Stereoselective synthesis and X-ray structure determination of novel 1,2-dihydroquinolinehydrazonopropanoate derivatives
Hendawy N. Tawfeek, Ahmed M. Tawfeek, Stefan Bräse, Martin Nieger, Essmat M. El-Sheref

TL;DR
Researchers developed a new method to create specific chemical compounds and confirmed their structures using advanced techniques.
Contribution
A novel aza-Michael addition method for synthesizing 1,2-dihydroquinolinehydrazonopropanoate derivatives with confirmed E-configuration.
Findings
The E-configuration of the synthesized compounds was confirmed via X-ray crystallography.
Crystal packing showed intermolecular and hydrogen bonds aligned parallel to the bc plane.
Structures were verified using FTIR, NMR, and mass spectrometry.
Abstract
A novel series of 1,2-dihydroquinolinhydrazonopropanoate have been synthesized via a convenient aza-Michael addition reaction between hydrazinylquinolinones and ethyl propiolate in ethanol under refluxing temperature. The structures for all obtained products were confirmed with FTIR, NMR spectrums, as well as mass spectrometry. In addition, the monoclinic structure for compounds 8a, 8c, and 8d was also confirmed via X-ray crystallography analyses. The E-configuration for the obtained products was confirmed form the X-ray analysis. On the other hand, the crystal packing shows that the intermolecular and hydrogen bonds between atoms are parallel to the bc plan.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
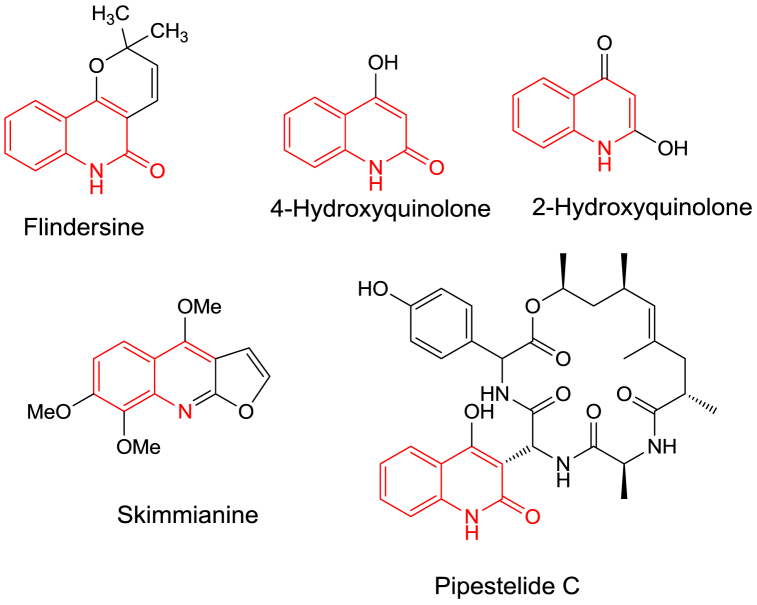 Figure 1
Figure 1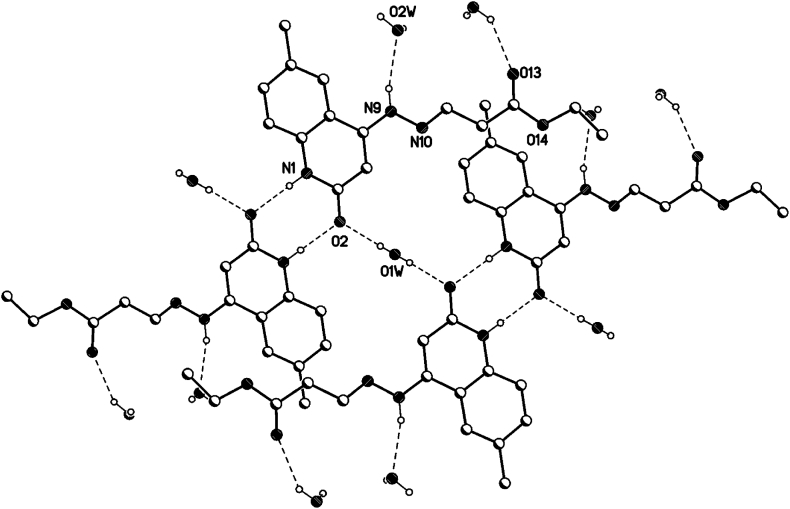 Figure 2
Figure 2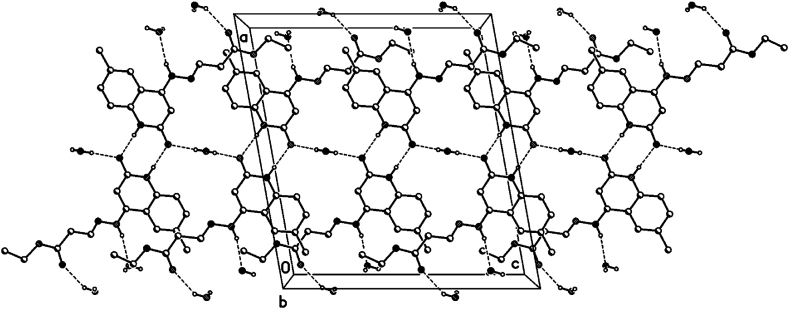 Figure 3
Figure 3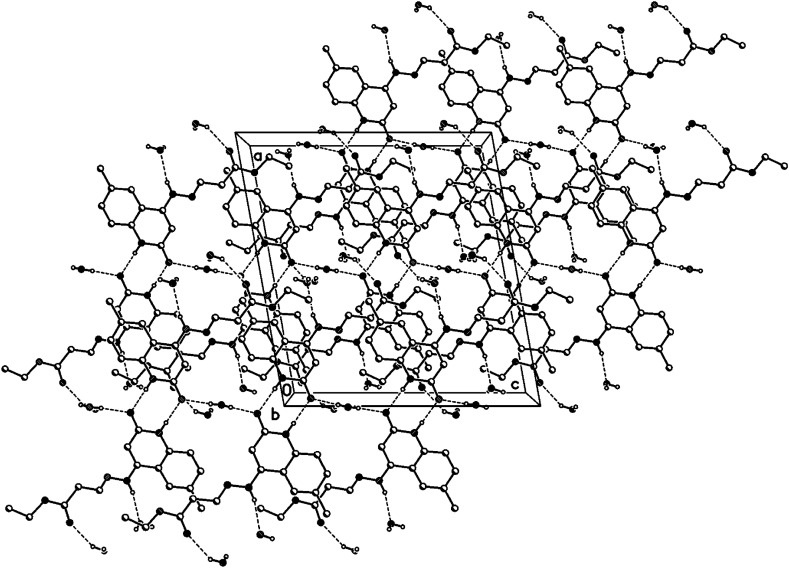 Figure 4
Figure 4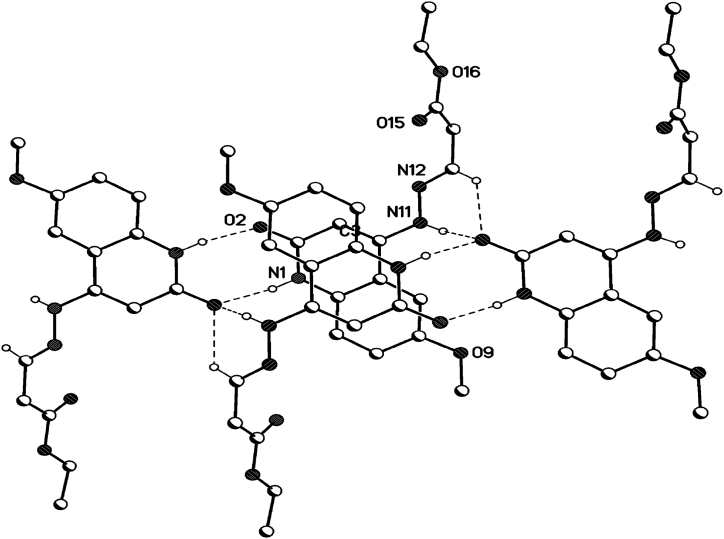 Figure 5
Figure 5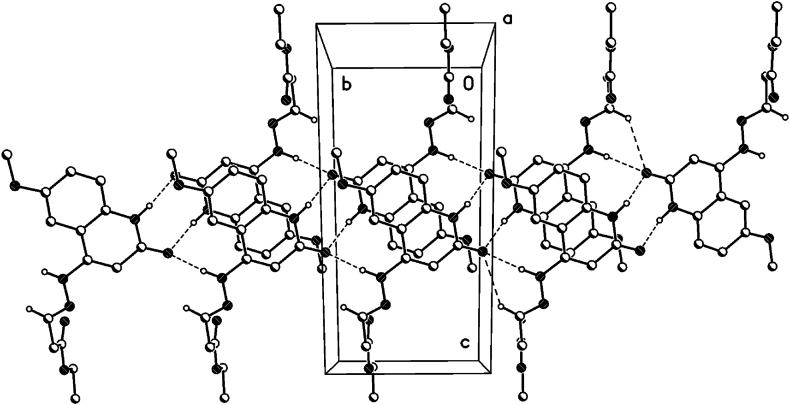 Figure 6
Figure 6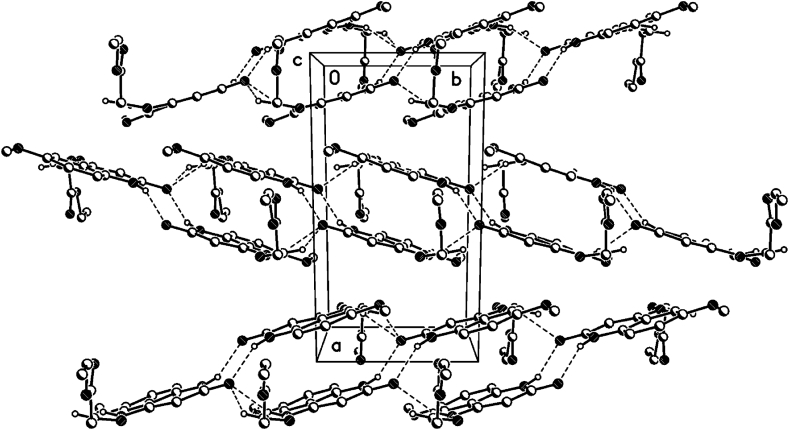 Figure 7
Figure 7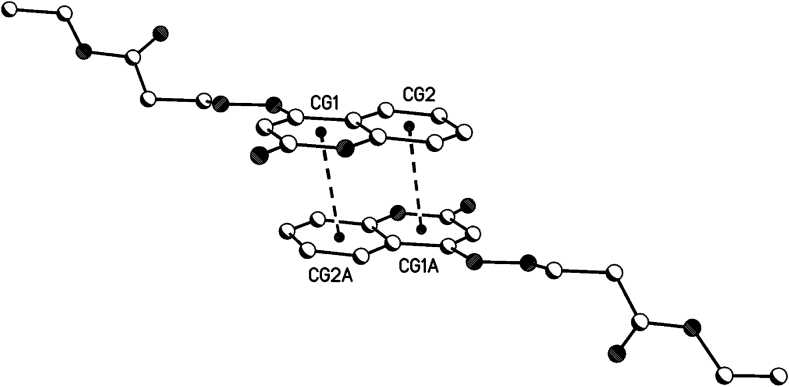 Figure 8
Figure 8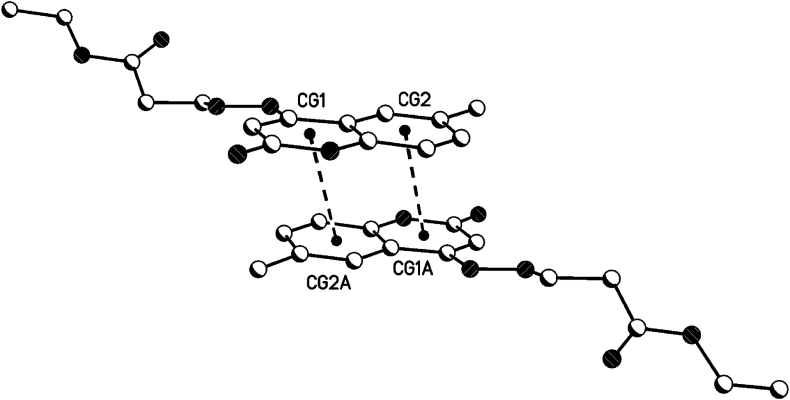 Figure 9
Figure 9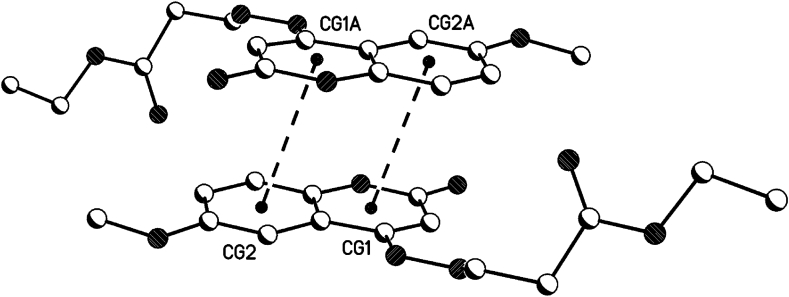 Figure 10
Figure 10 Figure 11
Figure 11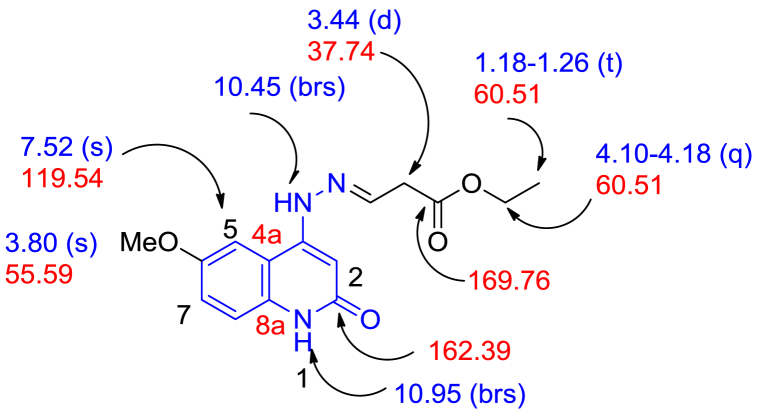 Figure 12
Figure 12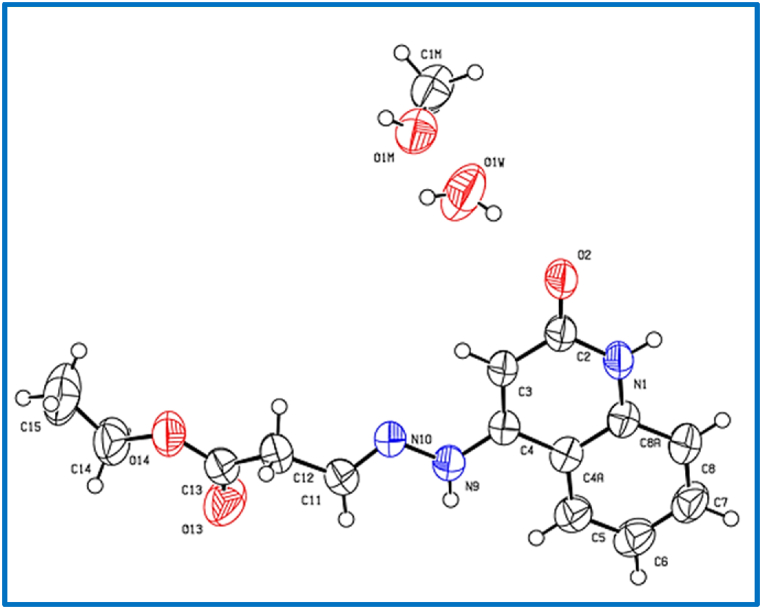 Figure 13
Figure 13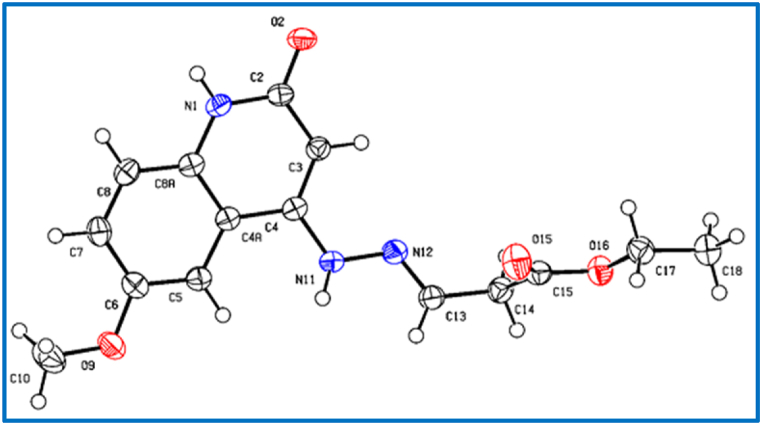 Figure 14
Figure 14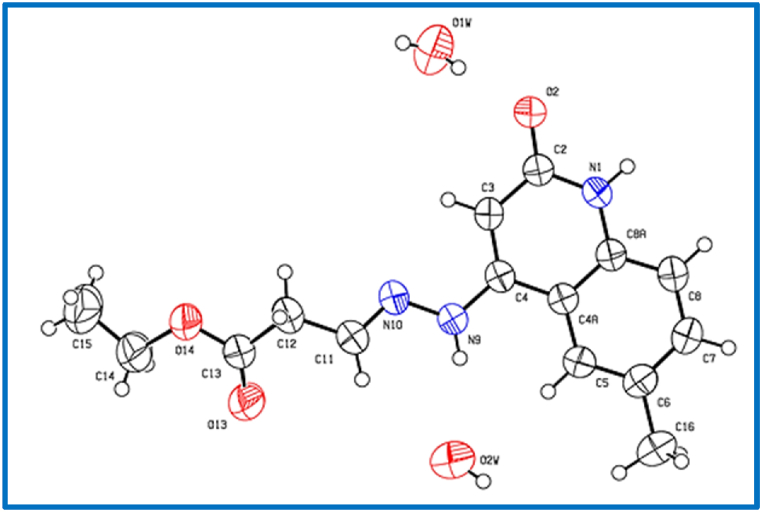 Figure 15
Figure 15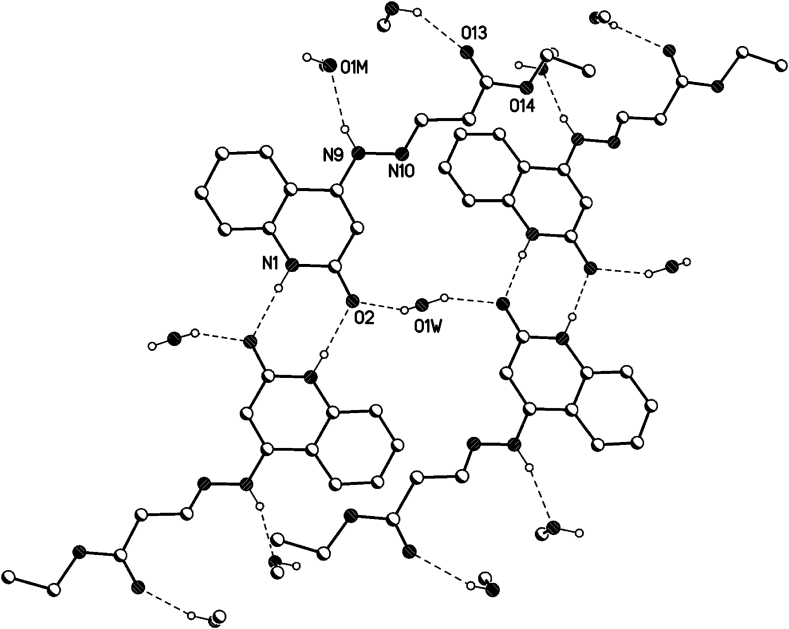 Figure 16
Figure 16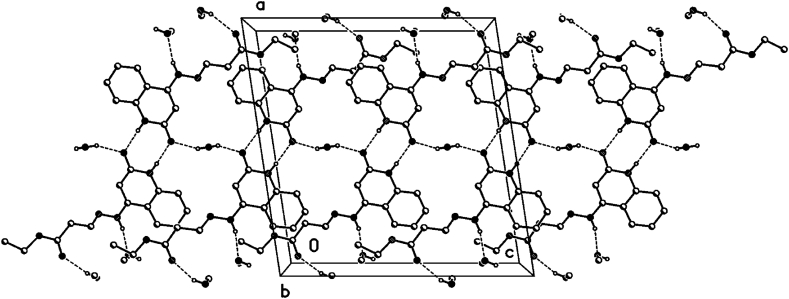 Figure 17
Figure 17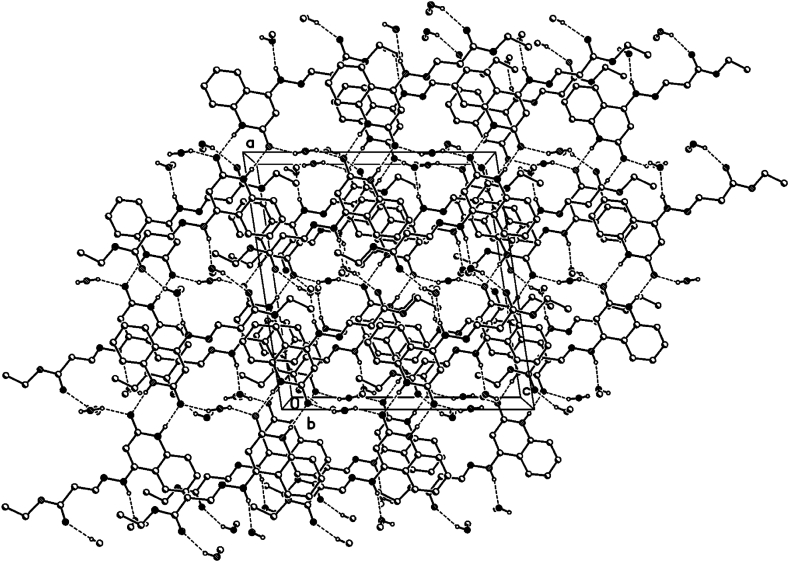 Figure 18
Figure 18 Figure 19
Figure 19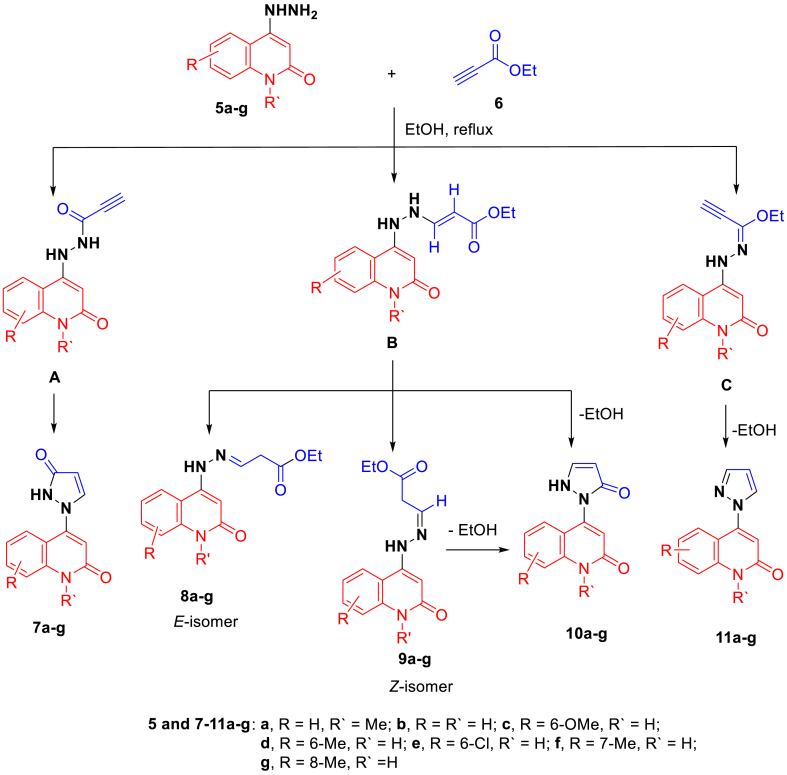 Figure 20
Figure 20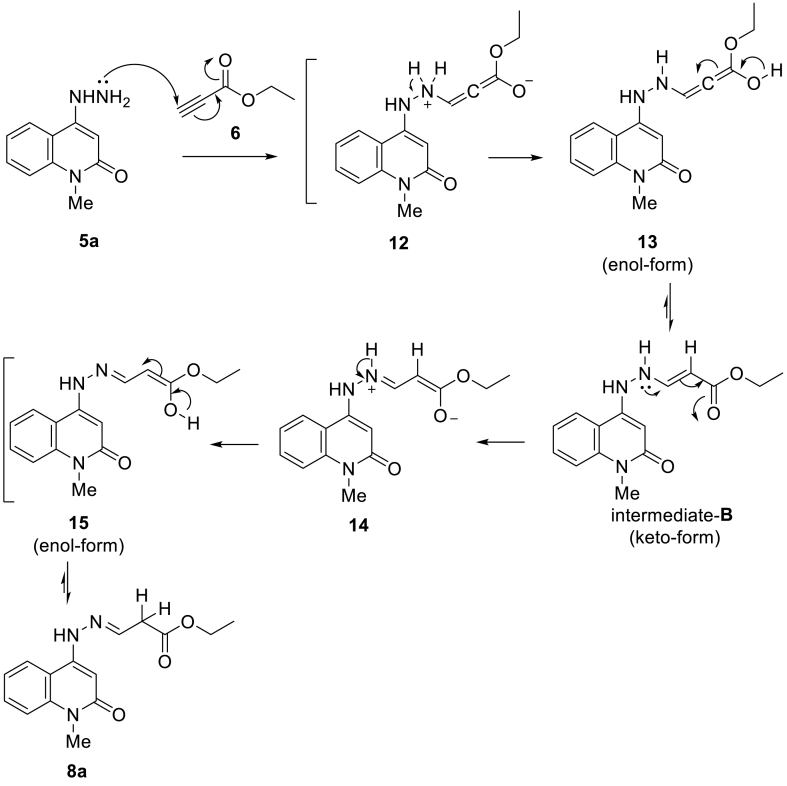 Figure 21
Figure 21Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and biological activity · Synthesis of Organic Compounds · Synthesis and Reactivity of Heterocycles
Introduction
1
As privileged structural subunits, quinolones have been witnessed in many biologically active compounds and natural products [[1], [2], [3]]. Moreover, 4-hydroxyquinolone, as well as Skimmianine, proven as an effective anti-cancer agent [4], and Flindersine, playing an antibacterial and antifungal activity [5], were examples of naturally extracting quinolone derivatives (Fig. 1). Quinoline derivatives possess peptide bonds like Pipestelide C that was isolated from a marine fungus (Fig. 1) [6].Fig. 1. Naturally occurring quinolone-related structures.Fig. 1
On the other hand, there were synthetic drugs based on the main quinolinone unit on their structures, such as L-701,324, which represented a selective antagonist at the glycine site of the NMDA receptor and counteracts haloperidol-induced muscle rigidity in rats [7], the other was benzothiadiazinyl quinolinedione described to be an inhibitor of the RNA-dependent RNA polymerase enzyme transcribed by the Hepatitis C virus (Fig. 2) [8].Fig. 2. Vital drugs, including quinolinone ring.Fig. 2
Douche et al. have reported the synthesis of imidazolylquinoline derivatives, which have been investigated as potential antiviral SARS-CoV-2 candidate [9].
The hydrazide-hydrazone moiety –NHN
<svg xmlns="http://www.w3.org/2000/svg" version="1.0" width="20.666667pt" height="16.000000pt" viewBox="0 0 20.666667 16.000000" preserveAspectRatio="xMidYMid meet"><metadata> Created by potrace 1.16, written by Peter Selinger 2001-2019 </metadata><g transform="translate(1.000000,15.000000) scale(0.019444,-0.019444)" fill="currentColor" stroke="none"><path d="M0 440 l0 -40 480 0 480 0 0 40 0 40 -480 0 -480 0 0 -40z M0 280 l0 -40 480 0 480 0 0 40 0 40 -480 0 -480 0 0 -40z"/></g></svg>
CH has been an important structure group for various biological activities, including antibacterial, antifungal, analgesic, anti-inflammatory, antidepressant, and anti-cancer activities [[10], [11], [12]]. Using the hydrazide-hydrazone moiety in conjunction with a quinoline system, supporting their activities as antimicrobial [13], antimycobacterial [14], anti-tubercular [15,16], anticonvulsant [17], and cytotoxic activity [18,19]. These biological activities are attributed to the numerous interesting and important properties of the hydrazide-hydrazone moiety, for example, their relatively higher metabolic stability towards proteases than amides and their tunable, labile nature in acidic pH [20].
Thus, the developing efficient strategies for constructing such frameworks has attracted the growing interest of synthetic chemists. The last decade has shown significant progress in quinolone derivatives synthesis and biological evaluation, owing to their wide application in medicinal chemistry.
Elbastawesy et al. have reported the synthesis of 6-substituted quinoline-2-one thiosemicarbazides, and their activities were evaluated invitro against the urease-producing R. mucilaginosa and Proteus mirabilis bacteria as fungal and bacterial [21].
Valencia et al. have reported the synthesis of quinolone-based thiosemicarbazones and investigated their invitro activities against Mycobacterium tuberculosis (M. tuberculosis) [22]. However, the reaction of hydrazinyl quinolone with tetracyanoethylene (TCNE) was carried out by Elbastawesy et al. to afford pyrazolyl quinolone derivatives and their biological activities as potential apoptotic antiproliferative agents targeting the EGFR inhibitory pathway have been investigated [23].
Recently, Al-Wahaibi et al. have constructed azaspiroquinolone derivatives via a one-pot reaction between hydrazinylquinolones, cyclic ketones, and thioglycolic acid, alongside studying the biological properties of the synthesized compounds as antiproliferative against four cancer cells [24].
Here, we have reported the reaction of hydrazonylquinolones as donating molecules with a π-deficient molecule ethyl propynolate, which possess a C
<svg xmlns="http://www.w3.org/2000/svg" version="1.0" width="20.666667pt" height="16.000000pt" viewBox="0 0 20.666667 16.000000" preserveAspectRatio="xMidYMid meet"><metadata> Created by potrace 1.16, written by Peter Selinger 2001-2019 </metadata><g transform="translate(1.000000,15.000000) scale(0.019444,-0.019444)" fill="currentColor" stroke="none"><path d="M0 520 l0 -40 480 0 480 0 0 40 0 40 -480 0 -480 0 0 -40z M0 360 l0 -40 480 0 480 0 0 40 0 40 -480 0 -480 0 0 -40z M0 200 l0 -40 480 0 480 0 0 40 0 40 -480 0 -480 0 0 -40z"/></g></svg>
C bond attached to an electron-withdrawing group. Our expected goal is to synthesize a pyrazoloquinolinone ring via nucleophilic attack of the hydrazinyl moiety into the triple bond followed by cyclization. Unfortunately, the analyses and the X-ray measurements clarified that 1,2-dihydroquinolinehydrazonopropanoate derivatives were formed without internal cyclization occurring. The reaction proceedes via nucleophilic addition of a nucleophile (QNHNH_2,_ donor) on the alkyne (HCCCOOCH_2_CH_3,_ Michael acceptor) in agree with aza-Michael addition reaction [25,26].
Experiments
2
Melting points were measured by a Stuart melting point apparatus and were uncorrected. The IR spectra were recorded using an FTIR Alpha 24 spectrophotometer as KBr pellets. The ^1^H and ^13^C NMR spectra were recorded in DMSO–d6 as a solvent on Varian Gemini NMR spectrometer at 400 and 100 MHz, respectively, using TMS as an internal standard. Chemical shifts were reported as δ values (ppm), while couplings constants (J) are measured in hertz (Hz). Some NMR spectra were measured in DMSO-d6 on a Bruker spectrometer (300 MHz for ^1^H and 75 MHz for ^13^C) at MICROANALYICAL CENTER, Cairo University. Mass spectra were recorded by a MAT 95 +FAB mass spectrometer in EI (70 eV) model. The reactions were closely monitored with TLC (thin-layer chromatography) on Merck alumina-backed TLC plates Pf_254_ using UV light.
Chemistry
2.1
The starting materials were prepared according to the reported articles [[27], [28], [29]], with slight modification in the preparation of hydrazinylquinolones 5a-g 4-chloroquinolones 4a-g and were refluxed with hydrazine hydrate for 3 h. Hydrzine hydrate acted as reagent and solvent. The formed precipitate was filtered, flushed several times with water, and used for the second step without further purifications.
Synthesis of quinolone hydrazonopropanoates
2.2
In a round bottomed flask (50 ml), substituted hydrazinyl quinolone-2-one (1 mmol) suspended in ethanol (10 ml) ethyl propiolate (1.2 mmol) was added, the mixture was refluxed for about 6 h. The reaction was monitored with TLC, until the starting spot disappeared. The formed precipitate was filtered off, flushed several times with hot ethanol, dried, and recrystallized using methanol furnished 1,2-dihydroquinolin ehydrazono propanoate derivatives 8a-g. Single crystals, suitable for X-ray crystallographic analyses for compounds 8b, 8c, and 8d, have been obtained using methanol as the choice solvent of crystallization.
(E)-Ethyl 3-(2-(1-methyl-2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate (8a)
2.3
Pale yellow crystals (methanol), (258 mg, 90 %), Mp 190 °C; IR (KBr): ν 3237 (NH), 3085 (aryl-CH), 2991 (str., ali-CH), 1736 (ester-CO) 1628 (amide-CO), 1602 (CN), 1554 (CC) cm^−1^. ^1^H NMR (300 MHz, DMSO-d6): δH = 1.22 (t, J = 6.9 Hz; 3H, CH_3_); 3.44 (s, 3H, N–CH_3_); 3.51–3.53 (d, J = 11.4 Hz; 2H, –CH_2_-CHN) [25,30]; 4.12–4.14 (q, J = 6.9 Hz; 2H, CH_2_O); 6.17 (s, 1H, H-3); 7.22–7.28 (m, 1H, quinolinone-H); 7.43–7.48 (m, 1H, quinolinone-H); 7.60–7.64 (m, 1H, quinolinone-H); 7.73–78 (m, 1H, quinolinone-H); 8.06 (t, J = 11.4 Hz; 1H, CHN); 10.55 (brs, 1H, hydrazono-NH) ppm. ^13^C NMR (75 MHz, DMSO-d6): δC = 14.14 (CH_3_-ethyl); 28.50 (N–CH_3_); 37.88 (CH_2_–CHN); 60.49 (-CH_2_O); 93.63 (C-3); 112.82 (C-4a); 115.16 (C-8); 120.96 (C-6); 122.35 (C-5); 131.09 (C-7); 140.05 (C-8a); 141.35 (C-4); 147.44 (CHN-); 162.03 (C-2); 169.83 (ester-CO) ppm. MS (EI, 70 eV): m/z (%) 288 [M^+^, 100].
(E)-Ethyl 3-(2-(2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate (8b)
2.4
Pale yellow crystals (methanol), (278 mg, 92 %), Mp 245 °C; ^1^H NMR (400 MHz, DMSO-d6): δH = 1.26 (t, J = 8.0 Hz, 3H, CH_3_); 3.48–3.51 (d, J = 16.0 Hz, 2H, –CH_2_-CHN) [30]; 4.16 (q, J = 8.0 Hz; 2H, CH_2_O); 6.08 (s, 1H, quinolinone-H-3); 7.10–7.18 (m, 1H, quinolinone-H); 7.26–7.31 (m, 1H, quinolinone-H); 7.45–7.55 (m, 1H, quinolinone-H); 7.78 (s, 1H, quinolinone-H); 8.08 (t, J = 16.0 Hz, 1H, CHN); 10.50 (brs, 1H, hydrazono-NH); 11.00 (brs, 1H, q quinolinone-NH) ppm. ^13^C NMR (100 MHz, DMSO-d6): δC = 14.05 (CH_3_-ethyl); 37.76 (CH_2_–CHN); 60.49 (-CH_2_O); 93.49 (C-3); 111.71 (C-4a); 115.93 (C-8); 120.61 (C-6); 121.87 (C-5); 130.35 (C-7); 139.32 (C-8a); 141.13 (C-4); 148.45 (CHN-); 162.79 (C-2); 169.72 (ester-CO) ppm.
(E)-Ethyl 3-(2-(6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate (8c)
2.5
Pale yellow crystals (methanol), (278 mg, 92 %), Mp 245 °C; IR (KBr): ν 3163 (NH), 3055 (aryl-CH), 2996 (str., ali-CH), 1727 (ester-CO) 1638 (amide-CO), 1604 (CN), 1546 (CC) cm^−1^. ^1^H NMR (400 MHz, DMSO-d6): δH = 1.26 (t, J = 8.0, 3H, CH_3_); 3.44 (d, J = 16.0, 2H, –CH_2_-CH = ); 3.80 (s, 3H, CH_3_O); 4.14 (q, J = 8.0 Hz; 2H, CH_2_O); 6.06 (s, H-3); 7.20 (m, 2H, quinolinone-H); 7.52 (s, 1H, quinolinone-H); 7.74 (t, J = 16.0 Hz, 1H, CHN) [30]; 10.45 (brs, 1H, hydrazono-NH); 10.95 (brs, 1H, quinolinone-NH) ppm. ^13^C NMR (100 MHz, DMSO-d6): δC = 14.05 (CH_3_-ethyl); 37.74 (CH_2_–CHN); 55.59 (CH_3_O); 60.51 (-CH_2_O); 93.05 (C-3); 112.01 (C-4a); 117.74 (C-8); 119.54 (C-5); 133.55 (C-7); 140.93 (C-8a); 143.31 (C-6); 148.9 (CHN); 153.63 (C-4); 162.39 (C-2); 169.76 (ester-CO) ppm. MS (EI, 70 eV): m/z (%) 303 [M^+^, 100].
(E)-Ethyl 3-(2-(6-methyl-2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate (8d)
2.6
Pale yellow crystals (methanol), (278 mg, 92 %), Mp 245 °C; ^1^H NMR (400 MHz, DMSO-d6): δH = 1.28 (t, J = 8.0 Hz, 3H, CH_3_); 3.44 (d, J = 12.0 Hz, 2H, –CH_2_-CHN); 4.09–4.18 (q, J = 8.0 Hz; 2H, CH_2_O); 6.07 (s, 1H, quinolinone-H-3); 7.13–7.20 (m, 1H, quinolinone-H); 7.28–7.33 (m, 1H, quinolinone-H); 7.73–7.82 (m, 2H, quinolinone-H and CHN); 10.51 (brs, 1H, hydrazono-NH); 10.98 (brs, 1H, quinolinone-NH) ppm. ^13^C NMR (100 MHz, DMSO-d6): δC = 14.03 (CH_3_-ethyl); 20.58 (CH_3_); 37.76 (CH_2_–CHN); 60.66 (CH_2_O); 93.60 (C-3); 111.61 (C-4a); 115.70 (C-8); 127.32 (C-5); 129.66 (C-7); 137.26 (C-6); 140.99 (C-8a); 148.32 (CHN); 162.78 (C2); 169.12 (ester-CO) ppm. Anal. Calcd for C_15_H_17_N_3_O_3_: C, 62.71; H, 5.96; N, 14.63. Found: C, 62.66; H, 5.88; N, 14.52.
Ethyl (E)-3-(2-(6-chloro-2-oxo-1,2-dihydroquinolin-4-yl)hydrazineylidene)propanoate (8e)
2.7
Pale yellow crystals (methanol), (267 mg, 87 %), Mp 181 °C; IR (KBr): ν 3232, 3152 (NH's), 3070 (aryl-CH), 2985 (str., ali-CH), 1734 (ester-CO) 1641 (amide-CO), 1595 (CN), 1544 (CC) cm^−1^. ^1^H NMR (300 MHz, DMSO-d6): δH = 1.23 (t, J = 7.2 Hz; 3H, CH_3_); 3.42–3.3.44 (d, J = 11.4 Hz; 2H, –CH_2_-CHN); 4.15 (q, J = 7.2 Hz; 2H, CH_2_O); 6.06 (s, 1H, H-3); 7.26–7.30 (m, 1H, quinolinone-H); 7.48–7.51 (m, 1H, quinolinone-H); 7.71–7.75 (t, J = 11.4 Hz; 1H, CHN); 10.56 (brs, 1H, hydrazono-NH); 11.20 (brs, 1H, quinolinone-NH) ppm. ^13^C NMR (75 MHz, DMSO-d6): δC δC = 14.10 (CH_3_-ethyl); 37.83 (CH_2_–CHN); 60.61 (CH_2_O); 94.33 (C-3); 112.92 (C-4a); 117.58 (C-8); 121.46 (C-5); 125.15 (C-7); 130.37 (C-6); 138.14 (C-8a); 141.78 (C-4); 147.79 (CHN); 162.80 (C2); 169.72 (ester-CO) ppm. MS (EI, 70 eV): m/z (%) 307 [M^+^, 100].
Ethyl (E)-3-(2-(7-methyl-2-oxo-1,2-dihydroquinolin-4-yl)hydrazineylidene)propanoate (8f)
2.8
Pale yellow crystals (methanol), (245 mg, 85 %), Mp 207 °C; IR (KBr): ν 3243, 3159 (NH's), 3067 (aryl-CH), 2936 (str., ali-CH), 1733 (ester-CO) 1630 (amide-CO), 1598 (CN), 1526 (CC) cm^−1^. ^1^H NMR (300 MHz, DMSO-d6): δH = 1.26 (t, J = 7.2 Hz; 3H, CH_3_-ethyl); 2.82 (s, 3H, CH_3_); 3.42 (d, J = 11.4 Hz, 2H, –CH_2_-CHN); 4.16 (q, J = 7.2 Hz; 2H, CH_2_O); 6.18 (s, 1H, H-3); 698 (m, 1H, quinolinone-H); 7.31 (m, 1H, quinolinone-H); 7.63 (t, J = 11.4 Hz; 1H, CHN); 7.84–7.87 (m, 2H, quinolinone-H); 10.48 (brs, 1H, hydrazono-NH); 11.18 (brs, 1H, quinolinone-NH) ppm. ^13^C NMR (75 MHz, DMSO-d6): δC = 14.12 (CH_3_-ethyl); 21.18 (CH_3_); 37.86 (CH_2_–CHN); 60.81 (CH_2_O); 92.88 (C-3); 112.19 (C-4a); 115.61 (C-8); 125.11 (C-6); 129.78 (C-5); 134.65 (C-7); 138.51 (C-8a); 141.83 (C-4); 148.62 (CHN); 162.30 (C2); 168.72 (ester-CO) ppm. MS (EI, 70 eV): m/z (%) 287 [M^+^, 100].
Ethyl (E)-3-(2-(8-methyl-2-oxo-1,2-dihydroquinolin-4-yl)hydrazineylidene)propanoate (8g)
2.9
Pale yellow crystals (methanol), (251 mg, 82 %), Mp 224 °C; IR (KBr): ν 3236, 3163 (NH's), 3076 (aryl-CH), 2996 (str., ali-CH), 1737 (ester-CO) 1625 (amide-CO), 1606 (CN), 1546 (CC) cm^−1^. ^1^H NMR (300 MHz, DMSO-d6): 1.24 (t, J = 7.2 Hz; 3H, CH_3_-ethyl); 3.44 (d, J = 12.0 Hz; 2H, –CH_2_-CHN) [30]; 2.39 (s, 3H, CH_3_); 4.16 (q, J = 7.2 Hz; 2H, CH_2_O); 6.06 (s, 1H, H-3); 7.09 (m, 1H, quinolinone-H); 7.35 (m, 1H, Ar–H); 7.76 (t, 12.0 Hz; 1H, CHN); 7.85 (m, 2H, quinolinone-H); 10.08 (brs, 1H, hydrazono-NH); 10.49 (brs, 1H, quinolinone-NH) ppm. ^13^C NMR (75 MHz, DMSO-d6): δC = 14.09 (CH_3_-ethyl); 17.63 (CH_3_); 37.83 (CH_2_–CHN); 60.55 (CH_2_O); 93.51 (C-3); 111.64 (C-4a); 119.77 (C-5); 120.47 (C-6); 123.80 (C-8); 131.69 (C-7); 137.64 (C-8a); 141.27 (C-4); 148.95 (CHN); 163.09 (C2); 169.77 (ester-CO) ppm. MS (EI, 70 eV): m/z (%) 307 (M^+^+H_2_O, 35); 289 (M^+^, 15), 190 (3).
Single crystal X-ray structure determination of 8b, 8c, and 8d
2.10
Single crystals generated by slow evaporation from methanol. A good crystal with a suitable size was selected for X-ray diffraction analysis.
The single-crystal X-ray diffraction study was carried out on a Bruker D8 Venture diffractometer with a PhotonII detector at 173(2) K or 298(2) K using Cu-Kα radiation (λ = 1.54178 Å). Dual space methods (SHELXT) [31] were employed for the structure solution, and refinement was carried out using SHELXL-2014 (full-matrix least-squares on F^2^) [32]. Hydrogen atoms were refined using a riding model (H(O) free, except MeOH in 8b). Semi-empirical absorption corrections were applied; for 8b, an extinction correction was applied.
Compound 8b
2.11
C_14_H_15_N_3_O_3_·0.5(CH_4_O)·0.5(H_2_O), Mr = 298.32 g mol^−1^, blocks yellow, size = 0.12 × 0.06 × 0.04 mm, monoclinic, C2/c (no.15), a = 16.981 (6) Å, b = 11.048 (4) Å, c = 16.479 (5) Å, β = 98.60 (2)°, λ = 1.54178 Å, V = 3056.8 (18) Å^3^, Z = 8, D_calcd_ = 1.296 Mg m^−3^, F(000) = 1264, μ = 0.80 mm^−1^, T = 298 K, 13838 measured reflections (2θ_max_ = 144.2^o°^) 2991 independent reflections [R_int_ = 0.049], 210 parameters and 148 restraints, R_1_ [for 2495 reflections with I > 2σ(I)] = 0.056 wR2 (for all data) = 0.177, S = 1.05, largest diff. peak and hole = 0.27 e Å^−3^/-0.23 e Å^−3^.
Compound 8c
2.12
C_15_H_17_N_3_O_4_, Mr = 303.32 g mol^−1^, plates yellow, size = 0.16 × 0.12 × 0.02 mm, monoclinic, space group P21/n (no.14), a = 14.6490 (9) Å, b = 7.0414 (5) Å, c = 15.5731 (10) Å, β = 115.737 (2)°, λ = 1.54178, Å V = 1447.00 (17) Å^3^, Z = 4, D_calcd_ = 1.392 Mg m^−3^, F(000) = 640, μ = 0.86 mm^−1^, T = 173 K, 14391 measured reflections (2θ_max_ = 144.4°), 2838 independent reflections [R_int_ = 0.056], 206 parameters, 2 restraints, R_1_ [for 2549 reflections with I > 2σ(I)] = 0.050, wR2 (for all data) = 0.144, S = 1.04, largest diff. peak and hole = 0.32 e Å^−3^/-0.32 e Å^−3^.
Compound 8d
2.13
C_15_H_17_N_3_O 1.5(H_2_O), Mr = 314.34, g mol^−1^, plates yellow, size = 0.35 0.25 × 0.20 mm, monoclinic, space group I (no.15), a = 17.8791 (3) Å, b = 11.1773 (2) Å, c = 16.4921 (2) Å, β = 100.178 (1)°, λ = 1.54178, Å V = 3243.92 (9) Å^3^, Z = 8, D_calcd_ = 1.287 Mg m^−3^, F(000) = 1336, μ = 0.80 mm^−1^, T = 298 K, 16724 measured reflections (2θ_max_ = 144.4°), 3185 independent reflections [R_int_ = 0.056], 214 parameters, 169 restraints, R_1_ [for 2799 reflections with I > 2σ(I)] = 0.069, wR2 (for all data) = 0.215, S = 1.08, largest diff. peak and hole = 0.42 e Å^−3^/-0.44 e Å^−3^.
CCDC 2256765 (8b), 2256766 (8c), and 2256767 (8d) contain supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif (deposited at the CSD April 17, 2023).
Results and discussion
3
To approach the synthesis of the targeted molecules, multi-synthetic steps had been carried out [[27], [28], [29]], as shown in Scheme 1. The reactions between 4-hydrazinylquinolin-2(1H)-ones 5a-g and ethyl propiolate (6) were carried out in ethanol under refluxing conditions to affored the corresponding 3-(2-(2-oxo-1,2-dihydroquinolin-4-yl)hydraz-ono)propanoate 8a-g (Scheme 2). The reactions were prolonged under basic conditions to promote the cyclization to the pyrazole ring. Unfortunately, the reaction was proceed to without cycliziation and give the only open-chain products 8a-g as a sole product.Scheme 1. Strategy for the synthesis of hydrazonoquinolone 5a-g.Scheme 1. Scheme 2Stereoselective interaction of hydrazinylquiolinone 5a-g with ethyl propiolate 6 and synthesis of 1,2-dihydroquinolinehydrazonopropanoate derivatives 8a-g.Scheme 2
The chemical composition of all the prepared compounds obtained are proved using modern spectroscopic methods such as: FTIR, ^1^H NMR, ^13^C NMR spectrum, mass spectrometry, elemental analysis, and as well as X-ray crystallography. Compound 8c is selected as an example, which is assigned as (E)-ethyl 3-(2-(6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate (Fig. 3). The IR spectra of compound 8c; have shown that the NH-group were observed at ν 3256 cm^−1^. Moreover, an aromatic-CH stretching vibrated at ν 3085 cm^−1^, and the aliphatic-CH at ν 2996 cm^−1^. Also, other peaks appeared at ν 1727, 1638, and 1604 cm^−1^, which assigned for (CO ester), (CO, quinolinone-C2), and CN, respectively. The ^1^H NMR spectra of compound 8c showed four singlet signals at δH 3.80, 6.06, 7.52, 10.45, and 10.95 ppm, which were assigned as methoxy group, quinolinone-H-3, quinolone-H-5, hydrazone-NH and quinolinone-NH, respectively. The other characteristic signals for the ethyl group at δH 1.26 (t, J = 8.0 Hz, 3H, CH_3_) and 4.14 ppm (q, J = 8.0 Hz, 2H, CH_2_O). The presence of the ethyl group was confirmed from the ^13^C NMR, which gives characteristic signals at δC 14.05 and 60.51 ppm for CH_3_ and CH_2_O- groups, respectively. Furthermore, two different carbonyl groups are resonated at δC 162.39 and 169.76 ppm, respectively, as quinolinone-C-2 and ester-CO. On the other hand, the mass spectrometry and elemental analysis confirmed that compound 8c has a molecular formula C_15_H_17_N_3_O_4_ with m/z = 303, which confirmed that compound 8c comes via interaction between 1 mol of 4-hydrazinyl-6-methoxyquinolin-2(1H)-one (5c) with 1 mol of ethyl propiolate (6) without any elimination as shown in Scheme 2.Fig. 3. Chemical structure for (E)- ethyl 3-(2-(6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl)hydrazinylidene)propanoate (8c).Fig. 3
Regarding the composition of ethyl propiolate (6), we notice that it contains two active centers, namely the carbonyl group (CO) and the triple bond (CC); therefore, it is possible to form analogues of the compound during the intermediates A-C. If the reaction proceeded via intermediate A, the pyrazolylquinolin-2(1H)-ones 7a-g would form; again, if the reaction proceeded through the intermediate B, our products 8a-g and Z-form 9a-g and pyrazolylquinolin-2(1H)-ones 10a-g, would be obtained, but if the reaction proceeded through the intermediate C, the reaction would give the corresponding pyrazolylquinolin-2(1H)-ones 11a-g. The possibility of forming these compounds is referred to as the stereoselectivity phenomena; the compounds 7a-g, 10a-g, and 11a-g were ruled out at first sight according to ^1^H NMR and ^13^C NMR spectrum, in addition to the mass spectrometry. In addition, to distinguish between the two isomeric structures 8a-g and 9a-g, which comes from the reaction between 4-hydrazinylquinolin-2(1H)-ones 5a-c and mole of ethyl propiolate (6) through the intermediate B as shown in Scheme 2.
The single crystals, suitable for X-ray measurements, were obtained from methanol. X-ray crystal structure analysis were performed for compound 8b, 8c and 8d (see Fig. 4, Fig. 5, Fig. 6), which indicate that our obtained products as E-isomer as shown in Fig. 3, Fig. 4, Fig. 5. Geometrical parameters are in the expected range for 1,2-dihydroquinolin-4-yl)hydrazono)propanoate (see Table 1). The 1,2dihydroquinoline and the hydrazono moieties are slightly twisted (7.4° (8b), 15.5° (8c), 8.3° (8d), see Table 1].Fig. 4X-Ray crystallographic molecular structure for (E)-ethyl 3-(2-(2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate (8b) (solvent omitted for clarity, displacement parameters are drawn at 30 % probability level).Fig. 4. Fig. 5X-Ray crystallographic molecular structure for (E)-ethyl 3-(2-(6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate (8c) (displacement parameters are drawn at 50 % probability level).Fig. 5. Fig. 6X-Ray crystallographic molecular structure for (E)-Ethyl 3-(2-(6-methyl-2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate (8d) (solvent omitted for clarity, displacement parameters are drawn at 30 % probability level).Fig. 6. Table 1Selected bond distances [Å], bond angles [°] and dihedral angles [°] for 8b, 8c and 8d [in brackets for 8c].Table 1[in brackets for 8c]8b8c8dN1–C21.362(2)1.3635(18)1.354(2)N1–H10.860.870(15)0.86C2–O21.259(2)1.2628(17)1.261(2)C2–C31.421(2)1.4255(19)1.423(3)C3–C41.364(2)1.3627(19)1.371(3)C4–C4a1.455(2)1.4558(19)1.445(2)C4a-C8a1.407)2)1.411(2)1.404(3)N1–C8a1.381(2)1.3816(19)1.381(3)C4–N9 [C4–N11]1.366(2)1.3824(18)1.377(2)N9–N10 [N11–N12]1.375(2)1.3753(16)1.366(2)N9–H9 [N11–H11]0.860.876(14)0.86N10–C11 [N12–C13]1.261(2)1.2659(19)1.263(3)C11–C12 [C13–C14]1.490(2)1.4966(19)1.494(3)C4–N9–N10 [C4–N11–N12]118.85(14)117.20(11)118.25(15)N9–N10–C11 [N11–N12–C13]117.38(15)115.86(12)117.45(17)N10–C11–C12 [N12–C13–C14]119.44(17)118.54(13)118.47(19)C3–C4–N9–N10 [C3–C4–N11–N12]−1.8(3)−1.1(2)−5.0(3)C4a-C4-N9-N10 [C4a-C4-N11-N12]177.51(15)178.63(11)174.41(17)C4–N9–N10–C11 [C4–N11–N12–C13}−169.71(18)156.97(13)−170.8(2)N9–N10–C11–C12 [N11–N12–C13–C14]−179.37(17)179.67(12)−179.88(19)angle between the normal of the l.S. planes of the 1,2dihydroquinoline and the hydrazono moiety [°]7.415.58.3
The X-ray data of compound 8c proves that (E)-ethyl 3-(2-(6-methyl-2-oxo-1,2-dihydroquinolin-4-yl)hydrazono)propanoate was formed exclusively from the reaction of 8c with ethyl propiolate. All the X-ray structure confirmed the E-configuration concerning N10=C11 (8b, 8d), N12=C13 (8c). As outlined from the X-ray structure, the configuration of all compounds in the E-form shows that pyrazole does not form because the E-form makes the electrophilic site far from the attack of the nucleophilic hydrazine-N^1^.
The crystal packing of 8b, 8c and 8d shows intermolecular hydrogen bonds as well as π-π interactions.
Intermolecular hydrogen bonds for 8b, 8c and 8d
3.1
All three compounds form centrosymmetric dimers via intermolecular hydrogen bonds (N1–H1⋯O2). There are additional intermolecular hydrogen bond for 8b and 8d with the disordered solvent (MeOH for 8b and water for 8d) for N9–H9⋯O(solvent) and O–H(solvent)…O13. In addition, 8b and 8d are intermolecular hydrogen bonds via the ordered water molecule (O1W on special position at the 2-fold axis, O1W–H1W⋯O2) linking the dimers. For 8c, there are additional intermolecular hydrogen bonds C13–H13⋯O2, N11–H11⋯O2 (see details below) [[33], [34], [35]].
Hydrogen bonds 8b
3.2
In the crystal packing of 8b (Fig. 7, Fig. 8, Fig. 9), the molecules are linked by intermolecular N9–H9⋯O1M, O1M-H1M···O13, O1W–H1W⋯O2, and N1–H1⋯O2 hydrogen bonds (Table 2) in a two-dimensional network parallel to the ac plane. The MeOH solvent molecule is disordered about a 2-fold axis (symmetry operator 1-x, y, 1.5-z), there is half a MeOH molecule in the asymmetric unit (or 0.5 MeOH molecule per formula moiety).Fig. 7. Intermolecular hydrogen bonds of compound 8b showing hydrogen bonds as dashed lines.Fig. 7. Fig. 8Crystal packing of compound 8b showing hydrogen bonds as dashed lines.Fig. 8. Fig. 9Crystal packing of compound 8b showing hydrogen bonds as dashed lines.Fig. 9. Table 2Selected hydrogen-bond geometry (Å, °) for 8b.Table 2D—H···A**D—HH···A**D···A**D—H···AN1–H1⋯O2^i^0.861.982.836 (2)175N9–H9⋯O1M^ii^0.862.072.900 (3)162O1M—H1M···O13^iii^0.822.192.823 (3)135O1W–H1W⋯O20.82 (1)1.93 (3)2.699 (2)155 (5)Symmetry codes: (i) -x+1, -y+1, -z+2; (ii) -x+1/2, y+1/2, -z+3/2; (iii) x+1/2, y-1/2, z.
Hydrogen bonds 8d
3.3
In the crystal packing of 8d (Fig. 10, Fig. 11, Fig. 12), the molecules are linked by intermolecular N9–H9⋯O2W, O2W–H2W1⋯O13, O1W–H1W⋯O2, and N1–H1⋯O2 hydrogen bonds (Table 3) in a two-dimensional network parallel to the ac plane. In 4 voids are 8 solvent water molecules (2 per void generated about a 2-fold axis (symmetry operator -x, y, 1.5-z, one water molecule per formula moiety). These water molecules are probably disordered.Fig. 10. Intermolecular hydrogen bonds of compound 8d showing hydrogen bonds as dashed lines.Fig. 10. Fig. 11Crystal packing of compound 8d showing hydrogen bonds as dashed lines.Fig. 11. Fig. 12Crystal packing of compound 8d showing hydrogen bonds as dashed lines.Fig. 12. Table 3Selected hydrogen-bond geometry (Å, °) for 8d.Table 3D—H···A**D—HH···A**D···A**D—H···AN1–H1⋯O2^i^0.861.982.839 (2)178N9–H9⋯O2W0.862.203.044 (3)165O1W–H1W⋯O20.83 (1)1.94 (2)2.736 (2)163 (5)O2W–H2W1⋯O13^ii^0.82 (1)2.20 (5)2.914 (3)146 (7)Symmetry codes: (i) -x+1, -y+1, -z+2; (ii) -x, y, -z+3/2.
Hydrogen bonds 8c
3.4
In the crystal packing of 8c (Fig. 13, Fig. 14, Fig. 15), the molecules are linked by intermolecular C13–H13⋯O2, N11–H11⋯O2, and N1–H1⋯O2 hydrogen bonds (Table 4) There are dimers in the b/c plane which are linked by bifurcular hydrogen bonds(C13–H13⋯O2, N11–H11⋯O2) forming an one-dimensional network along the a-axis, as well as π···π interactions along the a-axis (see below).Fig. 13. Intermolecular hydrogen bonds of compound 8c showing hydrogen bonds as dashed lines.Fig. 13. Fig. 14Crystal packing of compound 8c showing hydrogen bonds as dashed lines.Fig. 14. Fig. 15Crystal packing of compound 8c showing hydrogen bonds as dashed lines.Fig. 15. Table 4Selected hydrogen-bond geometry (Å, °) for 8c.Table 4D—H···A**D—HH···A**D···A**D—H···AN1–H1⋯O2^i^0.87 (2)2.01 (2)2.8219 (16)154 (2)N11–H11⋯O2^ii^0.88 (1)2.08 (2)2.9003 (16)155 (2)C13–H13⋯O2^ii^0.952.603.3397 (18)135Symmetry codes: (i) -x+1, -y+2, -z+1; (ii) x, y-1, z.
π···π interactions
3.5
For 1,2-dihydroquinoline moieties in all 3 compounds 8b, 8c and 8d, centrosymmetric dimers are generated by π···π interactions (see Table 5, Fig. 16, Fig. 17, Fig. 18). In 8b and 8d, these dimers linked the 2D-hydrogen bond networks in the a/c plane by π···π interactions in the direction of the b-axis. In 8c, these dimers linked the hydrogen bonded dimers in the b/c plane by π···π interactions in the direction of the a-axis (see also the 1D hydrogen bond network along the a-axis).Table 5. Geometrical parameters for the π-stacking moieties involved in the π···π interactions [Å, °] (Cg1 centroid of the ring N1–C2–C3–C4–C4a-C8a, Cg2 centroid of the ring C4a-C5-C6-C7-C8-C8a).Table 5. Compound8b8d8ccentroid distance between Cg1…C2ga/Cg2…Cg1a4.044.383.50Vertical distance from ring centroids Cg1/Cg2 to symmetry related 1,2-dihydroquinolin3.423.523.38angle between the centroid vector Cg1…C2ga/Cg2…Cg1a and the normal to the 1,2-dihydroquinolin plane147.9 (32.1)143.3 (36.7)165.0 (25.0)symmetry operator (a)0.5-x, 1.5-y, 2-z0.5-x, 1.5-y, 2-z1-x, 1-y, 1-zFig. 16π···π interactions of compound 8b (solvent and hydrogen atoms omitted for clarity).Fig. 16. Fig. 17π···π interactions of compound 8d (solvent and hydrogen atoms omitted for clarity).Fig. 17. Fig. 18π···π interactions of compound 8c (solvent and hydrogen atoms omitted for clarity).Fig. 18
The symmetry related planes of the 1,2-dihydroquinolin dimers are coplanar (centrosymmetric symmetry operator, see Table 5). Therefore, the angel between the symmetry related 1,2-dihydroquinolins are 0°.
Based on the (E)-configuration of the products as attempted by X-ray crystallographic determination, the plausible mechanism of the nucleophilic addition reaction (1,4-addition) which is mentioned as aza-Michael addition as mentioned in Scheme 3. The mechanism involved two main nucleophilic additional steps; on each step, the nucleophilic attack occurs via the lone pairs of electrons on the hydrazine nitrogen (N^2^), and the final product was obtained through the formation of intermediates 12–15. The steps of the mechanism involved two transformation of enol-form to keto-form structures: intermediate 13 (enol-form) to intermediate-B (keto-form) and intermediate 15 (enol-form) to the final product 8a as the more stable keto-form.Scheme 3. Postulated mechanism for the formation of compound 8a ((E)-1-methyl-4-(2-(3-oxohexylidene)hydrazinyl)quinolin-2(1H)-one).Scheme 3
Conclusion
4
We have reported in this article the synthesis of ethyl hydrazonoquinolone propanoate derivatives via a convenient reaction between substituted hydrazinyl-quinolinones and ethyl propiolate and determined the structure using advanced techniques of analyses and confirmed the correct structure using X-ray crystallographic analysis. Also, the X-ray discussion of the hydrogen bond among the molecules, as well as the pi-pi stacking interaction in the crystallatice.
Data availability statement
Data associated with the study has not been deposited into a publicly available repository. Data will be made available on request.
CRediT authorship contribution statement
Hendawy N. Tawfeek: Writing – original draft, Methodology, Conceptualization. Ahmed M. Tawfeek: Project administration. Stefan Bräse: Writing – review & editing, Formal analysis. Martin Nieger: Writing – review & editing, Formal analysis, Data curation. Essmat M. El-Sheref: Writing – review & editing.
Declaration of competing interest
No conflict of interest exists.
All authors confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Dr. Hendawy N. Tawfeek (Corresponding author) [email protected].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bessonova A.Components of Haplophyllum bucharicum Chem. Nat. Compd.36200032332410.1007/BF 02238348 · doi ↗
- 2Detsi A.Bardakaos V.Markopoulos J.Reactions of 2-methyl-3,1-benzoxazin-4-one with active methylene compounds: a new route to 3-substituted 4-hydroxyquinolin-2(1H)-ones J. Chem. Soc. Perkin. Trans.119962909291310.1039/p 19960002909 · doi ↗
- 3Ukrainets V.I.Bereznyakova I.N.Mospanova V.E.4-Hydroxy-2-quinolones 121. Synthesis and biological properties of 1-hydroxy-3-oxo-5,6-dihydro-3h-pyrrolo[3,2,1-ij]quinoline-2-carboxylic acid alkylamides Chem. Heterocycl. Compd.43200785686210.1007/s 10593-007-0136-4 · doi ↗
- 4Mizuta M.Kanamori H.Mutagenic activities of dictamnine and gamma-fagarine from dictamni radicis cortex (Rutaceae)Mutat. Res. Lett.144198522122510.1016/0165-7992(85)90054-54069140 · doi ↗ · pubmed ↗
- 5Duraipandiyan V.Ignacimuthu S.Antibacterial and antifungal activity of Flindersine isolated from the traditional medicinal plant, Toddalia asiatica (L.) Lam J. Ethnopharmacol.123200949449810.1016/jjep 20090202019481384 · doi ↗ · pubmed ↗
- 6Debbab A.Aly A.H.Lin W.H.Proksch P.Bioactive compounds from marine bacteria and fungi Microb. Biotechnol.320104456310.1111/j.1751-7915.2010.00179.x PMC 381576821255352 · doi ↗ · pubmed ↗
- 7Konieczny J.Ossowska K.Schulze G.Coper H.Wolfarth S.L-701,324, a selective antagonist at the glycine site of the NMDA receptor, counteracts haloperidol-induced muscle rigidity in rats Psychopharmacology (Berl)143199923524310.1007/s 00213005094210353425 · doi ↗ · pubmed ↗
- 8Dhanak D.Duffy K.Johnston V.K.Lin-Goerke J.Darcy M.Shaw A.N.Baohua G.Silverman C.Gates A.T.Earnshaw D.L.Casper D.J.Kaura A.Baker A.Greenwood C.Gutshall L.L.Maley D.Del Vecchio A.Macarron R.Hofmann G.A.Alnoah Z.Cheng H.-Y.Chan G.Khandekar S.Keenan R.M.Sarisky R.T.Identification and biological characterization of heterocyclic inhibitors of the hepatitis C virus RNA-dependent RNA polymerase J. Biol. Chem.2772002383223832710.1074/jbc.m 20556620012167642 · doi ↗ · pubmed ↗