Novel Heterozygous Missense Variants in Diacylglycerol Kinase Epsilon and Complement Factor I: Potential Pathogenic Association With Atypical Hemolytic Uremic Syndrome
Omar K Abughanimeh, Muhamed Baljevic, Alex Nester

TL;DR
A 69-year-old woman developed atypical hemolytic uremic syndrome after surgery, linked to new genetic mutations in DGKE and CFI, and fully recovered with eculizumab treatment.
Contribution
Identifies novel DGKE and CFI mutations as potential causes of atypical hemolytic uremic syndrome.
Findings
Novel heterozygous missense variants in DGKE and CFI were found in a patient with aHUS.
Treatment with eculizumab resolved the thrombotic microangiopathy without organ damage.
The case suggests a possible pathogenic role for these mutations in aHUS.
Abstract
Hemolytic uremic syndrome (HUS) is a thrombotic microangiopathy (TMA), which copresents with microangiopathic hemolytic anemia, thrombocytopenia, and kidney injury. While typical HUS is normally preceded by infections such as Shiga-toxin-producing Escherichia coli, atypical HUS (aHUS) has a genetic component that leads to dysregulation of the alternative complement pathway. We report a case of a 69-year-old female who developed aHUS after undergoing an elective knee surgery. Genetic testing revealed novel mutations affecting diacylglycerol kinase epsilon (DGKE) protein and complement factor I (CFI) that were not reported before as pathogenic. The patient was treated with eculizumab, leading to the complete resolution of TMA with no lasting organ damage.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
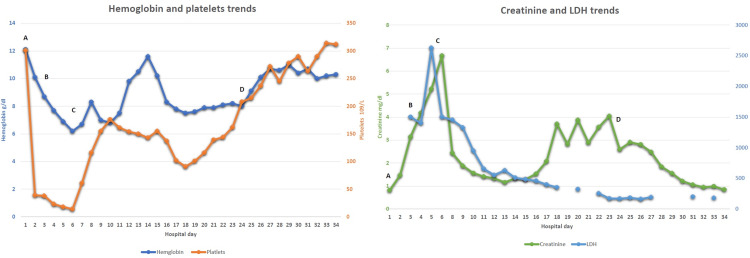 Figure 1
Figure 1| DGKE mutations | Mutation type | Reference |
| c.32C>A | Homozygous (premature termination codons) | Lemaire et al. [ |
| c.472InsT p.Trp158Leufs*8 | Homozygous (frameshift mutation) | Lemaire et al. [ |
| c.818G>C p.Arg273Pro | Homozygous (missense mutation) | Lemaire et al. [ |
| c.889-1G>A p.IVS5-1 | Homozygous (splice site mutation) | Lemaire et al. [ |
| c.486InsA p.Val163Serfs*3 | Heterozygous (frameshift mutation) | Lemaire et al. [ |
| c.188G>C p.Arg63Pro | Heterozygous (missense mutation) | Lemaire et al. [ |
| c.966G>A p.Trp322* | Homozygous (premature termination codons) | Lemaire et al. [ |
| c.1000C>T p.Gln334* | Homozygous (premature termination codons) | Lemaire et al. [ |
| c.888+40A.G | Homozygous for one family/heterozygous for one family (splice site mutation) | Mele et al. [ |
| c.231C>G, p.C77W | Heterozygous (missense mutation) | Li et al. [ |
| c.790_791delTG, p.C264Yfs*27 | Heterozygous (frameshift mutation) | Li et al. [ |
| c.1213–2A>G | Heterozygous (splice site mutation) | Miyata et al. [ |
| c.71delT | Heterozygous (frameshift mutation) | Miyata et al. [ |
| p.(Phe250Serfs*3) | Homozygous (nonsense) | Alabdulqader et al. [ |
| p.H536Qfs*16 | Homozygous (frameshift mutation) | Sánchez Chinchilla et al. [ |
| p.W322*/p.P498R | Heterozygous | Sánchez Chinchilla et al. [ |
| p.Q248H/p.G484Gfs*10 | Heterozygous | Sánchez Chinchilla et al. [ |
| c.562C>A p.Pro188Thr | Heterozygous (missense mutation) | Our case |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComplement system in diseases · Renal Diseases and Glomerulopathies · Erythrocyte Function and Pathophysiology
Introduction
Atypical hemolytic uremic syndrome (aHUS) is a rare thrombotic microangiopathy (TMA) attributed to genetic alterations of the alternative complement pathway [1]. These genetic mutations can present at any age [2]. Mutations causing aHUS can lead to either loss of function (affecting factor H and factor I) or gain of function (affecting C3 and factor B) [3]. In rare cases, new mutations are found that affect non-complement-related proteins such as diacylglycerol kinase epsilon (DGKE), plasminogen, and inverted formin-2. Mutation in the DGKE gene is very rare and typically presents in the first year of life [3]. DGKE is a protein involved in metabolism by affecting cell signaling. It is also responsible for the modulation of protein kinase C (PKC) activity. Hence, mutations affecting DGKE can result in the activation of PKC, which stimulates prothrombotic factors including tissue factor and von Willebrand factor [3].
Case presentation
A 69-year-old female with a history of hypertension, atrial fibrillation, chronic obstructive pulmonary disease (COPD), and chronic left knee septic joint following total knee arthroplasty, was admitted to the hospital for left knee cement spacer exchange. Even though the procedure was uncomplicated, she developed acute normocytic anemia (hemoglobin 8.7 g/dL), thrombocytopenia (53 x 10^3 ^µL), and acute kidney injury (creatinine 3.1 mg/dL) after three days of the surgery (Figure 1 shows the trend of relevant organ functions).
Trends of hemoglobin, platelets, LDH, and creatinineA: on the day of surgery; B: on the day of starting eculizumab; C: hemodialysis started; D: hemodialysis endedLDH: lactate dehydrogenase
Further workup showed schistocytes on peripheral smear, elevated lactate dehydrogenase (LDH; 1540 U/L, reference range: 98-192 U/L), undetectable haptoglobin, total bilirubin of 0.5 g/dl (reference range: 0.2-1.5 mg/dL), international normalized ratio (INR) of 1.8, fibrinogen 239 mg/dL (reference range: 160-450 mg/dL). Otherwise, white blood cell counts, liver enzymes, and electrolytes were within normal limits. A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) was sent given the concern of microangiopathic hemolytic anemias and the result showed 74% activity, suggesting atypical hemolytic uremic syndrome (aHUS).
The patient was started on eculizumab 900 mg weekly on the same day and hemodialysis after 48 hours (due to fluid overload and decreased urine output). Further studies revealed that the complement C3 level was 105 mg/dL (reference range: 90-180 mg/dL), complement C4 was 40 mg/dL (reference range: 12-45 mg/dL), CH50 was 87 CAE (reference range: 60-144 CAE), and complement factor B was 35.1 mg/dl. aHUS mutation panel revealed a heterozygous missense variant (c.562C>A (p.Pro188Thr)) in exon 3 of DGKE and a heterozygous missense variant (c.608C>T, p.Thr203Ile) in exon 4 of complement factor I (CFI). Her condition improved gradually on complement-directed therapy. She completed four weeks of weekly eculizumab followed by maintenance eculizumab 1200 mg every two weeks for six months. Her kidney function returned to normal levels after 24 days of complement-directed therapy, and she no longer required hemodialysis.
Discussion
Recessive DGKE mutations causing TMA were first described in 2013 by Lemaire et al. [4]. In their study, they identified recessive mutations in DGKE that co-segregated with aHUS in nine individuals who were all aged less than one year at the time of diagnosis. Later, Mele et al. reported another novel mutation, which was the first to be located beyond the exon-intron boundaries [5]. Table 1 summarizes DGKE mutations in aHUS reported in the literature so far.
There is no sufficient evidence to recommend an optimal management method for DGKE mutation-induced aHUS. There are some reported cases where patients responded to either eculizumab or plasma exchange while in others they did not [10]. Our patient was further found to have a heterozygous missense variant (c.608C>T, p.Thr203Ile) in exon 4 of CFI. In general, CFI mutations are rarely reported in the literature to be associated with aHUS [11]. Almalki et al. reported a case of aHUS that was believed to be caused by a heterozygous CFI variant, c.944G > A (p.Arg315Lys) [11]. The CFI mutation in our case had never been reported in an aHUS previously, while some other reports have proposed a possible link with age-related macular degeneration. [12,13]
While recognizing that aHUS pathogenic variants in DGKE are indeed typically inherited in an autosomal recessive or compound heterozygote manner (and that most heterozygotes are usually asymptomatic), our patient’s clinical presentation was in the setting of (a) chronic septic arthritis where secondary causes of TMA were not evident after an exhaustive infections workup (no evidence of fungemia or bacteremia with three separate blood cultures negative over seven days); (b) negative medical history for drug-induced causes; (c) only notable finding among a mutation profile that screened more than 20 different coagulation pathway genes coding for proteins known to be pathogenically associated with aHUS were variants of unknown significance in DGKE and CFI reported in this case. Unfortunately, more definitive data regarding each variant (e.g., minor allele frequency and in silico analysis) could not be provided due to technical limitations. Lastly, the dramatic clinical improvement following anti-complement therapy as well as the presence of heterozygous mutations in two genes known to be pathogenically involved with aHUS led us to postulate that either, alone or in combination as in this case, may be pathogenically relevant.
In general, aHUS has a poor prognosis with around 60% of patients progressing to end-stage renal disease (ESRD). It has an estimated mortality rate of 4-25% [5]. In this instance, complement-directed therapy was effective and prevented conditions associated with long-term organ damage such as ESRD. Considering the brisk clinical response to therapeutic challenge with anti-complement therapy, as well as previously noted associations of DGKE and CFI variants with aHUS, we believe that variants of unknown significance in this case potentially played a pathogenic role, and should be further examined for pathogenic potential at centers with adequate capabilities for minor allele frequency and in silico analysis, during subsequent screening efforts for patients with suspected aHUS.
Conclusions
aHUS is a rare, life-threatening disease with a genetic component. Mutations in DGKE are rare and usually present in the first years of life, though exceptions in the elderly can be found, leading to aHUS. Similarly, CFI mutations leading to aHUS are not commonly reported in the literature. There is currently no standard treatment for aHUS caused by DGKE or CFI mutations, but our case was successfully treated with eculizumab therapy.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Atypical hemolytic uremic syndrome: when the environment and mutations affect organ systems. a case report with review of literature Am J Hosp Med Abu Ghanimeh M Abughanimeh O Qasrawi A Qasem A 2017247412017 https://hdl.handle.net/10355/61508
- 2An unusual case of diffuse alveolar hemorrhage as a clinical manifestation of atypical hemolytic uremic syndrome: a case report Cureus Al-Shyoukh A Younis M Abughanimeh O Tahboub M Hamarshi MS 011201910.7759/cureus.5059 PMC 672189431516771 · doi ↗ · pubmed ↗
- 3The genetics of atypical hemolytic uremic syndrome Med Genet Feitz WJ van de Kar NC Orth-Höller D van den Heuvel LP Licht C 4004093020183093055110.1007/s 11825-018-0216-0PMC 6404389 · doi ↗ · pubmed ↗
- 4Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome Nat Genet Lemaire M Frémeaux-Bacchi V Schaefer F 5315364520132354269810.1038/ng.2590 PMC 3719402 · doi ↗ · pubmed ↗
- 5Characterization of a new DGKE intronic mutation in genetically unsolved cases of familial atypical hemolytic uremic syndrome Clin J Am Soc Nephrol Mele C Lemaire M Iatropoulos P 101110191020152585428310.2215/CJN.08520814 PMC 4455211 · doi ↗ · pubmed ↗
- 6A novel compound heterozygous mutation in DGKE in a Chinese patient causes atypical hemolytic uremic syndrome Hematology Li J Song Y Zhang Y 1011072520203209131810.1080/16078454.2020.1731969 · doi ↗ · pubmed ↗
- 7Atypical haemolytic uraemic syndrome in a Japanese patient with DGKE genetic mutations Thromb Haemost Miyata T Uchida Y Ohta T Urayama K Yoshida Y Fujimura Y 86286311420152601811110.1160/TH 15-01-0007 · doi ↗ · pubmed ↗
- 8A patient with a homozygous diacylglycerol kinase epsilon (DGKE) gene mutation with atypical haemolytic uraemic syndrome and low C 3 responded well to eculizumab: a case report BMC Nephrol Alabdulqader M Alfakeeh K 1402220213387907710.1186/s 12882-021-02352-8PMC 8056694 · doi ↗ · pubmed ↗