Combined Antitumor Effect of the Serine Protease Urokinase Inhibitor Upamostat and the Sphingosine Kinase 2 Inhibitor Opaganib on Cholangiocarcinoma Patient-Derived Xenografts
Faizal Z. Asumda, Nellie A. Campbell, Mohamed A. Hassan, Reza Fathi, Daniella F. Vasquez Rico, Melanie Kiem, Ethan V. Vang, Yo Han Kim, Xin Luo, Daniel R. O’Brien, Sarah A. Buhrow, Joel M. Reid, Michael J. Moore, Vered Katz Ben-Yair, Mark L. Levitt, Jennifer L. Leiting

TL;DR
This study shows that combining two drugs, upamostat and opaganib, can effectively reduce tumor growth in a mouse model of cholangiocarcinoma.
Contribution
The novel contribution is demonstrating the combined antitumor effect of upamostat and opaganib in a patient-derived xenograft model of cholangiocarcinoma.
Findings
Tumor volumes were significantly reduced in mice treated with upamostat, opaganib, or their combination.
The PAX165 xenograft model showed substantial expression of SPHK2, PRSS1, PRSS2, and PRSS3, which are targeted by the drugs.
Abstract
Cholangiocarcinoma (CCA) accounts for approximately 15% of primary liver cancers. CCA has a poor prognosis and, thus, more effective systemic treatments are needed. We tested opaganib and upamostat, drugs that target sphingosine kinase 2 and multiple serine proteases, primarily trypsins, which are highly expressed in CCA tumors. This study demonstrates the results of inhibiting these novel targets with these drugs individually and in combination in a patient-derived CCA xenograft mouse model. Upamostat is an orally available small-molecule serine protease inhibitor that is a highly potent inhibitor of trypsin 1, trypsin 2, trypsin 3 (PRSS1/2/3), and the urokinase-type plasminogen activator (uPA). These enzymes are expressed in many cancers, especially during tissue remodeling and subsequent tumor cell invasion. Opaganib (ABC294640), a novel, orally available small molecule is a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
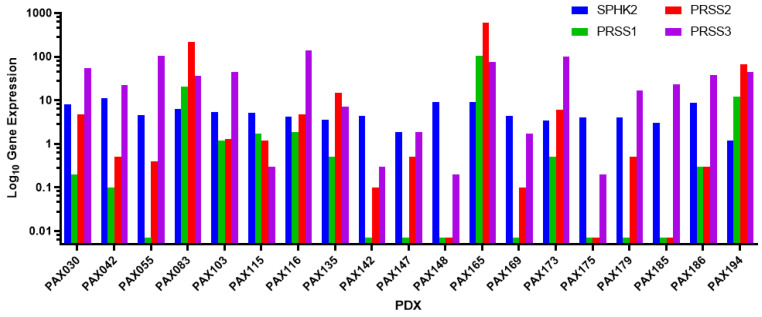 Figure 1
Figure 1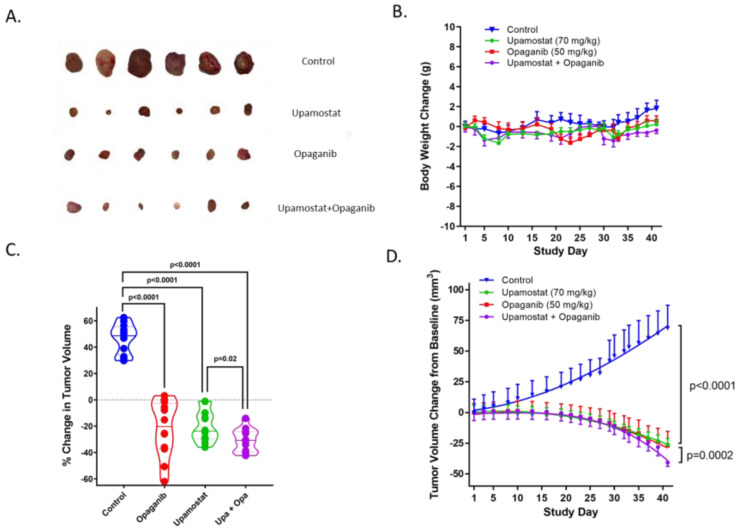 Figure 2
Figure 2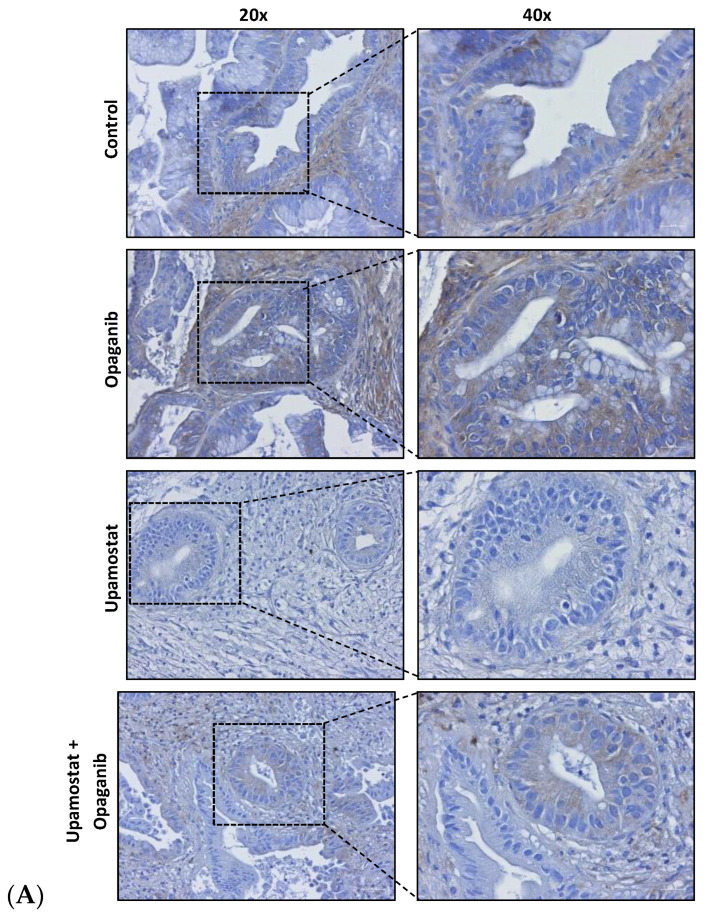 Figure 3
Figure 3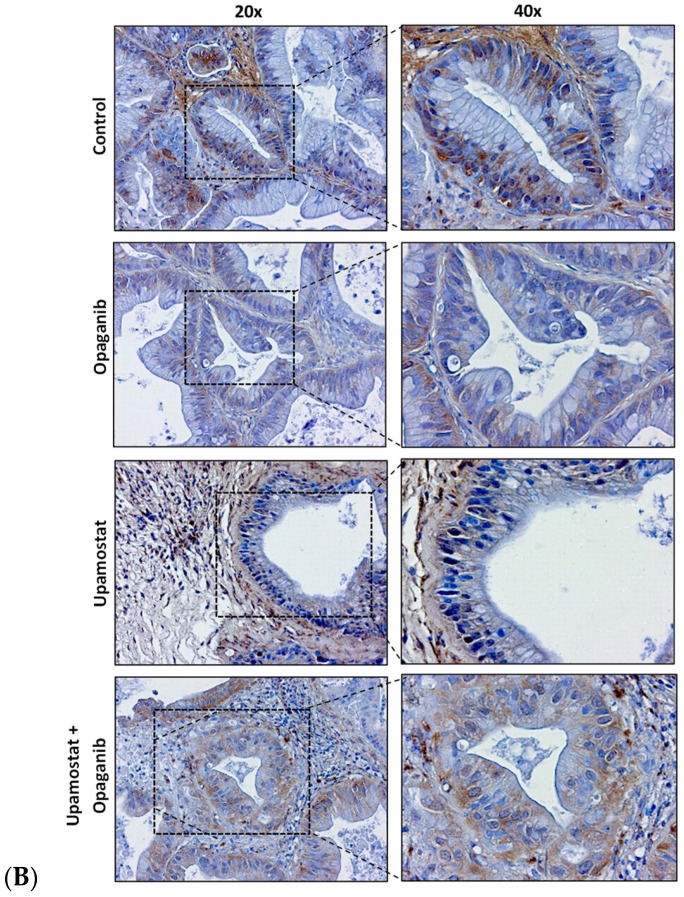 Figure 4
Figure 4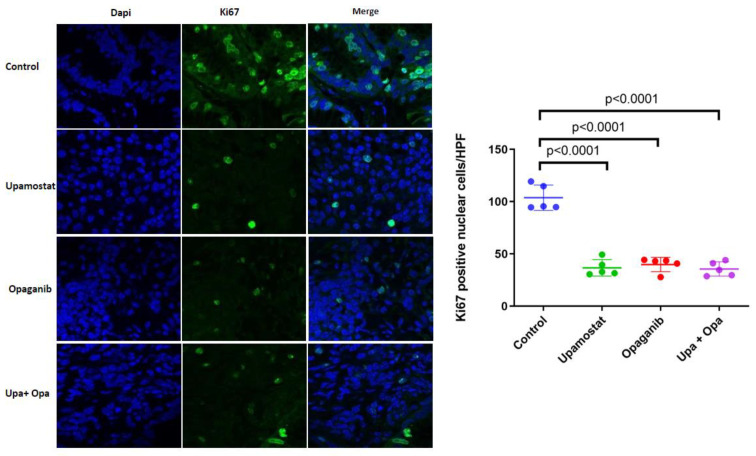 Figure 5
Figure 5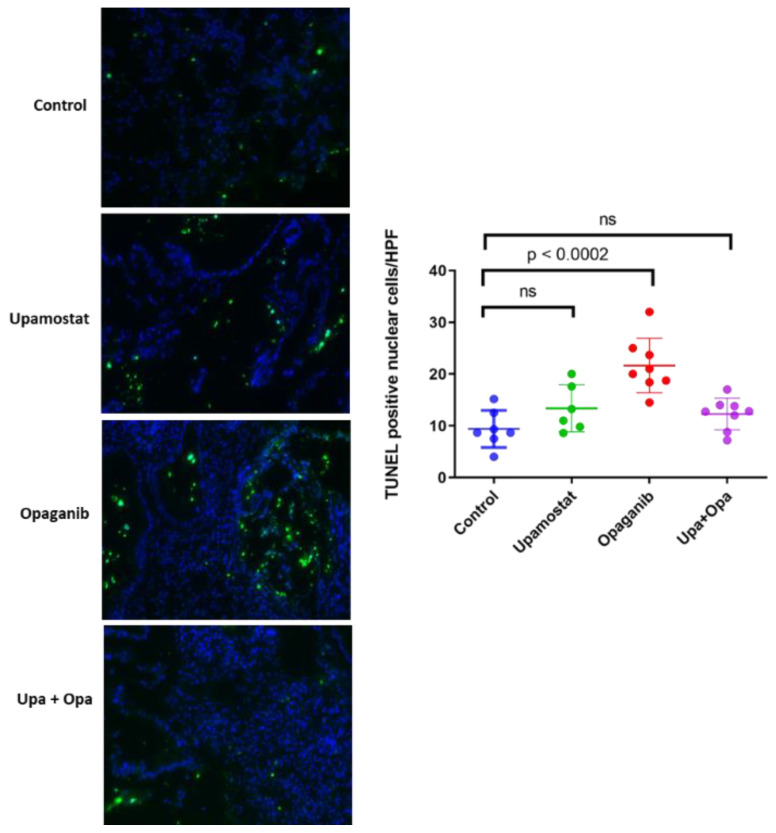 Figure 6
Figure 6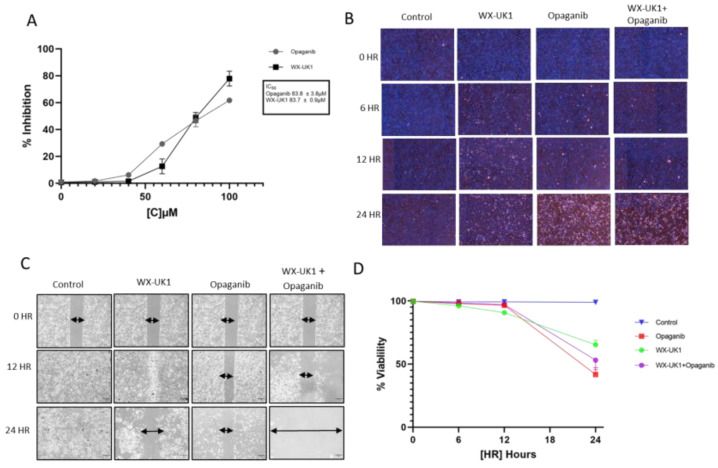 Figure 7
Figure 7- —RedHill Biopharma
- —National Cancer Institute
- —Mayo Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsProtease and Inhibitor Mechanisms · Coagulation, Bradykinin, Polyphosphates, and Angioedema · Peptidase Inhibition and Analysis
1. Introduction
Cholangiocarcinoma (CCA) is an epithelial tumor arising in the intrahepatic (iCCA), perihilar (pCCA), or distal biliary tree (dCCA). It is the second most common type of hepatic malignancy, accounting for 15% of all primary liver cancers and 3% of gastrointestinal cancers [1]. Due to the rising incidence of CCA in industrial countries and limited treatment options, CCA is of increasing public health importance. CCAs are asymptomatic at early stages, highly aggressive, and usually diagnosed at an advanced stage [2,3]. Curative treatment options, including surgical resection, are indicated for early-stage CCA. For unresectable tumors, palliative systemic treatment consisting of gemcitabine, platinum-based chemotherapy, and immune checkpoint inhibitor therapy is the current standard of care [3,4]. In about 10% to 15% of iCCA patients, FGFR2 fusions are identified as key oncogenic drivers [5]. Other clinically significant alterations include IDH1 mutations, mismatch repair deficiency, or NTRK mutations [5]. These patients, who constitute a minority of the CCA patient population, are candidates for targeted chemotherapy or immunotherapy, although neither therapeutic strategy is known to be curative [5]. Chemotherapy resistance as well as resistance to immunotherapy is frequent and existing therapeutic options are of limited effectiveness, thus exploring new targeted treatments is essential.
Upamostat (WX-671, Mesupron) is an orally available, small-molecule serine protease inhibitor that is a highly potent inhibitor of trypsin 1, trypsin 2, and trypsin 3 (PRSS1/2/3), as well as urokinase-type plasminogen activator (uPA) [6]. WX-UK1, the active metabolite of upamostat, is an inhibitor of S1 trypsin-like serine proteases and is used for in vitro studies [7,8]. Proteolytic enzymes like serine proteases are commonly overexpressed in solid tumors, including CCA, and mediate extracellular matrix (ECM) degradation surrounding the tumor. This is crucial in tissue remodeling, promoting cancer invasion and metastasis [9]. As a proteolytic serine enzyme complex, the plasminogen activation system (uPAS) is essential for tissue remodeling, cellular invasiveness, metastasis, ECM degradation, and tumor growth [10,11,12,13,14]. Blocking uPAS and plasmin formation leads to decreased growth and metastatic potential of tumor cells [15,16]. Multiple tumor models have demonstrated the in vivo anti-metastatic and antiproliferative activity of WX-UK1 and its prodrug upamostat [7].
Opaganib (ABC294640, Yeliva), a novel, orally available small molecule, is a first-in-class selective inhibitor of sphingosine kinase 2 (SPHK2). Sphingolipid-metabolizing enzymes control the dynamic balance of the cellular levels of important bioactive lipids, including the apoptotic compound ceramide and the proliferative compound sphingosine 1-phosphate (S1P), which is regulated by the sphingosine kinases SPHK1 and SPHK2, although SPHK2 appears to be more involved in cancer [17,18]. The SPHK2-specific inhibitor opaganib showed high efficacy in several preclinical cancer models and synergistic anticancer activity with chemotherapies or molecularly targeted therapies [19,20]. Anti-tumor activity of opaganib is mediated by multiple underlying mechanisms, including direct inhibition of cell proliferation [19], enhanced apoptosis of CCA cells through the upregulation of pro-apoptotic NOXA [21,22], and the induction of CCA cell autophagy [23].
Both upamostat and opaganib are in the clinical stage of development and are safe for human use [24,25,26]. We aimed to investigate the potential antitumor effect of upamostat and opaganib, individually and in combination, on CCA patient-derived xenografts (PDX) in nude mice using PAX165, a cholangiocarcinoma PDX that expresses substantial levels of SPHK2, PRSS1, PRSS2, and PRSS3.
2. Materials and Methods
2.1. In Vivo Treatment
The care and use of the animals for these studies were reviewed and approved by the Institutional Animal Care and Use Committee of the Mayo Clinic College of Medicine and Science.
2.1.1. Mouse–Human F1/F2 PDX Creation/Tumor Engraftment
According to the protocol approved by the Institutional Review Board (IRB) and the Institutional Animal Care and Use Committee (IACUC) of the Mayo Clinic College of Medicine and Science, tumors were minced into approximately 1 × 2 mm, coated with Matrigel (Corning, Corning, NY, USA) and engrafted into subcutaneous flank pockets of NOD/SCID mice (primary engraftment) [27,28,29]. Mice were treated with a one-time single dose of rituximab (0.1 mg/mL, anti-CD20, Genentech, South San Francisco, CA, USA) via intraperitoneal injection to minimize the development of lymphomas. All generated tumors were evaluated for histomorphology using H&E, comparing PDX tumors with original patient tumor slides by a Mayo Clinic GI pathologist. Time to tumor formation (TTF) was defined as the days from implantation to the first confirmed palpable tumor growth (approximately 3–4 mm). Time to tumor treatment (TTT) was defined as the number of days from implantation to when the tumor reached a volume of approximately 90–180 mm^3^ [27,28,29].
2.1.2. Opaganib and Upamostat Treatment of PDX Tumor-Bearing Mice
The CCA PDX tumor (PAX165), was selected from a panel of 19 CCA PDXs based on its significant expression of the known cellular targets of upamostat, and opaganib (SPHK2, PRSS1, PRSS2, and PRSS3). Mice were randomly divided into 4 groups of 18 mice each and treated by oral gavage with either 70 mg/kg of upamostat (RedHill Biopharma, Tel Aviv, Israel), 50 mg/kg of opaganib (RedHill Biopharma), a combination of both, or vehicle (phosphate buffer). Mice were treated once a day for 6 weeks and sacrificed by CO_2_ inhalation on the first day of the seventh week. Tumor volumes and body weights were measured three times a week using calipers and a balance. Tumor volume was calculated according to the formula (a × b^2^)/2, where a and b are the long and short tumor diameters, respectively. The mice were sacrificed by CO_2_ inhalation at 7 weeks after treatment initiation. Tumors were dissected from the mice and examined by H&E staining and immunohistochemistry. Additional tissues were collected for further pharmacokinetics (PK) analysis including 1.2 mL of whole blood (collected via cardiac puncture using heparinized syringes and processed for plasma), muscle, and liver tissue.
2.1.3. Pharmacokinetic (PK) Analysis
Mouse plasma samples were extracted in methanol. Separation of upamostat and its active metabolite WX-UK1, and opaganib, was accomplished via liquid chromatography using an Acquity UPLC BEH C18 analytical column. Upamostat, WX-UK1, and opaganib were detected via mass spectrometry (see Appendix A for the detailed protocol).
2.1.4. Ki67 Cell Proliferation Analysis
Consecutive cryosections (4 μm) of each tumor were fixed in acetone (10 min, RT) and incubated in H_2_O_2_ (10 min, RT, 0.03%) to block endogenous peroxidase activity. Proliferation rates were measured as the percentage of Ki67-positive cells in tumors. The presence of Ki67-positive tumor cells was analyzed using a confocal fluorescence laser scanning system.
2.1.5. TUNEL Assay for Cell Death Detection
Tissues were fixed in 4% PFA (20 min, RT), washed in 1× PBS (30 min, RT), and incubated in permeabilization solution (2 min, 4 °C). Positive control slides were incubated in 10 uL of DNase + 70 uL buffer (10 min, RT). TUNEL RXN mixture (50 uL enzyme solution, 450 uL label solution) was added to each sample, incubated with DAPI (1:5000 in 1× PBS, 5 min, RT), and analyzed under a fluorescence microscope.
2.1.6. Immunohistochemistry
Formalin-fixed paraffin-embedded (FFPE) tumor tissues from each of the treated mice were placed on glass slides. The tissues were deparaffinized and hydrated through xylene and a graded alcohol series. This was followed by permeabilization with 0.1% Triton X-100 in PBS, antigen retrieval with 10 mM sodium citrate buffer, and quenching of endogenous peroxidase activity with 0.3% H_2_O_2_. 5% BSA was used to block the tissues for 1 h at room temperature. Primary antibodies against SPHK2 (antibody: D2V3G) and the serine proteases [trypsin 1 and 3 (PRSS1, PRSS3), and putative trypsin 6 (PRSS3P2)] (antibody Abcam 200997) were used at concentrations of 1:500 and 1:1000 respectively, incubating overnight. The EnVision+ Dual Link System-HRP kit (Dako) was used as a secondary antibody. DAB Substrate Kit, Peroxidase (Vector Laboratories) was used to precipitate, at the location of the HRP, which was later visualized using light microscopy at 20× and 40× magnification.
2.2. In Vitro Assay and Staining
2.2.1. Cholangiocarcinoma Cell Line
The established intrahepatic cholangiocarcinoma (iCCA) cell line HuCCT1 was cultured in RPMI1640 medium (Gibco) supplemented with 5% fetal bovine serum and 0.1% Primocin (Invitrogen, Waltham, MA, USA) at 37 °C in a 5% CO_2_ incubator.
2.2.2. Cell Migration Assay
The cell migration assay was performed using the IBIDI wound healing assay. Using the 2-well µ-Dish 35 mm from IBIDI, cholangiocarcinoma cell lines were plated for 24 h. The culture inserts in the µ-Dish were removed to create the wounds. The wounds were photographed with a phase-contrast microscope at 0 and 12 h. Cell migration was quantitated by measuring the width of each wound. The experiments were repeated 3 times.
2.2.3. Cell Viability Assay
To determine the effects of different treatments on cell survival, HuCCT1 cells were seeded into 96-well plates in triplicate at densities between 2000 and 5000 cells/well. The cells were treated with varying concentrations of upamostat and opaganib for 0, 6, 12, and 24 h. Cell viability was measured and averaged. Hoechst solution (BioLegend, San Diego, CA, USA) was used to identify cells regardless of viability, whilst Propidium Iodide Solution (BioLegend) was used to stain the dead cells.
2.3. Data and Statistical Analysis
Statistical analysis was performed using GraphPad Prism 9. A p-value of 0.05 was considered statistically significant. Data from the PK study was acquired and analyzed by Waters MassLynx v4.1 software.
3. Results
3.1. PAX165 Expresses Significant Levels of Sphingosine Kinase 2 and Serine Proteases 1, 2 and 3
RNA sequencing data from a panel of 19 CCA PDXs established at the Mayo Clinic was analyzed to identify the most suitable PDX that expressed all the target genes at high levels [30]. The target proteins were sphingosine kinase 2, trypsin 1, trypsin 2, and trypsin 3. The respective genes for these proteins were SPHK2, PRSS1, PRSS2, and PRSS3. PAX165 had the highest expression of these proteins when compared to the other xenografts, making it the best for the study (Figure 1). Of note, sphingosine kinase 2 (SPHK2) and trypsin 3 (PRSS3), which are the main targets of opaganib and upamostat, respectively, were expressed at high levels.
3.2. Verification of Likeness between Original Tumor and Xenografts
Short tandem repeat (STR) analysis was performed to compare the characteristics of the second generation of the mouse xenograft (F2) and the xenografts implanted for the in vivo experiment (Table 1). This was done to confirm that the same tumor that was originally obtained from the patient was being used for this study.
To further validate genetic uniformity between the original tumor (F0) and the first-generation mouse xenograft (F1), H&E staining was performed. H&E staining revealed that the tumors were morphologically similar (Figure S1).
3.3. Opaganib and Upamostat Suppress CCA Tumor Growth in Mice
To assess the anti-tumor effects of opaganib and upamostat, tumor growth was measured in treated versus untreated mice. Upamostat and opaganib significantly suppressed tumor growth compared to the non-treated control group (p < 0.0001, Figure 2C,D). Combining both treatments resulted in greater growth inhibition, with a reduction in tumor volume, than upamostat alone or opaganib alone (p = 0.0002, Figure 2D). Tumor growth suppression mediated by opaganib and upamostat treatment was not accompanied by a decrease in body weight (Figure 2A,B).
3.4. Opaganib and Upamostat Modulate Drug Target Expression in CCA Tumors
Immunohistochemical staining was performed to evaluate the expression of trypsin 1/3 and SPHK2 in CCA tissues. The staining confirmed the specificity of opaganib and upamostat for their primary targets, sphingosine kinase, and serine protease, respectively. The upamostat treatment group showed lower numbers of trypsin 1/3 (PRSS1/3) positive cells (Figure 3A), whereas significantly fewer SPHK2-expressing cells were observed in the opaganib treatment group (Figure 3B).
3.5. Opaganib and Upamostat Inhibit Proliferation and Induce Apoptosis in CCA Tumors
Using PAX165, which expresses high levels of SPHK2 and trypsin 1/2/3 (PRSS1/2/3), immunofluorescence staining was performed to investigate the expression of Ki-67 cell proliferation markers in the treated tissues. In the Ki-67 assay, the control group showed a significant level of Ki-67-positive cells, indicating a high level of cellular proliferation. Conversely, in the treatment groups, Ki-67-positive cells were significantly reduced (p < 0.0001, Figure 4). This suggests that serine protease inhibition and/or sphingosine kinase 2 inhibition of upamostat and opaganib inhibit(s) cellular proliferation, leading to a decrease in the number of actively dividing cells.
To measure the effect of upamostat and opaganib on cell apoptosis, we conducted a TUNEL assay. We found that opaganib induced a significantly higher number of TUNEL-positive cells as compared to the control group, with a p < 0.0002 (Figure 5).
3.6. Pharmacokinetic (PK) Analysis Confirmed That Upamostat Is Metabolized to WX-UK1
A PK study was conducted to evaluate the distribution and metabolism of opaganib and upamostat. This showed an accumulation of upamostat and WX-UK1 in the tumor as well as in liver tissue. WX-UK1 also accumulated in muscle tissue, suggesting that upamostat is metabolized to WX-UK1. This is consistent with WX-UK1 being the active metabolite of upamostat (Table S1).
3.7. Cell Viability and Migration in CCA Cell Lines
The IC50 was assessed to determine the appropriate drug concentrations to test and analyze drug efficacy in cholangiocarcinoma cell lines. Inhibition analysis of opaganib and the upamostat active metabolite, WX-UK1, in the HuCCT1 CCA cell line, showed an IC50 of around 83 µM for both (Figure 6A).
Opaganib, WX-UK1 alone, and combination treatments were associated with decreased cell viability of CCA cell lines as indicated by increased fluorescence of dead cells (Figure 6B,D).
A migration assay was used to determine CCA cell proliferation after periods of 0, 12, and 24 h. HuCCT1 cells showed decreased migration when treated with a combination of WX-UK1 and opaganib (Figure 6C).
4. Discussion
CCA is a highly aggressive cancer with a poor prognosis, a high rate of tumor recurrence, and limited treatment options [31]. One approach to CCA treatment is to target specific highly expressed molecules that are critical for disease pathogenesis. The antitumor effects and safety of upamostat and its active metabolite WX-UK1 have been shown in multiple in vitro, preclinical, and human studies [8,32,33,34,35,36,37,38,39,40,41,42,43,44,45].
For this study of a CCA tumor PDX with the highest expression levels of our drug targets, we observed high levels of trypsin 3 (PRSS3) in the PAX165 tumor (Figure 1). Additionally, trypsin 3 was expressed at high levels in all the evaluated PDX tumors (Figure 3). This finding is particularly important and represents a novel observation in CCA tumors. Trypsin 3 expression is well established in the growth and metastasis of pancreatic tumors [46], but to the best of our knowledge, it has never been reported in CCA tumors. Serine proteases are known to be associated with cancer progression and metastasis [9]. Our group previously showed that WX-UK1, which is the active metabolite of upamostat (Mesupron), is a potent and specific inhibitor of five human proteases (trypsin-3, trypsin-2, trypsin-1, matriptase-1, and trypsin-6). Upamostat is the only known inhibitor of trypsin 3 that has reached clinical development [47].
Our group and others have previously demonstrated the overexpression of sphingosine kinase 2 (SPHK2) in CCA [22,23]. Using the first-in-class Sphk2 specific inhibitor (ABC294640, opaganib, Yeliva), we demonstrated in vitro inhibition of CCA cell proliferation [22,23]. Finally, Phase I studies of ABC294640 in solid tumors demonstrated the most significant response in CCA tumors [24]. Based on these preliminary data, we hypothesized that a combination of upamostat and opaganib may be therapeutically effective for reducing CCA solid tumor growth. In the present work, we selected a patient-derived CCA tumor based on the significant expression of the aforementioned targets and developed a mouse–human xenograft model of CCA. Herein, we show that upamostat and opaganib suppress CCA tumor growth in mice. Compared to the non-treated group, upamostat, and opaganib, each significantly reduced tumor growth. By combining both drugs, an even more significant tumor growth reduction was achieved. Of the two drugs, upamostat had the least effect on weight loss and both drugs in combination did not significantly cause weight reduction in mice. The significant reduction in tumor volume achieved with both drugs in combination likely stems from the divergent antitumor effects of each drug, which produces a more enhanced effect. Additionally, analysis of the Ki-67 and TUNEL assays reveal that both drugs, individually and in combination, significantly reduce proliferation, as well as possible involvement of opaganib in cellular apoptosis, respectively. The combination treatment did not seem to induce a significant increase in apoptotic cells; however, this could be because TUNEL detects only apoptotic cells. It is possible that most of the cells in the combination treatment group might have passed all the apoptotic stages detectable by TUNEL. Trypsin 1/3 and SPHK 2 expression were also evaluated in CCA tissues, and reduced numbers of the relevant target-positive cells were observed in the upamostat and opaganib groups, respectively. The reduction in staining could also be due to the drugs impeding the binding of the respective antibodies to the cells. This strongly suggests that upamostat and opaganib have high affinities for Trypsin 3 and Sphk2. A suggested approach for obtaining additional data for validation of treatment efficacy in the clinical trial setting could involve obtaining pre-and-post-paired treatment CCA tumor samples in patients to assess the key markers described in this paper.
Furthermore, an in vitro study demonstrated that the HuCCT1 CCA cell line is sensitive to opaganib and upamostat. WX-UK1, the active metabolite of upamostat and opaganib, has an inhibitory effect on cell viability and migration when the cells are treated individually or in combination. Treatment with WX-UK1 and opaganib individually both resulted in cell growth regression. The combination of both drugs produced a more potent effect, which suggests that studies to understand the complex mechanistic interactions between the different treatment groups are necessary.
5. Conclusions
In summary, we demonstrate that simultaneously targeting serine protease trypsin 3 and sphingosine kinase (SPHK 2) with upamostat and opaganib results in a more enhanced inhibition of CCA tumor growth in mice. The in vivo tumor growth reduction is associated with the inhibition of tumor cell proliferation and the induction of apoptosis. Importantly, when used in combination, we did not observe significant weight loss in mice. These studies support the clinical trial evaluation of the efficacy of combination treatment with both upamostat and opaganib for CCA.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sung H. Ferlay J. Siegel R.L. Laversanne M. Soerjomataram I. Jemal A. Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries CA Cancer J. Clin.202171209249 Available online: https://acsjournals.onlinelibrary.wiley.com/doi/epdf/10.3322/caac.21660(accessed on 24 April 2023)10.3322/caac.2166033538338 · doi ↗ · pubmed ↗
- 2Forner A. Vidili G. Rengo M. Bujanda L. Ponz-SarviséM. Lamarca A. Clinical Presentation, Diagnosis and Staging of Cholangiocarcinoma Liver Int.2019399810710.1111/liv.1408630831002 · doi ↗ · pubmed ↗
- 3Wang M. Chen Z. Guo P. Wang Y. Chen G. Therapy for Advanced Cholangiocarcinoma: Current Knowledge and Future Potential J. Cell Mol. Med.20212561862810.1111/jcmm.1615133277810 PMC 7812297 · doi ↗ · pubmed ↗
- 4Valle J.W. Lamarca A. Goyal L. Barriuso J. Zhu A.X. REVIEW|New Horizons for Precision Medicine in Biliary Tract Cancers Cancer Discov.2017794396210.1158/2159-8290.CD-17-024528818953 PMC 5586506 · doi ↗ · pubmed ↗
- 5Czauderna C. Kirstein M.M. Tews H.C. Vogel A. Marquardt J.U. Molecular Subtypes and Precision Oncology in Intrahepatic Cholangiocarcinoma J. Clin. Med.202110280310.3390/jcm 1013280334202401 PMC 8269161 · doi ↗ · pubmed ↗
- 6Oldenburg E. Schar C.R. Lange E. Plasse T.F. Abramson D.T. Fathi R. Towler E.M. Levitt M. Jensen J.K. Abstract 4200: New Potential Therapeutic Applications of WX-UK 1, as a Specific and Potent Inhibitor of Human Trypsin-like Proteases Cancer Res.201878420010.1158/1538-7445.AM 2018-4200 · doi ↗
- 7Setyono-Han B. Stürzebecher J. Schmalix W.A. Muehlenweg B. Sieuwerts A.M. Timmermans M. Magdolen V. Schmitt M. Klijn J.G.M. Foekens J.A. Suppression of Rat Breast Cancer Metastasis and Reduction of Primary Tumour Growth by the Small Synthetic Urokinase Inhibitor WX-UK 1Thromb. Haemost.20059377978610.1160/TH 04-11-071215841327 · doi ↗ · pubmed ↗
- 8Ertongur S. Lang S. Mack B. Wosikowski K. Muehlenweg B. Gires O. Inhibition of the Invasion Capacity of Carcinoma Cells by WX-UK 1, a Novel Synthetic Inhibitor of the Urokinase-Type Plasminogen Activator System Int. J. Cancer 200411081582410.1002/ijc.2019215170662 · doi ↗ · pubmed ↗