Aufarbeitung der Reaktionsmischung zurückgewonnen werden, deren Ursprung dabei vermutlich nicht in einem unvollständigen Umsatz von 2 a liegt, sondern in der als Nebenreaktion ablaufenden Retro‐En‐Reaktion.[12b] Im Gegensatz dazu konnten keinerlei, aus einer etwaigen α‐Sulfidierung resultierenden, Nebenprodukte beobachtet werden.[12a]
Im Folgenden sollte die breite Anwendbarkeit unserer Methode demonstriert werden. Zu diesem Zweck wurden zahlreiche Amide unter den beschriebenen Bedingungen umgesetzt und lieferten die gewünschten primären Amine in guten Ausbeuten (4 b–h, Schema 2). Von besonderer Bedeutung sind hierbei die morpholin‐ (4 d), weinrebamid‐ (4 e), und indolinylbasierten (4 f) Produkte, die für Folgereaktionen zur Synthese anderer Carbonylverbindungen geeignet sind. Die Anwesenheit eines Trifluoromethylsubstituenten nahe dem reaktiven Zentrum hatte keinen negativen Einfluss auf das Reaktionsergebnis (4 h). Die entwickelte Methode toleriert eine Vielzahl an funktionellen Gruppen einschließlich Alkene (4 i, 4 j), Alkine (4 k), Allylether (4 l), Ester (4 n), Nitrile (4 o) und Phthalimide (4 p). Überraschenderweise, wurde für ein Amid mit einer Ketofunktion eine vergleichsweise geringe isolierte Ausbeute (38 %) für das angestrebte aminierte Produkt 4 m erhalten. Als mögliche Ursache hierfür könnten unerwünschte Kondensierungsreaktionen in Frage kommen, die während der sauren Aufarbeitung zur Spaltung der N−S Bindung ablaufen. Verschiedene Heteroaromaten, sowie substituierte Aromaten wurden ebenso toleriert (4 q–t) und ein cyclopropylsubstituiertes Startmaterial 3 u konnte ebenso erfolgreich in das korrespondierende Produkt (4 u) überführt werden. Etwaige Nebenreaktionen, die aus einer Ringöffnung oder einer Zersetzung des Startmaterials resultieren, wurden nicht beobachtet. Bemerkenswerterweise konnte ein dreizehngliedriges Lactam in guter Ausbeute in das entsprechende α‐Aminoderivat (4 v) überführt werden.
Elektronenreiche Arylketone stellen ebenso geeignete Substrate für eine elektrophile Aktivierung dar,[11] was als Grundvoraussetzung für die vorliegende α‐Aminierungsstrategie betrachten werden kann. Hierbei resultierte die saure Aufarbeitung der Reaktionsmischung jedoch in Nebenreaktionen wie z. B. der Kondensation der generierten primären Aminofunktion mit der Ketogruppe des Startmaterials. Um dieses Problem zu umgehen wurde eine in situ Benzoylierung der freien NH_2_‐Gruppe durchgeführt, wodurch die Isolierung der korrespondierenden Reaktionsprodukte in Form der N‐geschützten Derivate ermöglicht wurde (5 a–f).
Die in dieser Arbeit dargestellte Reaktion sticht dadurch heraus, dass sie eine der wenigen Transformationen ist, die die direkte Installation einer freien NH_2_ Gruppe ermöglicht. Ein weiterer Vorteil der entwickelten Methode ist die Möglichkeit weiteren Nutzen aus der vielfältigen Chemie der erhaltenen Amine mittels anschließender in situ Funktionalisierungen zu schlagen. So sind freie NH_2_‐Gruppen beispielsweise für Peptidkupplungen geeignet und Amine 4 d, 4 v und 4 q konnten erfolgreich in situ in einem Ein‐Topf‐Verfahren mit verschiedenen Aminosäuren gekuppelt werden. Es sollte an dieser Stelle erwähnt werden, dass dies einer formalen und bis dato nicht beschriebenen “α‐Peptidierung” von zuvor unfunktionalisierten Amiden in einem einzigen Schritt gleichkommt, wobei die entsprechenden Peptide 6 a–d in exzellenten Ausbeuten erhalten werden (Schema 3A). Interessanterweise wurde Verbindung 6 d in sehr guter Ausbeute als 9 : 1 Mischung von Diastereomeren erhalten, wenn enantiomerenreines Sulfonamid (R)‐1 verwendet wurde.
Darüber hinaus konnte gezeigt werden, dass primäre Amine wie 4 r geeignete Substrate für Pictet–Spengler Cyclisierungen darstellen. In Abhängigkeit des Reaktionspartners (Paraformaldehyd oder Benzaldehyd) wurden hierbei die Piperidine 7 und 8 erhalten (Schema 3B).[13] Diese Transformation erlaubt die Verknüpfung des α‐Kohlenstoffatoms der Amidgruppe in 2 r mit der 3‐position des Heteroaromaten, wobei die eingeführte Aminogruppe quasi als “Stütze” agiert und so die gesamte Struktur versteift – eine potentiell hochgradig nützliche Transformation in der Wirkstoffentwicklung. Ketoamid 2 w wurde nachfolgend mittels einer Eintopf α‐Aminierung/reduktive Aminierungssequenz in Piperidin 9 überführt (Schema 3C).[14] Abschließend konnte ω‐chlorsubstituiertes Amid 2 x mittels einer tandem α‐Aminierung/Cyclisierung in sehr guter Ausbeute in Piperidin 10 überführt werden (Schema 3D).[15]
Aus mechanistischer Sicht gehen wir davon aus, dass zunächst ein nukleophiler Angriff des tert‐Butylsulfinamids auf die Keteniminiumspezies I oder deren Analogon (I′) erfolgt (Schema 4). Die so gebildete Sulfoniumspezies II durchläuft dann eine ladungsbeschleunigte [2,3]‐Umlagerung,[8j, 16] wobei Sulfenamid III gebildet wird.[17] Bemerkenswerterweise konnten sowohl Sulfenamide vom Typ III, als auch die primären α‐aminierten Produkte vor der abschließenden sauren Aufarbeitung mittels LC/MS detektiert werden. Die Aufarbeitung vervollständigt die angestrebte N−S Bindungsspaltung somit lediglich.
Zusammenfassend lässt sich festhalten, dass eine einfache Strategie zur direkten Einführung von primären Aminogruppen α zu Carbonylverbindungen unter milden Bedingungen entwickelt wurde. Die vorliegende Methode erlaubt eine Vielzahl an nachgeschalteten in situ Derivatisierungen, einschließlich Peptidkupplungen und Pictet–Spengler Cyclisierungen. Wir gehen davon aus, dass diese Methode einen allgemeinen Zugang zur Synthese von unnatürlichen α‐Aminosäurederivaten bietet und in Zukunft neuartige Wege für die Herstellung von Peptidomimetica sowie deren medizinischer Erforschung eröffnet.
Interessenkonflikt
Die Autoren erklären, dass keine Interessenkonflikte vorliegen.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1M. Serafini , S. Cargnin , A. Massarotti , G. C. Tron , T. Pirali , A. A. Genazzani , J. Med. Chem. 2021, 64, 4410–4429;33847110 10.1021/acs.jmedchem.1c 00181 PMC 8154580 · doi ↗ · pubmed ↗
- 2E. Vitaku , D. T. Smith , J. T. Njardarson , J. Med. Chem. 2014, 57, 10257–10274.25255204 10.1021/jm 501100 b · doi ↗ · pubmed ↗
- 3P. C. Meltzer , D. Butler , J. R. Deschamps , B. K. Madras , J. Med. Chem. 2006, 49, 1420–1432;16480278 10.1021/jm 050797 a PMC 2602954 · doi ↗ · pubmed ↗
- 4F. I. Carroll , B. E. Blough , P. Abraham , A. C. Mills , J. A. Holleman , S. A. Wolchenhauer , A. M. Decker , A. Landavazo , K. T. Mc Elroy , H. A. Navarro , M. B. Gatch , M. J. Forster , J. Med. Chem. 2009, 52, 6768–6781;19821577 10.1021/jm 901189 z · doi ↗ · pubmed ↗
- 5C. Bouteiller , J. Becerril-Ortega , P. Marchand , O. Nicole , L. Barre , A. Buisson , C. Perrio , Org. Biomol. Chem. 2010, 8, 1111–1120;20165802 10.1039/b 923255 a · doi ↗ · pubmed ↗
- 6M. C. Myers , J.-L. Wang , J. A. Iera , J.-K. Bang , T. Hara , S. Saito , G. P. Zambetti , D. H. Appella , J. Am. Chem. Soc. 2005, 127, 6152–6153.15853303 10.1021/ja 045752 y · doi ↗ · pubmed ↗
- 7“Electrophilic amination of carbanions, enolates, and their surrogates”: E. Ciganek , Organic Reactions, Vol. 72, Wiley, Hoboken, 2009, S. 1–366;
- 8A. M. R. Smith , K. K. Hii , Chem. Rev. 2011, 111, 1637–1656;20954710 10.1021/cr 100197 z · doi ↗ · pubmed ↗