Pins Gene Table v2.0: An Online Genome Database of 37 Pythium insidiosum Strains for Gene Content Exploration and Phylogenomic Analysis
Weerayuth Kittichotirat, Preecha Patumcharoenpol, Thidarat Rujirawat, Sithichoke Tangphatsornruang, Chompoonek Yurayart, Theerapong Krajaejun

TL;DR
This paper introduces an online database of 37 Pythium insidiosum strains to study their genes and evolution, helping understand how they cause disease in humans and animals.
Contribution
The novel contribution is the development of Pins Gene Table v2.0, a comprehensive database for comparative genomic analysis of P. insidiosum strains.
Findings
3156 P. insidiosum-specific genes are shared across genotypes and may contribute to disease in humans and animals.
112 genes matched known virulence proteins, potentially involved in vascular occlusion in pythiosis.
Clade-specific genes correlate with host preference, with clade-I strains more animal-specific and clade-II/III strains more human-specific.
Abstract
Unlike most pathogenic oomycetes, Pythium insidiosum infects humans and animals instead of plants. P. insidiosum has three clinically relevant genotypes/clades that cause a severe disease called pythiosis. To develop strategies for infection control, it is necessary to understand the biology and pathogenesis of this pathogen. Investigating the evolutionary mechanisms behind the host-specific adaptation is vital, and comparative genomic analysis can help with this. To facilitate genomic analysis, an online bioinformatics tool called P. insidiosum (Pins) Gene Table v2.0 was developed. This tool includes genomic data from 37 genetically diverse P. insidiosum strains and four related species. The database contains 732,686 genes, grouped into 80,061 unique clusters and further divided into core and variable categories at genus, species, and genotype levels. A high-resolution phylogenomic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
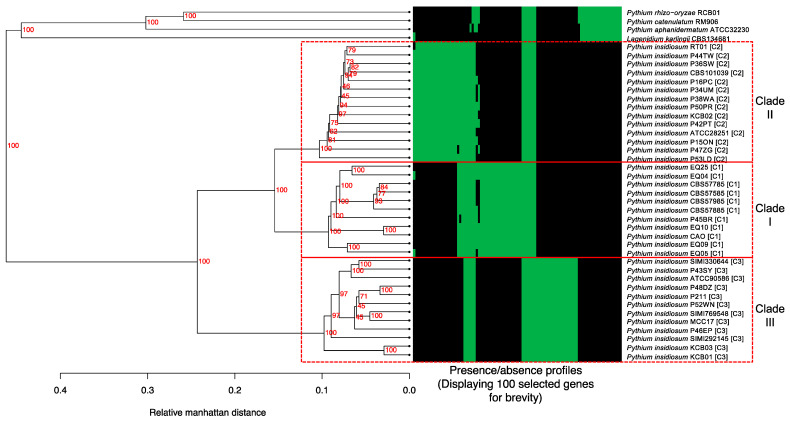 Figure 1
Figure 1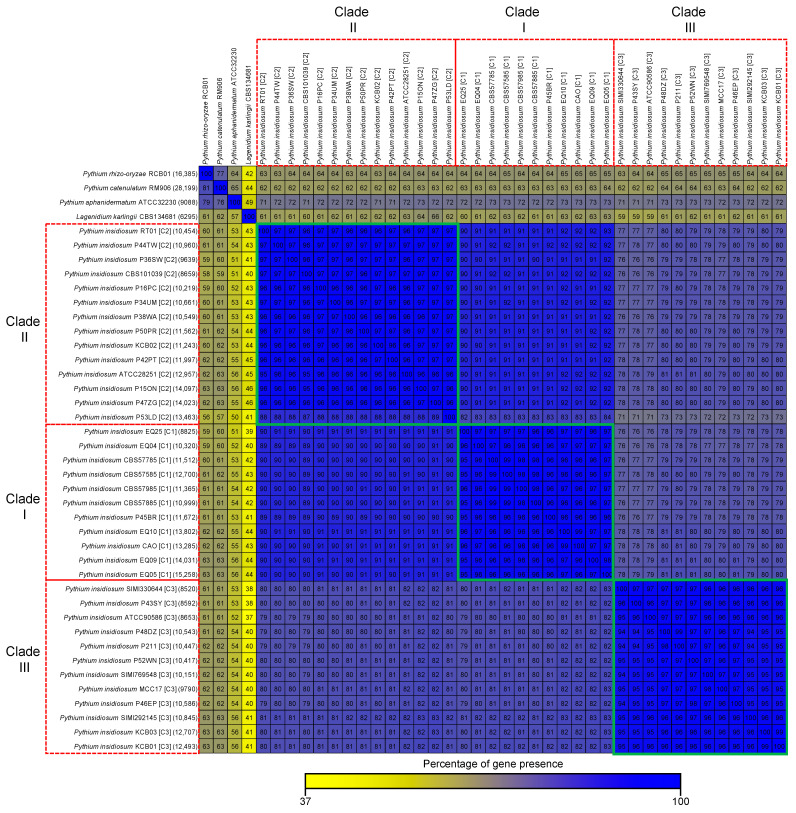 Figure 2
Figure 2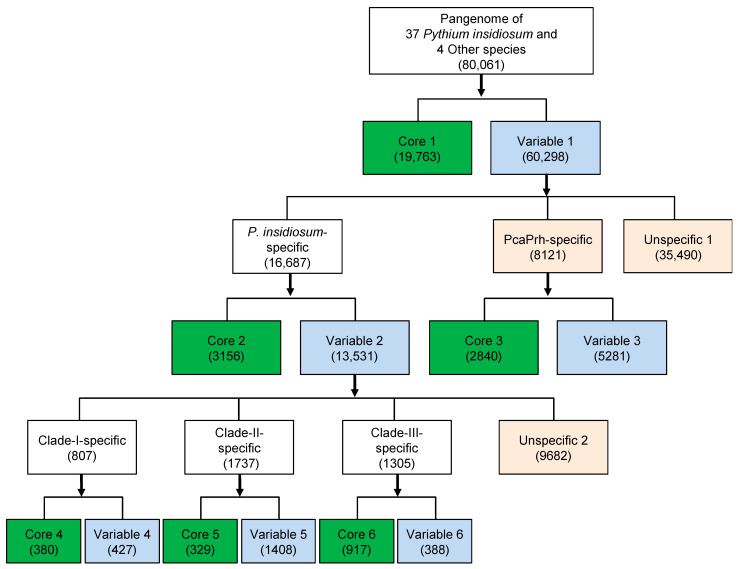 Figure 3
Figure 3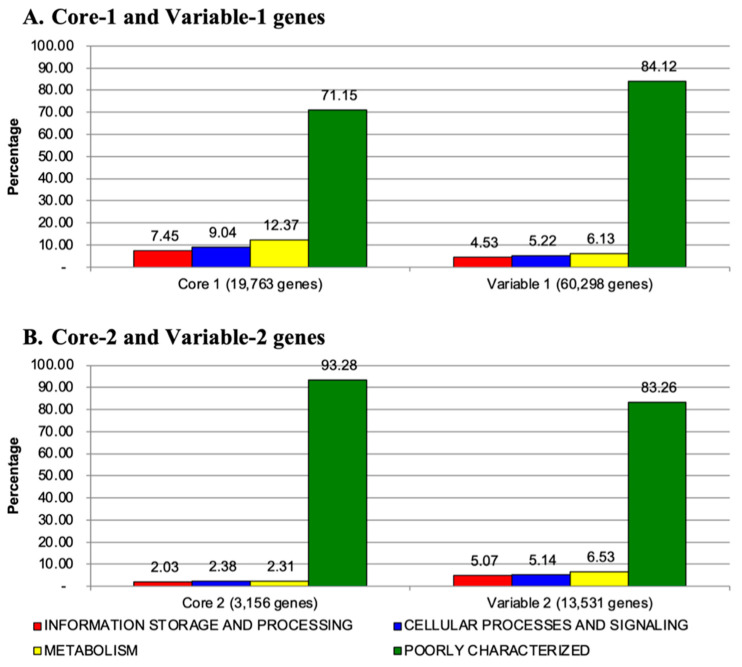 Figure 4
Figure 4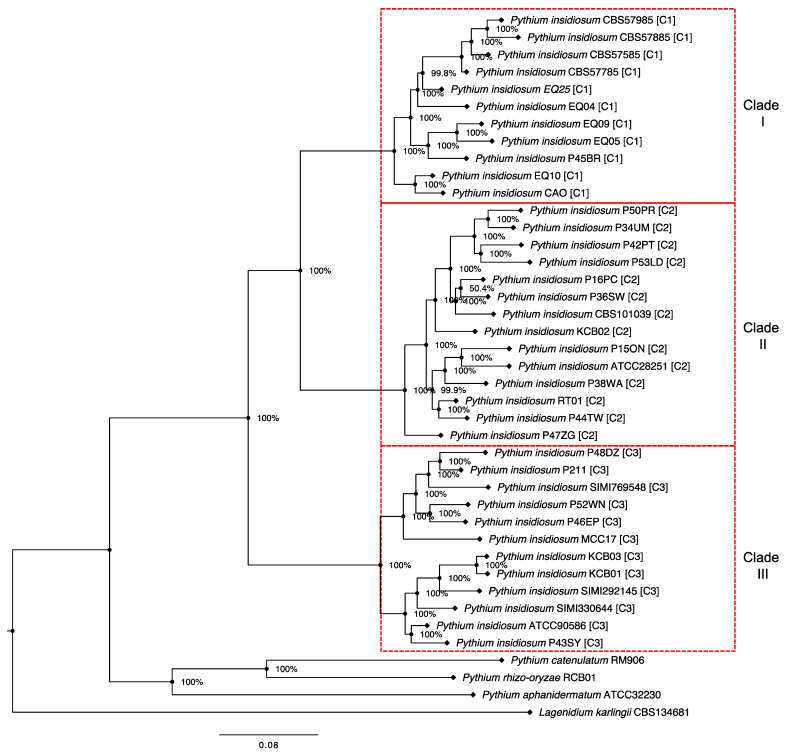 Figure 5
Figure 5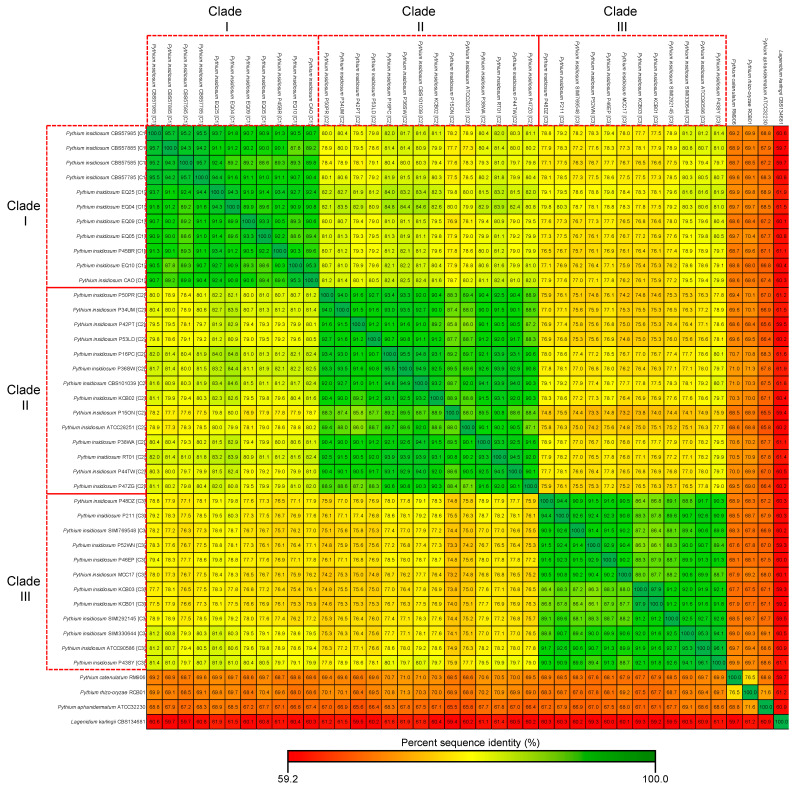 Figure 6
Figure 6- —National Science and Technology Development Agency and Mahidol University, Thailand
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInfectious Diseases and Mycology · Plant Pathogens and Fungal Diseases · Myxozoan Parasites in Aquatic Species
1. Introduction
Pythium insidiosum is an oomycete microorganism that belongs to the Stramenopiles/Sar clades of the Eukaryota Superkingdom (https://www.ncbi.nlm.nih.gov/taxonomy (accessed on 10 November 2023)). This organism is responsible for causing pythiosis, a severe infection that can affect humans and animals worldwide [1,2,3,4]. Unfortunately, diagnosing pythiosis can be challenging due to the lack of awareness about the disease and the absence of a reliable and easily accessible test [5,6,7,8,9,10,11,12,13,14]. Regular antifungal therapy is usually ineffective against this infection, so surgical removal of the affected organ (typically the eye or leg) is often necessary to prevent the disease from progressing and leading to death [5,15,16]. Developing a practical approach for detecting and treating pythiosis is crucial. To achieve this goal, we need to gain a better understanding of the biology and pathogenesis of P. insidiosum. Unfortunately, this organism remains poorly understood, despite the fact that it is unique among oomycetes in that it primarily infects humans and animals rather than plants [17]. Comparative genomic analysis can reveal the evolutionary mechanisms behind the host-specific adaptation in P. insidiosum. Investigating these mechanisms is essential to a better understanding of the pathogen.
In the previous study [18], we introduced the P. insidiosum (Pins) Gene Table v1.0 as an online bioinformatics tool to help researchers in the comparative genomic study of this species. The software included the genome contents of 10 strains of P. insidiosum, categorized into three phylogenetic clades: three strains in clade-I (prevalent in the Americas), five strains in clade-II (prevalent in Asia and Australia), and two strains in clade-III (mostly prevalent in Thailand) [19]. While this tool was helpful, it only represented a small fraction of the genomic diversity of P. insidiosum. To address this limitation, we utilized the high-throughput genome sequencing platform MGISEQ-2000RS (MGI Tech Co., Ltd., Shenzhen, China) to generate additional genome data from 37 P. insidiosum strains. These strains were isolated from various hosts and geographic locations, and their genomic data was incorporated into the Pins Gene Table v2.0. We also included the genome sequences of four other oomycetes species (i.e., Pythium rhizo-oryzae, Pythium catenulatum, Pythium aphanidermatum, and Paralagenidium karlingii) for comparison with P. insidiosum. It is worth noting that P. rhizo-oryzae and P. catenulatum inhabit the same environment as P. insidiosum but do not cause infections in humans or animals [20,21,22]. Identifying species-specific genes could help us understand the pathogenicity of P. insidiosum.
Among the genome-related information on P. insidiosum reported so far [18,23,24], the Pins Gene Table v2.0 is currently the most comprehensive genome database available. It was developed to promote genomic comparison, phylogenomic analyses, and pathogenicity exploration of this pathogenic species. The software’s tabular format makes it easy to use and understand, even for those with limited experience in bioinformatics programming. In this article, we will describe how the Pins Gene Table v2.0 was developed and provide some examples for navigating, comparing, and analyzing the genomes to better understand the biology and pathogenesis of P. insidiosum.
2. Methods and Materials
2.1. Generation and Recruitment of Genome Data from P. insidiosum and Related Species
Thirty-seven strains of P. insidiosum and four related oomycete species (i.e., P. catenulatum strain RM906, P. rhizo-oryzae strain RCB01, Pythium aphanidermatum strain ATCC32230 and Paralagenidium karlingii strain CBS134681) maintained in our laboratory collection (Table 1) were analyzed for species identity and clade classification (i.e., clade I, II, and III) using rDNA sequence analysis [19,25]. The organisms were grown in Sabouraud dextrose broth, shaking at 37 °C for 7–10 days. Hyphal materials were separated from the broth culture, and genomic DNA (gDNA) was extracted using the method of Lohnoo et al. [26]. A gDNA library for each organism was constructed using an MGI Eazy FS Library Prep Kit (MGI Tech Co., Ltd., Shenzhen, China) and the manufacturer’s protocol. Finally, shotgun genome sequences were generated through 150-bp paired-end sequencing by an MGISEQ-2000RS sequencer (MGI Tech Co., Ltd., Shenzhen, China). Obtained reads were cleaned using MegaBOLT V2.4.0 before de novo sequence assembly using SPAdes v3.14.0 [27] and the default setting (the k-mer size was adjusted to 21, 33, 55, 77, and 99).
All recruited draft genomes were analyzed for basic parameters, such as total bases, number of contigs, N50, and GC content. Augustus v3.3.3 [28] was used to predict open reading frames (ORFs) in the genomes. Draft genomes from two out of 37 P. insidiosum strains (i.e., ATCC90586 and P43SY (also known as Pi057C3)) have been reported elsewhere [29]. All newly assembled genome sequences have been assigned an accession number and deposited in the National Center for Biotechnology Information (NCBI) and DNA Data Bank of Japan (DDBJ) databases.
2.2. Clusters of Homologous gene for Genome Content Comparison
We compared the gene content of 37 different P. insidiosum strains and four other oomycete species listed in Table 1 to examine their genomic variability. Our previously published protocol [30] was used to group the genes together based on various sequence similarity parameters at both DNA and protein levels. To group genes into the same cluster, we employed the following criteria: BLAST E-value of 10^−6^, pairwise sequence identity of at least 30%, and pairwise sequence alignment coverage for both query and subject of at least 50%. These loose criteria enable the grouping of distant homologs and reduce the chances of false positives in detecting group- or genome-specific genes present only in a subset of genomes. If a gene is still not found in a genome using these criteria, it is probably absent rather than present but significantly diverged from its corresponding orthologous genes found in other genomes. The final result of the homologous gene cluster analysis is displayed in a table format, referred to as the Pins Gene Table v2.0. Each row in the table represents a gene, while the columns represent the genomes from all 41 organisms used in this study. Each cell in the table provides information on homologous genes or genomic regions found in the corresponding genome.
2.3. Phylogenomic Analysis of P. insidiosum and Other Oomycetes Based on Core Genes
For the core gene-based phylogenomic analysis, we selected 115 genes identified in all 37 P. insidiosum strains and four other oomycete species. We ensured that these genes did not have length variations of more than 45% relative to the longest representative gene in each homologous gene cluster. We aligned the nucleotide sequences of each core gene from all genomes using ClustalW version 2 with default parameters [31]. After aligning, we removed gaps and concatenated the results to produce a single multiple-sequence alignment file. We used FastTree2 to create a maximum likelihood tree and carried out a bootstrap analysis to test the reliability of the tree [32]. Finally, the phylogenetic tree was visualized using FigTree v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 15 November 2023)).
2.4. Hierarchical Clustering of the Gene presence Profile Data
We created gene presence profile data for each of the 41 genomes from our Gene Table result. Specifically for each genome, the value of 1 or 0 was used to represent whether or not each of the gene cluster was found. If a gene cluster was not predicted, but a homologous genomic region was found, we still assigned a value of 1 (present). We then used MeV 4.8.1 with default parameters to perform hierarchical clustering of the genomes based on their gene presence profile data [33]. Finally, we used FigTree software to draw the hierarchical clustering tree result.
2.5. Functional Annotation of the Identified Genes
We used our Gene Table result to obtain genes found in all genomes (Core genes) and exclusively identified in P. insidiosum but not in other oomycetes (P. insidiosum-specific). The protein sequences of these genes were subjected to a BLAST search against the NCBI non-redundant protein database (https://www.ncbi.nlm.nih.gov/ (accessed on 15 November 2023)) and the SwissProt database (https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz (accessed on 15 November 2023)). The annotated proteins were assigned into one of the four Clusters of Orthologous Groups (COG) functional categories: (i) information storage and processing, (ii) cellular processes and signaling, (iii) metabolism, and (iv) poorly characterized or hypothetical proteins [34]. The so-called P. insidiosum-specific genes were further compared against the MvirDB database [35] to identify any putative virulence factors present in P. insidiosum similar to those found in other known pathogens (BLAST E-value cutoff of 10^−6^).
3. Results and Discussion
3.1. Genome Data of P. insidiosum Strains and Related Species
Table 1 summarizes the genome data of 37 P. insidiosum strains and four related oomycete species in this study. P. insidiosum was collected from humans (n = 18), animals (i.e., horses and dogs; n = 14), and the environment (n = 5); they represent the clinically relevant genotypes of the pathogen, including clade-I (primarily prevalent in the Americas; n = 11), clade-II (mostly prevalent in Asia and Australia; n = 14), and clade-III (prevalent in Thailand; n = 12). The clade-I strains of P. insidiosum are strongly associated with pythiosis in animals, while the clade-II and -III strains can infect humans and animals. Among the closely related oomycetes, P. karlingii and P. aphanidermatum can occasionally infect humans or animals [36,37,38]. P. rhizo-oryzae and P. catenulatum [20,21,22] were included in this study for genome comparison because they inhabit the same environment as P. insidiosum. However, there is no report of their infection in humans or animals.
This study employed the MGI short-read platform for genome sequencing of all the organisms. The gDNA samples extracted from all organisms were quantity- and quality-validated to ensure the genome data obtained were comparable. The genome sizes and the number of predicted genes in P. insidiosum appeared diverse, ranging from 42.7 to 75.4 Mb (average: 59.8 Mb) and 12,008 to 22,015 genes (average: 17,006 genes), respectively. The contig sequences were compared with the NCBI nucleotide database, and no significant hit with other species was detected. This suggests that the generated genome sequence data did not possess any contamination. P. insidiosum clade-III strains had a larger average genome size (63.9 Mb), followed by clade-I (60.5 Mb) and clade-II (55.6 Mb) strains. The clade-II strains possessed a markedly lower gene number (16,202 genes on average) compared with clade-I (17,384 genes) and clade-III (17,598 genes) strains. The non-pathogenic Pythium species (i.e., P. rhizo-oryzae and P. catenulatum) had a remarkably larger genome size (97.4 Mb on average) and higher gene number (41,901 genes on average) compared with P. insidiosum (59.8 Mb and 17,006 genes on average). In contrast, two other oomycete species (i.e., P. karlingii and P. aphanidermatum), which occasionally infect humans or animals, had a compatible genome size (average: 58.3 Mb) but a strikingly lower gene number (average: 9825 genes) compared with P. insidiosum.
Despite the variation in genome sizes and gene numbers, the GC contents within P. insidiosum were similar, ranging from 56.9 to 57.7%, with an average of 57.4%. The other recruited oomycete species showed relatively lower GC contents (<54%) in P. karlingii and P. aphanidermatum and relatively higher GC contents (>59%) in P. rhizo-oryzae and P. catenulatum. The GC content of 57–58% could be considered a characteristic of P. insidiosum. The differences in genome sizes and gene contents among P. insidiosum strains and related oomycete species might link to the different phenotypes and virulence.
3.2. Classification of Homologous Genes within the Oomycetes
A total of 732,686 genes can be identified in the genomes of P. insidiosum (37 strains) and other recruited species (i.e., P. catenulatum, P. rhizo-oryzae, P. aphanidermatum, and P. karlingii), as shown in Table 1. These genes can be grouped into 80,061 unique homologous gene clusters using a stringent threshold, such as BLAST E-value of 10^−6^, sequence identity of at least 30%, and sequence alignment coverage of at least 50%. All homologous gene cluster data were stored in the Pins Gene Table v2.0, which can be accessed online at https://202.28.6.19/cgi-bin/gt/viewer?organism=pythium&build=211019 (accessed on 15 January 2024) and can be operated using the explicit instruction of the previous software version [18,39]. The number of genes in any single genome (ranging from 12,008 to 22,015) was markedly smaller than the number of homologous gene clusters (n = 80,061), indicating that no single genome had its genes classified in all clusters. For instance, the genome of P. catenulatum RM906 contained the highest gene number (n = 47,975), which can be allocated into 28,199 clusters (35.2% of all clusters). The genome of P. insidiosum P52WN contained 22,015 genes (the highest among this species), which can be assigned to only 10,417 clusters (13.0% of all clusters). These findings suggest a high variation in the gene contents among these microorganisms, especially within P. insidiosum strains.
3.3. Gene Contents of P. insidiosum and Other Oomycetes
To examine the similarities and differences in gene content among P. insidiosum strains and other oomycete species, we analyzed the presence and absence of 80,061 homologous gene clusters across their genomes. The gene presence/absence profiles were used to perform a hierarchical clustering analysis to obtain a dendrogram, which revealed a clear separation of all P. insidiosum strains from other species (Figure 1). Moreover, P. insidiosum also formed three major clades, corresponding to the rDNA-based genotypes (Table 1), in which clade-I and -II had more similar gene contents than clade-III. A heat map of 100 representative genes showed a firm agreement with the dendrogram-based grouping of the organisms (Figure 1). These results indicate that gene content variation was essential in the evolutionary divergence of P. insidiosum from other oomycete species and within the same species.
To measure the similarity of the gene content of different genomes, we compared all the genes in each genome with all the genes in the other genomes (Figure 2). We calculated the percentages of genes shared between one genome (in each row) and the other genomes (in the columns) to summarize the gene content similarities. P. insidiosum strains from the same clade had a high gene content similarity, from 87% to 99% (average: 96%), as seen in Figure 2 (green squares). However, P. insidiosum strains from different clades had a lower gene content similarity, from 71 to 93% (average: 83%). The gene content similarity between P. insidiosum and other oomycetes was relatively lower, from 37% to 73% (average: 64%), showing that P. insidiosum had a different and unique set of genes that might be related to its ability to cause disease in humans and animals.
3.4. Core and Variable Genes of P. insidiosum
Unlike most pathogenic oomycetes, which affect plants, P. insidiosum infects humans and animals. Data generated here (Table 1; Figure 1 and Figure 2) showed that different strains of P. insidiosum had a high degree of genomic variation, which may reflect their evolutionary history and adaptation to different hosts (i.e., humans, horses, dogs) and geographic environments (i.e., Americas, Asia, and Australia). However, to cause an infection in humans and animals, such different P. insidiosum strains should contain preserved genes with pathogenic functions distinct from genes that are variably present in the other oomycetes. To investigate this, we grouped 80,061 homologous gene clusters from P. insidiosum and other oomycetes into core and variable categories at different levels (i.e., genus, species, and clade/genotype). The genes were divided into Core-1 and Variable-1 (Figure 3). Core-1 genes were considered housekeeping genes (n = 19,763) presented in all organisms. Variable-1 genes were accessory genes (n = 60,298) that vary among genomes. Variable-1 genes were subdivided into three categories: P. insidiosum-specific, PcaPrh-specific, and Unspecific-1. P. insidiosum-specific genes (n = 16,687) were unique to P. insidiosum and not found in other species. PcaPrh-specific genes (n = 8121) were present only within the two non-pathogenic Pythium species (P. catenulatum and P. rhizo-oryzae). Unspecific-1 genes (n = 35,490) were not unique to P. insidiosum, P. catenulatum, and P. rhizo-oryzae and may be shared with other species, including P. karlingii and P. aphanidermatum.
The P. insidiosum-specific genes can be split into Core-2 and Variable-2. Core-2 genes (n = 3156) were common to all P. insidiosum genomes, whereas Variable-2 genes (n = 13,531) were variable among genomes of this species. Similarly, the PcaPrh-specific genes were subgrouped into Core-3 (n = 2840) and Variable-3 (n = 5281) genes. The gene classification in P. insidiosum and four other oomycetes was summarized in Figure 3. Core-2 genes are significant because some may be responsible for the species-specific virulence required for the pathogenesis of pythiosis in humans and animals. Moreover, Core-2 genes may be potential targets for detecting different P. insidiosum strains to improve pythiosis diagnosis and for developing an anti-P. insidiosum therapy (i.e., drug and vaccine designs) to promote pythiosis prevention and treatment.
The COG functional classification [34] was adopted to annotate the Pangenome-level genes (Core-1 and Variable-1; Figure 4A) and P. insidiosum-specific genes (Core-2 and Variable-2; Figure 4B). Most Core-1 (71.2%), Variable-1 (84.1%), Core-2 (93.3%), and Variable-2 (83.3%) genes were assigned as genes encoding poorly characterized or hypothetical proteins. The lesser portion of the gene products in each group was functionally involved in either (i) information storage and processing (2.0–7.5%), (ii) cellular processes and signaling (2.4–9.0%), or (iii) metabolism (2.3–12.4%). Overall, the Core-1 genes can be functionally annotated at the highest frequency (28.9%), partly due to the genes in this group being shared among all different organisms and then considered housekeeping genes, which are usually better studied than other types of genes. In contrast, a tiny portion of the Core-2 genes (6.7%) can be annotated, indicating the functions of these P. insidiosum-specific genes, potentially involved in the pathogenesis of pythiosis, are largely unknown and require exploration.
3.5. Evolutionary Relationship of P. insidiosum and Other Oomycetes
We identified 115 single-copy core genes (a subset of Core-1; Figure 1) in all P. insidiosum strains and four other oomycetes. A BLAST search showed that 77 (67%) of these single-copy core genes had known functions, while 38 (33%) were assigned as poorly characterized or hypothetical proteins (Supplementary Table S1). These core genes might be related to the basic features of the oomycetes but not to the mechanisms of infection in humans or animals. These core genes were adopted to investigate the evolutionary relationship between different P. insidiosum strains and some closely related oomycetes. We concatenated the 115 genes, aligned them, and removed the gaps, resulting in a 77,751-base-long sequence. A maximum likelihood-based phylogenetic tree was then built from this alignment (Figure 5). The tree showed that P. insidiosum strains formed three groups (clade-I, II, and III) distant from other oomycete species. The phylogenetic resolution was high as each P. insidiosum strain had its own branch, suggesting they had diverged from a common ancestor.
As shown in Figure 5, the phylogenetic tree of different P. insidiosum strains reveals that the 115 single-copy core gene sequences were more similar within each clade than between different clades. This observation aligned with the percent sequence identities of these core-gene sequences calculated between pairs of genomes of 37 P. insidiosum strains and four other oomycetes (Figure 6). For instance, the average percent core-gene sequence identity was 91.4% (range: 88.7–95.7%) within P. insidiosum clade-I strains, 91.0% (range: 85.8–95.5%) within clade-II strains, and 90.5% (range: 86.1–97.9%) within clade-III strains. However, when strains from different clades were compared, the average percent of core-gene sequence identity markedly lowered to 78.5% (73.2–84.8%). Furthermore, when P. insidiosum was compared with other oomycetes, the average percent sequence identity dropped to only 66.6%. Within P. insidiosum, clade-I and -II strains are more closely related to each other than to clade-III strains, suggesting a significant divergence of clade-III from the other strains during the evolutionary history of this pathogenic species.
The distribution of P. insidiosum genotypes is strongly linked to geographic location [19,25]. For instance, the Clade-I genotype is exclusively found in American strains, while the isolated Thai strains are always classified as either the Clade-II or -III genotype [19,25]. This study examined the P45BR strain, originally isolated from a dog infected in Thailand. Through hierarchical clustering-based and single-copy core gene-based phylogenomic analyses, we found that this Thai strain is closely related to Clade-I strains from the Americas (Figure 1 and Figure 5). Our finding supports the recent report by Chindampron et al. of another Thai dog infected with a Clade A_th_ (equivalent to Clade-I) strain [40] and confirms that Clade-I strains are already present in Thailand. Regarding the affected hosts, we have observed that Clade-I strains have been found almost exclusively in animals in the Americas, where human cases are rare [1,19,25]. At the same time, humans in Thailand have always been infected with Clade-II and -III strains [1,19,25]. Counting the Clade-I strains isolated from Thai dogs but never found in Thai patients, it is conceivable that Clade-I strains are more specific to animals, while Clade-II and -III strains are more specific to humans. The clade-specific genes of P. insidiosum (i.e., Core 4–6 and Variable 4–6; Figure 3) may be responsible for the host-specific virulence.
3.6. Exploring P. insidiosum Pathogenicity
A comparative genomic approach, using the Pins Gene Table v2.0, was employed to explore the pathogenicity of P. insidiosum. P. catenulatum and P. rhizo-oryzae inhabit the same aquatic environment as P. insidiosum [41] but never appear to cause an infection in humans and animals. Phylogenomic analyses distantly separated P. catenulatum and P. rhizo-oryzae from P. insidiosum (Figure 1 and Figure 4). The genes presented only in P. catenulatum or P. rhizo-oryzae (assigned in the PcaPrh-specific group; n = 8121; Figure 3) were less likely to play a role in an infection in humans and animals. In contrast, the core genes presented in all P. insidiosum strains (Core-2 genes; n = 3156; Figure 3), but not in the non-pathogenic Pythium species, might be centrally involved in the pathogenesis of pythiosis.
To elaborate more on the pathogenicity of P. insidiosum, we compared the 3156 Core-2 genes with the MvirDB database containing about 30,000 records of microbial virulence proteins and toxins, each assigned a virulence factor ID (VFID) [35]. We found that 112 (3.5%) of the Core-2 genes had significant matches with 66 VFIDs exhibiting sequence identity ranges from 19.5% to 50.0%, with an average sequence identity of 33.3% (Supplementary Table S2). This result indicates that they are potential virulence factors shared by genetically diverse P. insidiosum strains. This finding also suggests that P. insidiosum might have adopted an array of different pathogenesis mechanisms from other pathogens via lateral gene transfer events. However, with the low sequence identities relative to known virulent genes from other pathogens, additional investigations are needed before this conclusion can be made. Among the MvirDB database hits, several potential virulence factors were homologous to the reticulocyte binding protein (VFID26643; matched by the gene IDs p-cluster126489, p-cluster200695, p-cluster208509, p-cluster534610, and p-cluster644124), the hemagglutinin/adhesin (VFID7841; matched by p-cluster501203 and p-cluster534263), the venom prothrombin activator (VFID31481; matched by p-cluster496872 and p-cluster707160), and the hemoglobin-binding protein (VFID2575; matched by p-cluster474142). These highlighted virulence factors might conceivably link to a pathological feature of pythiosis where infection occurs in arteries, leading to blood clot formation and arterial occlusion [5,15]. Further experiments are required to validate their biological function involved in the pathogenesis of pythiosis.
4. Conclusions
An online database called Pins Gene Table v2.0 was developed to facilitate the genomic analysis of P. insidiosum. The MGI-based next-generation sequencing technology was used to generate genomic data for 37 genetically diverse strains of P. insidiosum isolated from different hosts and geographic locations and four other oomycetes species. The database contains 732,686 genes, which can be grouped into 80,061 unique homologous gene clusters. These gene clusters were categorized into core and variable categories at different levels (i.e., genus, species, and clade/genotype). A high-resolution phylogenomic relationship among P. insidiosum strains and other oomycetes was projected through hierarchical clustering and core gene analyses. We identified 3156 P. insidiosum-specific genes that were shared among all genotypes/clades and may be responsible for causing disease in humans and animals. After comparing these species-specific genes against the MvirDB database, 112 had significant matches with 66 known virulence proteins, some of which (i.e., reticulocyte-binding protein, hemagglutinin/adhesin, prothrombin activator, and hemoglobin-binding protein) might be involved in blood clot formation and arterial occlusion, a pathological feature of pythiosis. The correlation of genotypes, geographic origins, and affected hosts suggests that clade-I strains are more specific to animals, while clade-II/III strains are more specific to humans. The clade-specific genes might link to host preference. In summary, Pins Gene Table v2.0 is a comprehensive genome database that is easily accessible for users with minimal bioinformatics experience to navigate, compare, and analyze the genomes of this pathogen.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yolanda H. Krajaejun T. Global Distribution and Clinical Features of Pythiosis in Humans and Animals J. Fungi 2022818210.3390/jof 8020182 PMC 887963835205934 · doi ↗ · pubmed ↗
- 2Gaastra W. Lipman L.J. De Cock A.W. Exel T.K. Pegge R.B. Scheurwater J. Vilela R. Mendoza L. Pythium insidiosum: An Overview Vet. Microbiol.201014611610.1016/j.vetmic.2010.07.01920800978 · doi ↗ · pubmed ↗
- 3Mendoza L. Ajello L. Mc Ginnis M.R. Infection Caused by the Oomycetous Pathogen Pythium insidiosum J. Mycol. Med.19966151164
- 4Thianprasit M. Chaiprasert A. Imwidthaya P. Human Pythiosis Curr. Top. Med. Mycol.1996743549504058 · pubmed ↗
- 5Chitasombat M.N. Jongkhajornpong P. Lekhanont K. Krajaejun T. Recent Update in Diagnosis and Treatment of Human Pythiosis Peer J 20208 e 855510.7717/peerj.855532117626 PMC 7036273 · doi ↗ · pubmed ↗
- 6Elshafie N.O. Hanlon J. Malkawi M. Sayedahmed E.E. Guptill L.F. Jones-Hall Y.L. Santos A.P. Nested PCR Detection of Pythium sp. from Formalin-Fixed, Paraffin-Embedded Canine Tissue Sections Vet. Sci.2022944410.3390/vetsci 908044436006359 PMC 9412607 · doi ↗ · pubmed ↗
- 7Behera H.S. Barik M.R. Das S. Sharma S. Simple Polymerase Chain Reaction Assay to Differentiate between Fungal and Pythium insidiosum Keratitis Clin. Exp. Ophthalmol.20214963063210.1111/ceo.1393933931931 · doi ↗ · pubmed ↗
- 8Kulandai L.T. Lakshmipathy D. Sargunam J. Novel Duplex Polymerase Chain Reaction for the Rapid Detection of Pythium insidiosum Directly from Corneal Specimens of Patients with Ocular Pythiosis Cornea 20203977577810.1097/ICO.000000000000228432118672 · doi ↗ · pubmed ↗