Characterization of the genomic sequence of a circo-like virus and of three chaphamaparvoviruses detected in mute swan (Cygnus olor)
Sarah François, Sarah C. Hill, Christopher M. Perrins, Oliver G. Pybus

TL;DR
This study identifies and characterizes four new single-stranded DNA viruses found in mute swans from the UK.
Contribution
The discovery of a circo-like virus in birds expands its known host range beyond mammals.
Findings
A circo-like virus was identified in mute swans, previously only found in mammals.
Three chaphamaparvoviruses were detected through viromic analysis of swan fecal samples.
The complete genomic sequences of all four viruses were characterized.
Abstract
We report the complete genomes of four ssDNA viruses: a circular replication-associated protein-encoding single-stranded DNA virus belonging to a clade previously detected only in mammals, and three chaphamaparvoviruses, which were detected by viromic surveillance of mute swan (Cygnus olor) fecal samples from the United Kingdom.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
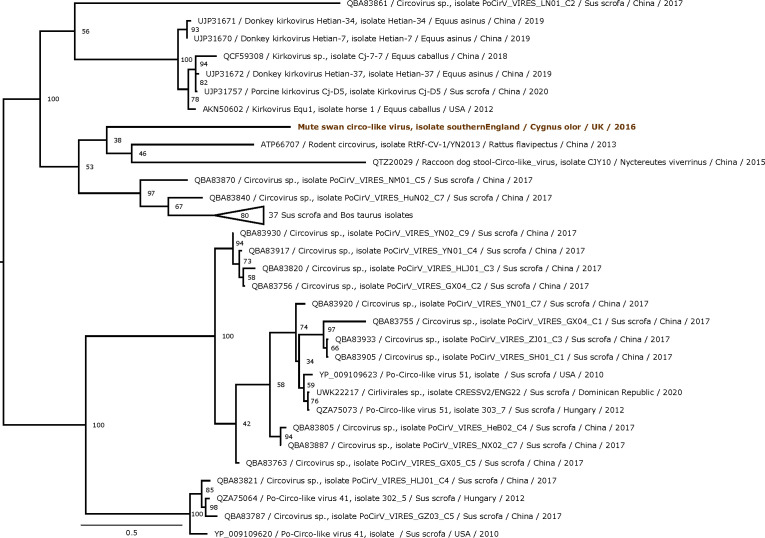 Fig 1
Fig 1| Virus | Genome | Coverage | Putative proteins | Closest identified relatives | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (nt) | %GC | Average | Number | Sample | Name | Size (nt) | Size (AA) | Virus name | Accession | AA pairwise identity | Host name | |
| Chaphamaparvovirus anseriform7 | 4,370 | 41.9 | 50 | 2,019 |
| NS1 | 2,007 | 669 |
| 79.60% |
| |
| NS2 | 594 | 198 | Wood duck chaphamaparvovirus |
| 73.20% |
| ||||||
| NS3 | 438 | 146 | Chestnut teal chaphamaparvovirus 1 |
| 68.30% |
| ||||||
| VP | 1,671 | 557 |
| 61.90% |
| |||||||
| Chaphamaparvovirus anseriform8 | 4,296 | 39.5 | 230 | 9,206 |
| NS1 | 2,052 | 684 |
| 50.50% | Unspecified bird | |
| NS2 | 621 | 207 | Chestnut teal chaphamaparvovirus |
| 50.50% |
| ||||||
| NS3 | 429 | 143 | Chestnut teal chaphamaparvovirus |
| 49.30% |
| ||||||
| VP | 1,626 | 542 |
| 45.90% | Unspecified bird | |||||||
| Chaphamaparvovirus anseriform9 | 4,432 | 39.5 | 206 | 8,343 |
| NS1 | 2,007 | 669 | Mute swan feces-associated chapparvovirus 6 |
| 72.60% |
|
| NS2 | 606 | 202 | Chestnut teal chaphamaparvovirus 1 |
| 62.10% |
| ||||||
| NS3 | 447 | 149 | Chestnut teal chaphamaparvovirus 1 |
| 66.00% |
| ||||||
| VP | 1,689 | 563 | Mute swan feces-associated chapparvovirus 6 |
| 69.90% |
| ||||||
- —UKRI | Biotechnology and Biological Sciences Research Council (BBSRC)
- —UK International Coronavirus Network
- —Wellcome Trust (WT)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAnimal Virus Infections Studies · Viral gastroenteritis research and epidemiology · Virus-based gene therapy research
ANNOUNCEMENT
Our knowledge of viruses infecting wild birds remains scarce, which is detrimental to poultry health and wildlife conservation (1, 2).
We processed seven mute swan (Cygnus olor) non-invasive samples collected in United Kingdom between 2016 and 2019 [for details, see reference (3)]. About 0.5 mL of feces was collected into a tube containing 1 mL of Universal Transport Media. Tubes were shaken and kept on ice in the field, and stored at −80°C.
Viromes were obtained as described in reference (4). We followed manufacturers’ instructions and default parameters except where otherwise noted. Samples were homogenized by a bead beater, filtered through a 0.45 µm filter, digested by DNaseI and RNaseA incubation at 37°C for 1.5 h. DNA and RNA were extracted using a QIAamp Viral RNA Mini Kit. Reverse transcription was performed using a SuperScript IV VILO kit, cDNAs were purified by a QIAquick PCR Purification Kit, and dsDNA was synthesised by Klenow DNA polymerase I. DNA was amplified by random PCR amplification (Q5 Hot Start High-Fidelity kit). PCR products were purified using a NucleoSpin gel and PCR clean-up kit. Libraries were prepared using a NEB NEXT Ultra II DNA Library prep kit, and sequenced on a NovaSeq6000 in 2 × 150 bp paired-end mode.
Adaptors were removed and reads were filtered for quality (q30 and length >45 nt) using cutadapt 2.19 (5), and 153,109,590 paired-end reads were assembled into contigs by MEGAHIT 1.2.9 (6). Taxonomic assignment was achieved using DIAMOND 0.9.30 against the NCBI nr protein database (7). Genome coverage was assessed by mapping using Bowtie2 3.5.1 (local sensitive) (8). Open reading frames (ORFs) were identified using ORF finder (length cutoff >300 nt) on Geneious Prime 2022.0.2 (9), and were annotated by blastp query-centered alignment against RefSeq viral database on 18 September 2023.
We reconstructed the complete circular genome of mute swan circo-like virus (MSCLV; length: 3,663 nt; GC content: 35.6%; average coverage depth: 298; 9,968 mapped reads, SRR26091305) and confirmed it through Sanger sequencing of PCR amplicons using GoTaq HotStar kit with overlapping primers. Chromatograms were checked for disparities. MSCLV genome contained a replication-associated protein gene (918 nt – predicted amino acid sequence: 306 aa), a capsid protein gene (507 nt – 169 aa), and a putative origin of replication marked by a conserved nonamer motif (TACTAAAGTA) flanked by a stem-loop structure (10). The closest relatives of MSCLV are pig-infecting circo-like viruses (11) [Po-Circo-like virus isolate CZH12 (MW881210) with which MSCLV shared 50.8% replication-associated protein pairwise identity; and Po-Circo-like virus HN39-01 (OP302752), 28.4% capsid protein identity] (Fig. 1). Based on the most conserved species demarcation threshold for circular replication-associated protein-encoding single-stranded DNA virus families (i.e., 77% genome-wide identity), MSCLV putatively belongs to a divergent species (12).
Maximum likelihood phylogenetic tree based on the capsid protein of the MSCLV and its 65 closest relatives. Protein sequences used in phylogenetic analyses were obtained by blastx from the NCBI nr database (18 September 2023). Proteins were aligned using MAFFT 7.450 with the L-INS-i algorithm. Maximum likelihood trees were estimated using RAxML 8.2.11, under the LG + G + I + F protein evolution model. Branch support was evaluated using 100 bootstrapped replicates. Trees were mid-point rooted and visualized with MEGAX 10.2.6. Bootstrap values (100 replicates) >30% are indicated at each node. The scale bar corresponds to expected amino acid substitutions per site. The sequence obtained from our sample is in bold red.
We report the complete CDS (coding sequence) of three members of the mammal and bird infecting Chaphamaparvovirus genus (Parvoviridae family, Hamaparvovirinae subfamily, 10.6084 /m9.figshare.24777786). Their closest relatives are bird-associated chaphamaparvoviruses from wild Anatidae samples, with which they shared between 50.5% and 79.6% non-structural protein 1 (NS1) protein identity (Table 1). Based on the Parvoviridae family species demarcation threshold (i.e., 85% NS1 protein identity), these viruses could belong to novel species (13).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Olsen B, Munster VJ, Wallensten A, Waldenström J, Osterhaus ADME, Fouchier RAM. 2006. Global patterns of influenza A virus in wild birds. Science 312:384–388. doi:10.1126/science.112243816627734 · doi ↗ · pubmed ↗
- 2François S, Pybus OG. 2020. Towards an understanding of the avian virome. J Gen Virol 101:785–790. doi:10.1099/jgv.0.00144732519942 PMC 7641393 · doi ↗ · pubmed ↗
- 3Hill SC, François S, Thézé J, Smith AL, Simmonds P, Perrins CM, van der Hoek L, Pybus OG. 2022. Impact of host age on viral and bacterial communities in a waterbird population. ISME J 17:215–226. doi:10.1038/s 41396-022-01334-436319706 PMC 9860062 · doi ↗ · pubmed ↗
- 4François S, Filloux D, Fernandez E, Ogliastro M, Roumagnac P. 2018. Viral metagenomics approaches for high-resolution screening of multiplexed arthropod and plant viral communities. Methods Mol Biol 1746:77–95. doi:10.1007/978-1-4939-7683-6_729492888 · doi ↗ · pubmed ↗
- 5Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EM Bnet j 17:10. doi:10.14806/ej.17.1.200 · doi ↗
- 6Li D, Liu CM, Luo R, Sadakane K, Lam TW. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics 31:1674–1676. doi:10.1093/bioinformatics/btv 03325609793 · doi ↗ · pubmed ↗
- 7Buchfink B, Xie C, Huson DH. 2014. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi:10.1038/nmeth.317625402007 · doi ↗ · pubmed ↗
- 8Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi:10.1038/nmeth.192322388286 PMC 3322381 · doi ↗ · pubmed ↗