Draft genome sequences of three Carnobacterium maltaromaticum isolates from two low pH bogs in Denmark
Taya Tang, Jørgen J. Leisner

TL;DR
This paper presents draft genomes of three Carnobacterium maltaromaticum bacteria isolated from acidic bogs in Denmark.
Contribution
The novelty is the sequencing and reporting of three new draft genomes from C. maltaromaticum isolates in low pH bogs.
Findings
Three draft genome sequences of C. maltaromaticum were obtained from low pH bogs in Denmark.
The Illumina MiSeq platform was used for genome sequencing.
Abstract
Carnobacterium maltaromaticum is a lactic acid bacterium that is widely distributed in the environment, including freshwater. Here, we report the draft genome sequences of three C. maltaromaticum isolates from low pH Danish bogs, using the Illumina MiSeq platform.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
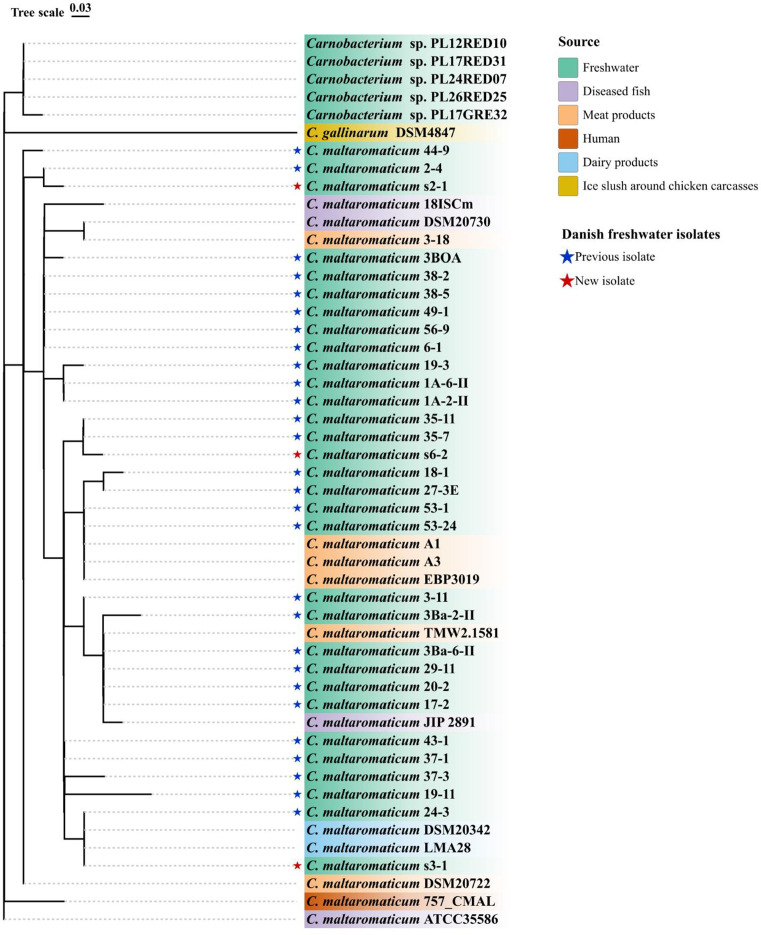 Fig 1
Fig 1| Variable | s2-1 | s3-1 | s6-2 |
|---|---|---|---|
| Location | Site 1 | Site 2 | Site 2 |
| Type | Forest bog | Forest bog | Forest bog |
| pH | 4.35 | 4.40 | 4.30 |
| Geographic coordinates | 55°45 | 55°49 | 55°49 |
| Genome characteristics | |||
| Assembly size (bp) | 3,728,548 | 3,498,272 | 3,930,617 |
| N50 (bp) | 181,003 | 188,278 | 246,871 |
| L50 (bp) | 7 | 6 | 8 |
| GC (%) | 34.10 | 34.34 | 34.14 |
| Number of contigs | 73 | 47 | 76 |
| Number of CDS | 3,372 | 3,141 | 3,566 |
| Number of tRNAs | 57 | 63 | 62 |
| Number of rRNAs | 8 | 7 | 12 |
| Number of sequences | 645,632 | 449,296 | 222,660 |
| Total number of reads (bp) | 193,689,600 | 134,788,800 | 66,798,000 |
| Genome coverage | 55× | 38× | 19× |
| Bacteriocins | BM1 | BM1 | None |
| BioProject accession |
|
|
|
| BioSample accession |
|
|
|
| SRA accession for raw reads |
|
|
|
| GenBank accession |
|
|
|
- —China Scholarship Council (CSC)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Microbial Community Ecology and Physiology · Mycobacterium research and diagnosis
ANNOUNCEMENT
The lactic acid bacterium Carnobacterium maltaromaticum is widely distributed in Danish freshwater environments during the winter season but underrepresented in bogs and poor fens (<pH 5.15) (1). Here, we show that C. maltaromaticum could be readily isolated from such habitats when culture-dependent sampling was done during the summer season and by using an enrichment procedure including an increased volume of sample relative to enrichment medium.
Ten milliliters of water samples were obtained on 30 May 2023, from two sites (Table 1) (1). One or three milliliters of samples were mixed 1:1 with all purpose Tween (APT) broth (Difco, Sparks, MD, USA) and incubated anaerobically at 15°C for 3 d before plating on de Man, Rogosa, Sharpe (MRS) agar (Oxoid, Thermo Fisher Scientific, Waltham, USA) anaerobically at 15°C for 3–5 d. Single colonies of Gram-positive and catalase-negative isolates (s2-1 from site 1; s3-1 and s6-2 from site 2) were cultivated in 3-mL APT broth under anaerobic and static conditions at 15°C for 3 d. Bacterial cells were harvested by centrifugation, and DNA extraction was performed using a DNeasy Blood and Tissue Kit, according to the manufacturer’s instructions (Qiagen GmbH, Hilden, Germany). DNA concentrations were measured by NanoDrop One Spectrophotometer (Thermo Fisher Scientific). Library preparation was done using the Nextera DNA Flex Library Prep Kit (Illumina, San Diego, USA). Sequencing on an Illumina MiSeq platform was performed as described by Li et al. (2), generating paired-end reads of 2 × 300 bp. An initial quality control involved FastQC v.0.11.9 (3) and trimming low-quality bases (Q <20) and adapter sequences with Cutadapt v.4.4 (4). Trimmed reads were assembled using SPAdes v.3.13.1 (5) with assembly quality assessed by QUAST 5.2.0 (6). Genome annotation was performed with Prokka v. 1.13 (7). Within the assembled contigs, the 16S rRNA gene regions were identified using Barrnap v.0.9 (8). The corresponding sequences were extracted using Bedtools v2.30.0 (9). Identification of the isolates was performed by comparing their 16S rRNA gene sequences via BLAST against the NCBI 16S rRNA database. A phylogenetic tree based on single-nucleotide polymorphism (SNP) variants in the whole core genomes was constructed using sequences of the 3 isolates from this study, 28 freshwater isolates of C. maltaromaticum from our previous study (BioProject: PRJNA990564, BioSample: SAMN36274541 to SAMN36274568) (1) and 19 published genome sequences of C. maltaromaticum, Carnobacterium gallinarum, and Carnobacterium sp. retrieved from the NCBI database. SNPs were identified by mapping reads against the reference C. maltaromaticum ATCC 35586 genome (GCF_000238575.1) via CSI Phylogeny v1.4 (10) and visualized in ChiPlot (11). Finally, we probed the presence of genes encoding bacteriocins by AntiSMASH 6.0 (12) and BAGEL 4 (13). All bioinformatic tools were run with default parameters.
Sequence statistics of the C. maltaromaticum genomes are summarized in Table 1, and a phylogenetic tree shows the close relatedness of the three isolates from this study to other C. maltaromaticum strains (Fig. 1). Genes encoding carnobacteriocin BM1 and the corresponding immunity protein were found in the s2-1 and s3-1 genomes. No genes encoding structures of other bacteriocins were detected.
A maximum-likelihood phylogenetic tree based on SNP variants in the whole core genomes of the Carnobacterium spp. strains. C. maltaromaticum ATCC 35586 was used as the reference genome to map and screen the SNPs.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tang T , Martinenghi LD , Hounmanou YMG , Leisner JJ . 2023. Distribution and ecology of the generalist lactic acid bacterium Carnobacterium maltaromaticum in different freshwater habitats: metabolic and antagonistic abilities. Environ Microbiol. doi:10.1111/1462-2920.16508 37750577 · doi ↗ · pubmed ↗
- 2Li H , Ramia NE , Borges F , Revol-Junelles A-M , Vogensen FK , Leisner JJ . 2021. Identification of potential citrate metabolism pathways in Carnobacterium maltaromaticum. Microorganisms 9:2169. doi:10.3390/microorganisms 9102169 34683489 PMC 8537297 · doi ↗ · pubmed ↗
- 3Andrews S . 2010. Fastqc: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom.
- 4Martin M . 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EM Bnet j 17:10. doi:10.14806/ej.17.1.200 · doi ↗
- 5Prjibelski A , Antipov D , Meleshko D , Lapidus A , Korobeynikov A . 2020. Using spades de novo assembler. Curr Protoc Bioinformatics 70:e 102. doi:10.1002/cpbi.102 32559359 · doi ↗ · pubmed ↗
- 6Gurevich A , Saveliev V , Vyahhi N , Tesler G . 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. doi:10.1093/bioinformatics/btt 086 23422339 PMC 3624806 · doi ↗ · pubmed ↗
- 7Seemann T . 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi:10.1093/bioinformatics/btu 153 24642063 · doi ↗ · pubmed ↗
- 8Seemann T . 2021. Barrnap 0.9: rapid ribosomal RNA prediction. https://github.com/tseemann/barrnap/09