Assembly and annotation of 2 high-quality columbid reference genomes from sequencing of a Columba livia × Columba guinea F1 hybrid
Emily T Maclary, Carson Holt, Gregory T Concepcion, Ivan Sović, Anna I Vickrey, Mark Yandell, Zev Kronenberg, Michael D Shapiro

TL;DR
This paper presents high-quality genomes for two dove species, improving resources for studying their genetics and evolution.
Contribution
The study provides new, high-quality genome assemblies for Columba livia and Columba guinea using hybrid sequencing.
Findings
The C. livia genome assembly Cliv_3 is more complete and accurate than previous versions.
The C. guinea genome is the first for this species and enhances avian comparative genomics.
The assemblies improve tools for studying genomic traits in C. livia and related species.
Abstract
Pigeons and doves (family Columbidae) are one of the most diverse extant avian lineages, and many species have served as key models for evolutionary genomics, developmental biology, physiology, and behavioral studies. Building genomic resources for columbids is essential to further many of these studies. Here, we present high-quality genome assemblies and annotations for 2 columbid species, Columba livia and Columba guinea. We simultaneously assembled C. livia and C. guinea genomes from long-read sequencing of a single F1 hybrid individual. The new C. livia genome assembly (Cliv_3) shows improved completeness and contiguity relative to Cliv_2.1, with an annotation incorporating long-read IsoSeq data for more accurate gene models. Intensive selective breeding of C. livia has given rise to hundreds of breeds with diverse morphological and behavioral characteristics, and Cliv_3 offers…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
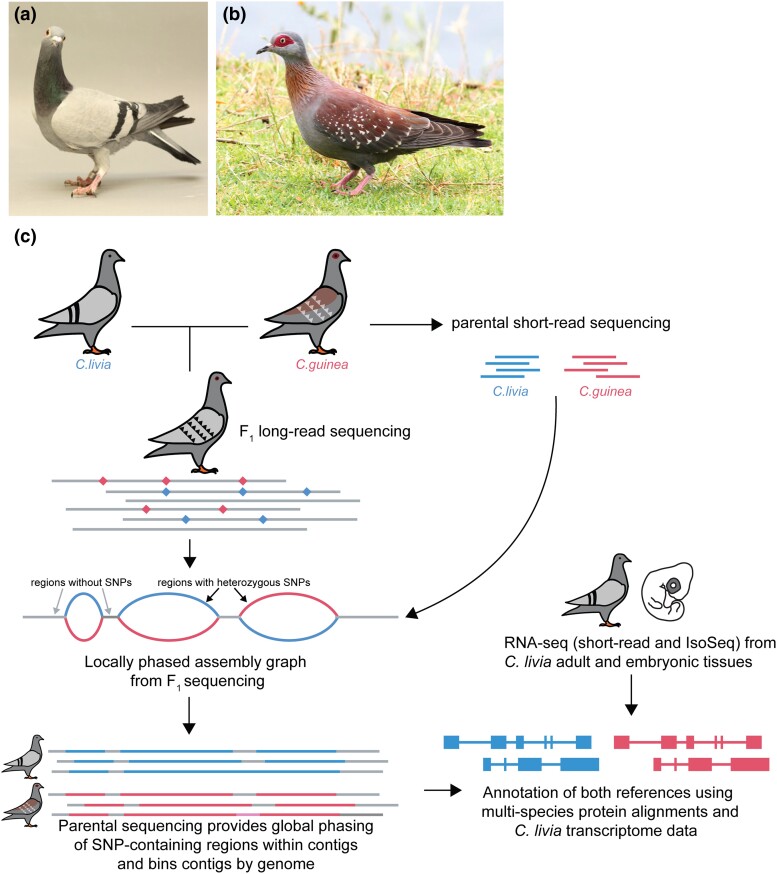 Fig. 1
Fig. 1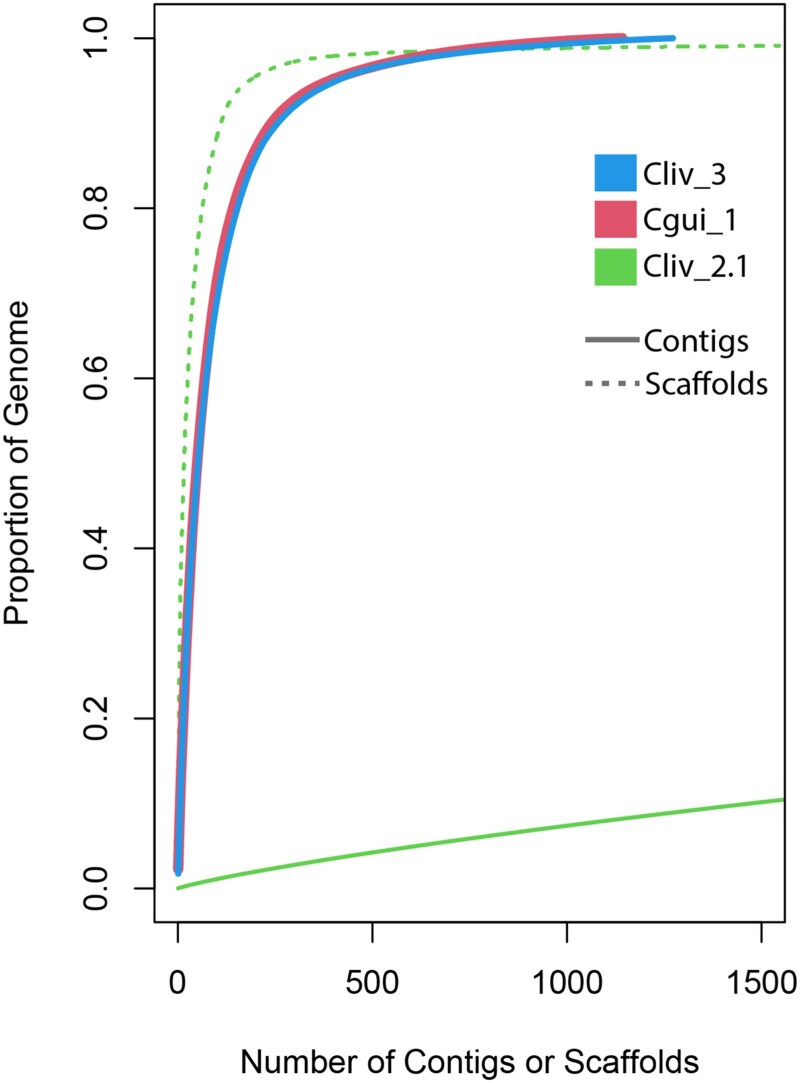 Fig. 2
Fig. 2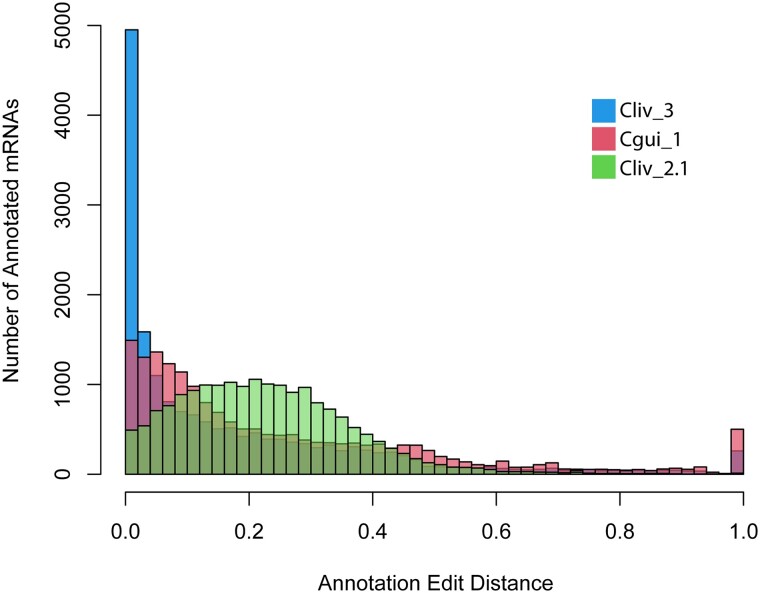 Fig. 3
Fig. 3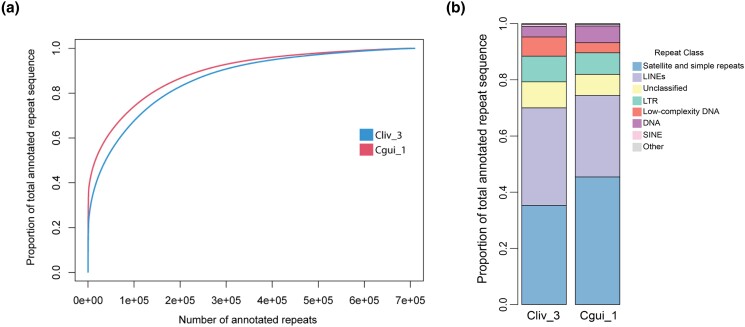 Fig. 4
Fig. 4| Genome | Genome size | Number of contigs | Contig N50 | Annotated genes | mRNA models |
|---|---|---|---|---|---|
| Cliv_3 | 1.28 Gb | 1273 | 6.97 Mb | 16,853 | 18,431 |
| Cgui_1 | 1.30 Gb | 1147 | 7.61 Mb | 17,966 | 17,966 |
| Cliv_2.1 | 1.11 Gb | 107, 177 | 26 kb | 15,392 | 18,966 |
| Genome | BUSCO: Aves (aves_odb10 database) | BUSCO: Vertebrates (vertebrata_odb10 database) | ||||||
|---|---|---|---|---|---|---|---|---|
| Complete | Complete (duplicated) | Fragmented | Missing | Complete | Complete (duplicated) | Fragmented | Missing | |
| Cliv_3 | 97.0% | 0.4% | 0.4% | 2.2% | 95.6% | 0.7% | 1.6% | 2.0% |
| Cgui_1 | 96.9% | 0.4% | 0.5% | 2.2% | 95.5% | 0.8% | 1.6% | 2.1% |
| Cliv_2.1 | 96.4% | 0.3% | 0.8% | 2.4% | 90.0% | 0.6% | 5.1% | 4.2% |
| Linkage group | Number of contigs | Size (base pairs) | Corresponding |
|---|---|---|---|
| 1 | 65 | 213,903,937 | 1 (196.45 Mb) |
| 2 | 59 | 163,618,457 | 2 (149.54 Mb) |
| 3 | 31 | 122,001,762 | 3 (110.64 Mb) |
| 4 | 12 | 79,237,614 | 4 |
| 5 | 17 | 69,363,043 | 5 (59.51 Mb) |
| 6 | 6 | 55,545,771 | 7 (36.38 Mb) |
| 7 | 7 | 46,431,064 | 6 (36.22 Mb) |
| 8 | 5 | 35,246,159 | 8 (29.58 Mb) |
| 9 | 2 | 13,779,628 | 18 (11.62 Mb) |
| 10 | 2 | 15,514,691 | 20 (14.27 Mb) |
| 11 | 3 | 29,989,141 | 9 (23.72 Mb) |
| 12 | 4 | 20,545,355 | 13 (17.91 Mb) |
| 13 | 3 | 17,672,897 | 14 (15.33 Mb) |
| 14 | 2 | 21,279,300 | 11 (19.64 Mb) |
| 15 | 2 | 21,666,617 | 4 |
| 16 | 6 | 7,135,027 | 24 (6.48 Mb) |
| 17 | 2 | 22,657,501 | 12 (20.12 Mb) |
| 18 | 2 | 11,857,108 | 17 (11.09 Mb) |
| 19 | 3 | 22,666,117 | 10 (20.45 Mb) |
| 20 | 3 | 14,961,191 | 15 (12.70 Mb) |
| 21 | 3 | 8,468,065 | 21 (6.97 Mb) |
| 22 | 1 | 8,321,153 | 26 (5.35 Mb) |
| 23 | 1 | 6,512,245 | 23 (6.25 Mb) |
| 24 | 4 | 6,093,282 | 22 (4.96 Mb) |
| 25 | 1 | 11,162,903 | 19 (10.46 Mb) |
| 26 | 3 | 6,907,984 | 27 (5.23 Mb) |
| 27 | 1 | 5,550,848 | 28 (5.44 Mb) |
| 28 | 3 | 3,442,929 | 25 (3.07 Mb) |
| 29 | 5 | 2,852,076 | 34 (3.47 Mb) |
| Z | 70 | 84,521,162 | Z (86.04 Mb) |
| Other | 14 | 2,563,644 | NA |
- —National Institutes of Health10.13039/100000002
- —H.A. & Edna Benning Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and phenotypic traits in livestock · Genetic and Environmental Crop Studies · Genetic diversity and population structure
Introduction
The domestic pigeon (Columba livia) is a unique model system for understanding the molecular and developmental basis of phenotypic diversity. C. livia has been under intensive artificial selection by pigeon fanciers for thousands of years, giving rise to more than 350 breeds with extensive variation in a variety of complex traits, including body size, beak morphology, feather color and morphology, and behavior (Stringham et al. 2012; Domyan and Shapiro 2017; Nimpf et al. 2019; Shao et al. 2019). In many cases, the magnitude of phenotypic diversity among C. livia approaches species-level diversity among wild bird species (Darwin 1868; Baptista et al. 1997). Thus, C. livia offers extraordinary phenotypic variation and experimental accessibility as a model for comparative genetics and developmental biology, both of which rely on a high-quality genome assembly and annotated gene models.
While domestic pigeons themselves are a spectacularly diverse single species, the family Columbidae is also one of the most diverse and geographically distributed extant avian lineages (Soares et al. 2016). Beyond C. livia, other doves and pigeons have served as important focal species for studies of physiology, behavior, and evolution (Johnson et al. 2010; Nimpf et al. 2019; Shao et al. 2019; Boyd et al. 2022; Burns-Cusato and Cusato 2022; Phillmore et al. 2022; Wasserman et al. 2023). Developing genomic resources for additional columbids provides resources for understanding avian diversity and ecological adaptation and critical tools for experimental biology. Columba guinea, the speckled pigeon, is of particular interest as a study species for comparative genomics because C. guinea and C. livia have partially overlapping geographical ranges and are interfertile. C. livia and C. guinea can hybridize to produce fertile male F_1_s and potentially fully fertile backcrosses after only a few generations (Taibel 1949). We previously found that C. guinea contributed to the evolution of domestic and free-living populations of C. livia through the introgression of a potentially advantageous pigmentation trait (Vickrey et al. 2018).
Prior work developed reference genomes for C. livia (Shapiro et al. 2013; Damas et al. 2017; Holt et al. 2018). However, updated sequencing technology has paved the way for substantial improvements in contiguity and completeness, as seen in a recently released C. livia assembly (Wang et al. 2023). Additionally, new assembly methods can produce haplotype-resolved assemblies from heterozygous individuals (Nurk et al. 2020; Cheng et al. 2021). Here, we report 2 genome assemblies from a single C. guinea × C. livia F_1_ hybrid individual: the first assembly and annotation for C. guinea and an updated C. livia assembly with major improvements in contiguity, a new annotation incorporating full-length transcriptome evidence, and scaffolding based on extensive genetic linkage data.
Methods
Animal husbandry
Pigeons were housed in accordance with protocols approved by the University of Utah Institutional Animal Care and Use Committee (protocols 19-02011 and 22-03002). An F_1_C. livia × C. guinea hybrid (band ID MDS 687) was bred in-house from a male domestic rock pigeon (Racing Homer breed; band ID MDS 004) and a female speckled pigeon (band ID MDS 529). Blood was collected from adult animals by brachial wing venipuncture.
Genome sequencing and assembly
DNA was extracted from whole blood using the Qiagen DNEasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA) for the parental F_0_C. guinea (female, band ID MDS 529) and C. livia (male, band ID MDS 004) individuals. Samples were treated with RNAse during extraction. Isolated F_0_ DNA was submitted to the University of Utah High Throughput Genomics Shared Resource for library preparation using the Illumina Tru-Seq PCR-Free Library Kit. The resulting libraries were sequenced on the Illumina NovaSeq platform. Whole blood from 1 male F_1_C. livia × C. guinea hybrid sample (band ID MDS 687) was submitted to the University of Delaware DNA Sequencing and Genotyping Center for DNA extraction, library preparation, and sequencing on the PacBio Sequel II system.
We used a standard short-read trio binning pipeline with hifiasm (Cheng et al. 2021). Parental k-mers were identified and counted with yak (v. r55; https://github.com/lh3/yak) (Cheng et al. 2021) using the settings -k 29 -b20 -t 12. The parental k-mer databases and PacBio hifi reads were then provided to hifiasm (v. 0.16.1-r375; https://github.com/chhylp123/hifiasm) (Cheng et al. 2021) for assembly of both genomes using the command “hifiasm -o.asm -t64 -1 maternal_kmer_database.yak -2 paternal_kmer_database.yak hifi_reads.fq”.
Assemblies were screened for contamination by genetic material from other organisms using FCS-GX (v. 0.3.0) (NCBI; https://github.com/ncbi/fcs/wiki/FCS-GX). We identified and removed one 7.5-kb contig, h2tg000968l, in the C. livia assembly and one 7.5-kb contig, h1tg000935l, in the C. guinea assembly predicted to be fungal in origin. Assemblies were screened for sequencing adaptor contamination using FCS-adaptor (https://github.com/ncbi/fcs); adaptors at the end of contigs were trimmed. One C. livia contig (h2tg000073l) with an internal adaptor was predicted to contain contiguous sequence based on both linkage data and contiguous sequence assembled across the same region in C. guinea. For this contig, we used NCBI BLASTN (v. 2.13.0) (Camacho et al. 2009) to identify reads from both the C. guinea and C. livia haplotype spanning the adaptor sequence. Adaptor sequence was excised and replaced with sequence from spanning reads with the C. livia haplotype. Other contigs with internal adaptors were split at the adaptor sequence. Based on these screens, contamination represented <0.001% of sequence in both genome assemblies.
We assessed the completeness of both assemblies with BUSCO (v. 5.3.2), using both the avian-specific aves_odb10 database and the more generalized vertebrate vertebrata_odb10 database (Manni et al. 2021).
IsoSeq transcriptome sequencing
One C. livia embryo at the equivalent of Hamburger–Hamilton (Hamburger and Hamilton 1951) stage 25 was isolated from a fertilized egg and dissected in PBS. The head and body were separated and stored in RNAlater (Invitrogen, Cat. #AM7021). Total RNA was extracted from the whole head using the QIAGEN RNEasy Plus Kit (QIAGEN, Cat. #74030). PacBio SMRTbell libraries were prepared, sequenced, and processed into hifi reads by the DNA Sequencing Center at Brigham Young University. Adapters were trimmed from hifi reads using lima (v2.5.0). Poly-A tails and artificial concatemers were removed using the isoseq3 (v3.4.0) “refine” command, and isoforms were clustered using isoseq3 cluster.
Genome annotation
A repeat library generated for the previously published Cliv_2.1 annotation set (GCA_000337935.2) was used to generate a repeat annotation of both genomes, supplemented by RepBase RepeatMasker Edition version 20181026 (Bao et al. 2015; Holt et al. 2018) (http://www.repeatmasker.org).
MAKER (Holt and Yandell 2011) was used to annotate gene models for both C. livia and C. guinea. Protein evidence used by MAKER includes annotated zebra finch proteins downloaded from RefSeq (GCF_003957565.2), annotated chicken proteins downloaded from RefSeq (GCF_016699485.2), and all UniProtKB/Swiss-Prot proteins (International Chicken Genome Sequencing Consortium 2004; Boutet et al. 2007; Warren et al. 2010). IsoSeq sequences from C. livia were provided to MAKER as transcript evidence in FASTA format. Previously published Cliv_2.1 mRNA-seq data (Holt et al. 2018) were aligned using STAR (Dobin et al. 2013) against both the C. livia and C. guinea reference genomes and then assembled using StringTie (Pertea et al. 2015) and provided to MAKER as aligned GFF3 formatted transcript features. We then followed the published MAKER Alternate Protocol 1 with StringTie models being provided as both transcript evidence (est_gff) as well as gene prediction models (pred_gff) (Campbell et al. 2014).
Ab initio gene prediction through MAKER was configured to use Augustus (Stanke et al. 2006), trained according to the published MAKER Support Protocol 1 (Campbell et al. 2014) using UniProtKB/Swiss-Prot models first aligned by BLASTP (Camacho et al. 2009) and Exonerate (Slater and Birney 2005). These models were then converted by MAKER into gene models based on maximum open reading frame in the alignments. These protein alignment-based models were converted to GenBank format using MAKER's zff2genbank.pl script. We then followed Augustus's training documentation to produce final training models for C. livia and C. guinea. After the initial annotation, all Augustus ab initio models were processed through InterProScan (Jones et al. 2014) to rescue models that contain known protein domains but were initially rejected by MAKER in accordance with MAKER Basic Protocol 5 (Campbell et al. 2014) to produce the maximum annotation set.
The C. livia MAKER “max” models were compared to Gallus gallus BLASTX protein alignments using the BEDTools “intersect” command (Quinlan and Hall 2010). This process identified transcripts with exons overlapping 2 or more G. gallus protein models, which may represent false merges of 2 or more protein products in the final C. livia annotation set. These models were then manually curated in WebApollo (Lee et al. 2013). A total of 742 MAKER “max” models were split into 2 or more models in the final annotation. All split models were again processed through InterProScan as above to identify known protein domains. The C. guinea MAKER models were not manually curated.
Whole-genome comparisons
The Cliv_3 and Cgui_1 assemblies were aligned to each other using D-GENIES with Minimap (v. 2.24) to generate a genome-wide dotplot, PAF format alignments, and BAM format alignment files to calculate coverage (Cabanettes and Klopp 2018; Li 2018). The Cliv_3 assembly was also aligned to Cliv_2.1 scaffolds for comparison using the same approach. The Cliv_2.1 assembly has a small number of scaffolds that span multiple Cliv_3 contigs. To evaluate the utility of Cliv_2.1 reference genome to fill gaps in the Cliv_3 assembly, we used quickmerge to merge Cliv_2.1 contigs or Cliv_2.1 scaffolds with the Cliv_3 assemblies (Chakraborty et al. 2016) and determine if the breaks between Cliv_3 contigs contained N tracts or contiguous sequence in Cliv_2.1 scaffolds.
Reciprocal alignments from Minimap were used to screen for putative large-scale (>100 kb) duplications, deletions, and inversions. Sequence regions with no alignments between species were identified as possible deletions in the species used as the “query” sequence. Tandem Repeats Finder (Benson 1999) was used to identify tandem repeats in nonaligning sequence. Repetitive intervals identified by Tandem Repeats Finder were merged with repetitive intervals annotated by RepeatMasker. The BEDTools “subtract” function (v. 2.28.0) (Quinlan and Hall 2010) was used to compare nonaligning regions of the Cliv_3 and Cgui_1 assemblies to the coordinates of repeat intervals to identify any large nonrepetitive regions that did not align between species (Benson 1999). Putative duplications were identified using the BEDTools “genomecov” function to assess coverage in BAM format alignments. To identify possible inversions, PAF format alignments were filtered to exclude regions of low sequence identity and regions with multiple alignments, grouped by Cgui_1 contig, Cliv_3 contig, and relative orientation using the BEDTools “groupby” function and then manually examined for changes in relative orientation within pairs of aligned contigs.
Comparison of Cgui_1 and Cliv_3 gene content
Cgui_1 and Cliv_3 gene models were compared using NCBI BLASTP to identify transcript pairs that were reciprocal best BLASTP hits (Camacho et al. 2009). For transcripts without reciprocal best BLASTP hits (“unmatched genes”), we used LiftOff (v. 1.6.3) to map GFF format annotations onto the reciprocal species genome (Shumate and Salzberg 2021). LiftOff mappings were compared to each annotation using the BEDTools “intersect” function to identify exon overlap (Quinlan and Hall 2010). For a small subset of representative gene arrays identified by LiftOff, NCBI BLASTN (Camacho et al. 2009) was used to compare sequence identity and exon boundaries for genes within the array.
Comparison of repeat annotation
GFF format repeat annotations were collapsed into nonredundant, nonoverlapping intervals using the BEDTools “cluster” and “groupby” functions (Quinlan and Hall 2010). Summed interval lengths were used to calculate total repeat content and cumulative distributions of repeat element size. Repeat-type classifications from the GFF annotations were used for all analysis of repeat-type distribution.
Terminal intervals were defined as the first and last 5 kb of sequence from each contig. For comparisons of repeat distribution, 10,000 random 5-kb intervals were generated using the BEDTools “makewindows” function (Quinlan and Hall 2010). Intervals were not filtered to remove overlapping windows. Terminal and random intervals from each species were scanned for repeat content using BEDTools “intersect” to identify the number of base pairs covered by annotated repeats. Repeat composition of intervals was calculated based on the number of base pairs covered by each repeat type.
Identification and evaluation of tandem 28-mer repeats
Tandem Repeats Finder (Benson 1999) was used to characterize a repetitive element identified in the Cliv_3 and Cgui_1 repeat annotations as “MuDR2-TC.” Examination of sequence within the repeats annotated as “MuDR2-TC” identified a tandem repeating 28-mer with the consensus sequence TGTCACAAACCCCATTGGACAGCGTGTG. Jellyfish (v. 2.3.0) (Marçais and Kingsford 2011) was used to count 28-mers in several avian genome assemblies (Columbina picui, GCA_013397635.1; Patagioenas fasciata, GCA_002029285.1; Pterocles gutturalis, GCA_000699245.1; Streptopelia turtur, GCA_901699155.2; and Tauraco erythrolophus, GCA_000709365.1) and unassembled Illumina short-read genomic shotgun sequences (Patagioenas oenops, SRS1476223; Columba palumbus, SRS1416881; Columba rupestris, SRS346866; Patagioenas speciosa, SRS1476204; Macropygia mackinlayi, SRS1476214; Streptopelia decaocto, SRS1476207; Streptopelia picturata, SRS1476196; and Zenaida asiatica, SRS1476211) to screen for this 28-mer repeat in other columbid and outgroup genomes (B10K consortium 2019; Beijing Genomics Institute 2012, 2013a, b; Boyd 2016; Rice 2016; Wellcome Sagner Institute 2018).
Linkage map construction and anchoring to revised assembly
Genotyping and linkage map construction for 5 F_2_ intercrosses of C. livia were carried out as previously described (Domyan et al. 2016; Boer et al. 2021; Maclary et al. 2021). Briefly, genotyping by sequencing (GBS) data were generated from founders and F_2_ populations. GBS reads were trimmed using CutAdapt (Martin 2011) and then mapped to the Cliv_2.1 reference genome reads using Bowtie2 (Langmead and Salzberg 2012). Genotypes were called using Stacks2 (v. 2.5.3) by running “refmap.pl” (Catchen et al. 2013). We constructed genetic maps using R/qtl v1.46-2 (www.rqtl.org; Broman et al. 2003).
To compare the linkage maps to the Cliv_3 assembly, each 90-bp locus containing a genetic marker was parsed from the Stacks2 output file “catalog.fa.gz” and queried to the Cliv_3 assembly using nucleotide–nucleotide BLAST (v2.6.0+) with the parameters -max_target_seqs 1 -max_hsps 1 (Camacho et al. 2009). Hits were filtered to retain alignments with an E-value of <4E^−24^, a threshold that permits multiple SNPs in each short sequence.
We used an Old German Owl × Racing Homer (OGO × Hom) linkage map as our primary reference for anchoring (Boer et al. 2021). A total of 5,978 markers from the OGO × Hom linkage map mapped to the Cliv_3 assembly with an E-value of <4E^−24^ and hits on 245 Cliv_3 contigs. We then identified markers mapping to 66 additional contigs in our 4 other crosses. We used linkage data from contigs shared between maps to determine placement for these 66 contigs within the OGO × Hom map. Markers within contigs were ordered based on base pair position, and the relative orientations of contigs with >3 markers spanning at least 30 kb were determined based on calculated recombination frequencies in linkage maps.
RagTag scaffolding
C. livia genome contigs were further scaffolded using the RagTag v2.1.0 scaffold function, with the C. guinea genome used as the reference (Alonge et al. 2019). The order and orientation of C. livia contigs within RagTag scaffolds were then compared to linkage map anchoring results. RagTag scaffolding provided ordering and orientation information for 31 additional contigs. Where possible, RagTag results were used to orient contigs with fewer than 3 GBS linkage markers or poorly spaced linkage markers. Contig orientations based on RagTag were also compared to oriented contigs from linkage map anchoring and regions where RagTag and linkage map orientation disagree are noted in Supplementary Table 2.
Comparison to chicken genome assembly
Cliv_3 proteins were compared to G. gallus GRCg7b reference proteins using NCBI BLASTP with the parameters -max_target_seqs 1 -max_hsps 1 (Camacho et al. 2009). Alignments were filtered to keep only pairs that were reciprocal best hits. Reciprocal best hits were then grouped by the linkage group of origin for the Cliv_3 protein and the chromosome of origin for the G. gallus protein, and the proportion of reciprocal best hit proteins shared between each Cliv_3 linkage group and G. gallus chromosome was calculated to identify correspondences. Gene order was not considered.
Results and discussion
Genome assemblies for C. livia and C. guinea
We generated 58,482,263,241 bp of circular consensus sequences from an F_1_ hybrid C. livia × C. guinea sample, with an average sequence length of 15.8 kb, for an estimated coverage of 22.8× per haplotype. We additionally generated over 96 Gb of C. livia short-read sequencing and 95 Gb of C. guinea short-read sequencing from F_0_ individuals, for an approximate genome coverage of 75× per parental sample. We used hifiasm to assemble haplotype-resolved C. livia and C. guinea genomes from an F_1_ hybrid individual (Fig. 1). The final C. livia assembly is 1,283,322,617 bp in length and consists of 1,273 contigs, while the C. guinea assembly is 1,302,215,470 bp and has 1,147 contigs (Table 1). Whole-genome alignment of the Cliv_3 and Cgui_1 assemblies show clear 1:1 alignments for the majority of both genomes (85 and 84% of the Cliv_3 and Cgui_1 assemblies, respectively; Supplementary Fig. 1).
Genome assembly and annotation strategy. a, b) Representative images of C. livia a) and C. guinea a). C. livia photo: Sydney Stringham. C. guinea photo: Roy Lowry, CC BY 4.0, via iNaturalist.co.uk. c) Genome assembly strategy for haplotype-resolved C. livia and C. guinea genomes and annotations from an F1 hybrid.
The Cliv_3 and Cgui_1 assemblies have contig N50s of 6.97 and 7.61 Mb, respectively. Therefore, the overall contiguity of the Cliv_3 assembly shows significant improvement over Cliv_2.1, which has a contig N50 of only 26 kb (Table 1), and compares favorably to a recent C. livia PacBio-based assembly that has a slightly higher contig N50 of 7.8 Mb, but a larger number of contigs (1,949) (Wang et al. 2023). The Cliv_2.1 assembly contains several very large scaffolds, but they have numerous gaps, and the assembly is dominated by smaller scaffolds (Table 1; Fig. 2). We examined Cliv_2.1 scaffolds that span multiple Cliv_3 contigs and found that gaps between Cliv_3 contigs that are spanned by Cliv_2.1 scaffolds always encompass 1 or more tracts of Ns (missing or ambiguous sequence). Thus, despite having several larger scaffolds, the Cliv_2.1 assembly does not contain contiguous sequence that can be used to resolve gaps between Cliv_3 contigs.
Cliv_3 and Cgui_1 assemblies show major improvements in contiguity over Cliv_2.1.
BUSCO analysis of highly conserved gene content of the new genome assemblies confirms both improved completeness and reduction in fragmentation of gene models compared to the Cliv_2.1 assembly. We found a 5% increase from Cliv_2.1 to Cliv_3 in the number of complete single-copy BUSCOs from the vertebrata_odb10 database and a reduction in fragmented and missing BUSCOs (Table 2).
We examined whole-genome alignments of Cliv_3 and Cgui_1 for evidence of duplications, deletions, and inversions. We did not identify any contiguous >100-kb regions with increased alignment coverage in either species, suggesting an absence of large duplications. We did identify several regions of >100 kb that did not align between species; however, >92% of this sequence content is repetitive. The lack of cross-species alignment in these regions could be due to their repetitive nature or a consequence of species-specific repeat expansion and does not necessarily reflect large species-specific deletions or gains of unique sequences. Alignments between Cliv_3 and Cgui_1 show several large inversions (Supplementary Table 1), which could be characteristic of one or the other species, the Racing Homer C. livia breed, or the specific parents of the hybrid individual sequenced. Analysis of broader samples of both C. guinea and C. livia would be required to distinguish among these possibilities.
While contiguity is greatly improved in Cliv_3 and Cgui_1 relative to Cliv_2.1, the new genomes are not assembled to the chromosome level. We examined sequence at the termini (sequences at either end) of Cliv_3 and Cgui_1 contigs to determine if sequence characteristics contribute to assembly challenges and found an enrichment of annotated repeat elements compared to random 5-kb intervals (2-tailed t-test, P < 1E^−15^). In addition to high repeat content, the terminal 5 kb of Cliv_3 and Cgui_1 contigs showed changes in repeat composition compared to size-matched random windows across the genome, with enrichment of simple and satellite repeats (Supplementary Fig. 2). Furthermore, only 0.27% of Cliv_3 repeat tracts genome-wide are >5 kb in length. Contig termini represent <1% of the Cliv_3 genome, but 39% of these long repeat elements overlap the 5-kb terminal intervals of contigs; 26% of terminal intervals show >99% coverage by a repeat tract longer than 5 kb. Long simple repeats, satellites, and tandem repeat tracts can be particularly challenging for genome assembly, especially when repeat length exceeds read length or divergence between repeats is not sufficient to resolve them by phasing (Tørresen et al. 2019; Nurk et al. 2020; Peona et al. 2021). These 5-kb intervals at the termini of contigs may represent fractions of larger repeat elements that we are unable to assemble because their lengths exceed common sequencing read lengths. We also noted that contigs from C. livia and C. guinea did not always terminate at orthologous sequences: in some instances, a single Cliv_3 contig spanned 2 Cgui_1 contigs, and vice versa, suggesting the presence of species-specific repeat expansions may have impacted genome assembly and contiguity.
Linkage-based scaffolding of the Cliv_3 assembly
To evaluate the relative positions and orientations of Cliv_3 contigs, we used linkage data from F_2_ crosses and contigs from the Cgui_1 assembly that spanned gaps in the Cliv_3 assembly. We were able to assign 342 Cliv_3 contigs covering 1,151 Mb of sequence (89.7% of the Cliv_3 assembly) to 37 autosomal linkage groups and 1 Z chromosome linkage group (Table 3; Supplementary Table 2). Several of the smaller (<20 Mb) linkage groups in our anchored map consist of only 1 or 2 Cliv_3 contigs, suggesting that some contigs represent nearly complete chromosomes or chromosome arms.
Based on gene content, we identified Cliv_3 linkage groups that correspond to 29 chicken chromosomes (Table 3). Prior analyses showed that the ortholog of chicken chromosome 4 is split into 2 chromosomes in the rock pigeon (Derjusheva et al. 2004; Damas et al. 2017) and our linkage groups also support this split. We found at least 1 Cliv_3 linkage group corresponding to each chicken autosome >4 Mb (n = 26) and the Z chromosome. We were unable to identify a linkage group corresponding to several chicken microchromosomes <4 Mb, perhaps due to genomic rearrangements between species.
Genome annotations for Cliv_3 and Cgui_1
The Cliv_3 annotation consists of 16,853 gene models encoding 18,431 unique mRNAs (Table 1). The Cgui_1 annotation consists of 17,966 gene models and mRNAs (Table 1; only 1 isoform is annotated for each gene because we did not use species-specific transcriptome evidence). In addition to the short-read RNA-sequencing data used for the Cliv_2.1 annotation, the Cliv_3 annotation incorporates full-length IsoSeq transcript data as evidence. We assessed annotation edit distance (AED), a measure of concordance between gene models and transcript evidence alignments, for the Cliv_3 and Cgui_1 data sets. AED scores were significantly lower for the Cliv_3 and Cgui_1 annotations than for Cliv_2.1, indicating improved concordance between MAKER annotation models and supporting C. livia RNA-seq evidence (Fig. 3).
AEDs for Cliv_3, Cgui_1, and Cliv_2.1 assemblies. Cliv_3 and Cgui_1 show significantly lower AED scores (P < 1E−15, t-test) than Cliv_2.1, indicating improved concordance between annotation models and RNA-seq data in the new annotations.
We compared the MAKER C. livia and C. guinea annotation sets using reciprocal best BLASTP hit analysis and identified 13,821 matched gene models between the 2 annotations. Models not matched by reciprocal best BLASTP hits tend to have higher AED scores, indicating reduced concordance with protein and mRNA model evidence (Supplementary Fig. 3a). For the models not matched by best reciprocal BLASTP hits, we looked for correspondence between C. livia and C. guinea genes by using LiftOff (Shumate and Salzberg 2021) for reciprocal mapping between species. Ninety-four percent of nonmatched Cliv_3 genes mapped to the Cgui_1 assembly, while 85% of nonmatched Cgui_1 genes mapped to the Cliv_3 assembly. Most of these genes show shared intron–exon structure with a model from the alternate species assembly, but these correspondences were not identified by best BLASTP analysis for several reasons, including species-specific mutations that disrupted annotation. For example, 40% of Cgui_1 gene models that mapped to the Cliv_3 assembly did not have valid open reading frames, despite high sequence conservation between the 2 assemblies allowing for the identification of orthologous exons. We also found evidence of fragmentation, in which a single gene model in 1 species is broken into several gene models in the other (Supplementary Fig. 3b). Interestingly, many of these nonmatched and fragmented genes were also part of tandem arrays of genes with high sequence identity and conserved intron/exon structure (Supplementary Fig. 3b). Some of these nonmatched gene models may also arise from species-specific expansion or contraction of tandem gene arrays.
Differences in repeat content between C. livia and C. guinea
We found an expansion of annotated repeats in C. guinea compared to C. livia. In C. guinea, annotated repeats account for 20.8% of the genome assembly, while they account for only 16.8% in C. livia (see Supplementary Tables 3 and 4 for repeat annotations). The C. guinea genome is specifically enriched for longer repeat tracts (Fig. 4a). We also found changes in repeat composition (Fig. 4b). The Cliv_3 assembly is enriched for low complexity DNA, while the Cgui_1 assembly is enriched for simple and satellite repeats and DNA repeat elements. The higher DNA repeat content in Cgui_1 was particularly striking: this assembly contains nearly 2-fold more sequence annotated as DNA repeat elements compared to Cliv_3 (16.15 Mb in Cgui_1 vs 8.23 Mb in Cliv_3).
Repeat annotations differ between C. livia and C. guinea. a) Cumulative distribution plot of C. livia and C. guinea repeats. The Cgui_1 genome is enriched for longer repeat tracts. b) Distribution of repeat classifications in Cliv_3 and Cgui_1 assemblies.
The Cgui_1 repeat annotation showed substantial enrichment for a DNA element annotated as “MuDR2-TC,” which consists of tandem 28-mer repeats. BLAST hits to the NCBI nr database and screening for the 28-mer sequence in other avian genome assemblies and shotgun sequencing reads showed that this element is restricted to the monophyletic group that includes the columbid genera Columba and Streptopelia (Supplementary Fig. 4). The 28-mer repeats are frequently interspersed with PR1 elements that are found in C. livia centromeres (Solovei et al. 1996) and thus may represent centromeric sequence. However, the 28-mer repeat does not exclusively colocalize with PR1 sequences. Centromeric satellite sequence in columbid species is not well characterized. Unlike telomeric sequence, centromere satellite repeats are diverse and evolve rapidly between species, and changes in centromeric sequence may drive speciation (Fukagawa 2013; Melters et al. 2013). Whether this lineage-specific repetitive element in Columba and Streptopelia may play a role in the diversification of dove and pigeon species remains an open question.
Advantages of parallel assembly and annotation of hybrid genomes
Annotation of a single assembly typically does not allow the types of comparisons that we are able to make between Cliv_3 and Cgui_1. Trio assemblies from other F_1_ hybrids either have not included annotations (Rice et al. 2019) or have focused analyses on specific genes of interest within the annotation (Low et al. 2020; Greenhalgh et al. 2022). As such, there is little precedent for comparing differences in annotations between closely related species with genome assemblies and annotations completed using the same pipeline. It is unclear how much variation in gene content should be expected between closely related species, particularly for tandem gene arrays or other repetitive elements, or how much cross-species comparisons could be affected by differences in assembly and annotation pipelines. While genome annotation pipelines and computational gene prediction perform well for conserved genes in nonrepetitive regions that are well supported by protein and RNA-seq evidence, the prevalence of nonmatched gene models between the Cliv_3 and Cgui_1 assemblies, including fragmented models and arrays of gene models in repetitive regions, highlights both the continued need for manual curation and refinement of genome assemblies and the importance of focusing on well-conserved orthologs for assessment of genome completeness and interspecies comparisons.
Conclusions
Here, we present a new long-read genome assembly for C. livia and the first genome assembly for C. guinea, both derived from a single hybrid individual. The Cliv_3 assembly shows substantial increases in contiguity over the Cliv_2.1 assembly and resolves numerous gaps, and contiguity is similar in the Cgui_1 assembly. These increases in contig length and resolution of gaps will improve variant detection for comparative genomics in both species, including detection of structural variants or deletions that were previously hampered by short scaffold lengths or gaps that impair short-read mapping. The addition of the C. guinea genome will improve understanding of genomic diversity among columbids, a highly diverse and species-rich avian lineage. Furthermore, our updated Cliv_3 annotation shows improved concordance with RNA-seq and protein alignments, and the addition of long-read IsoSeq data provides new information about isoform diversity.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alonge M , Soyk S, Ramakrishnan S, Wang X, Goodwin S, Sedlazeck FJ, Lippman ZB, Schatz MC. 2019. Ra GOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 20(1):224. doi:10.1186/s 13059-019-1829-6.31661016 PMC 6816165 · doi ↗ · pubmed ↗
- 2B 10K consortium . 2019. Columbina picui genome assembly. Gen Bank. GCA_013397635.1.
- 3Bao W , Kojima KK, Kohany O. 2015. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA.6(1):11. doi:10.1186/s 13100-015-0041-9.26045719 PMC 4455052 · doi ↗ · pubmed ↗
- 4Baptista LF , Trail PW, Horblit HM. 1997. Handbook of the Birds of the World. Barcelona: Lynx Edicions. (J del Hoyo, A Elliott, & J Sargatal, Eds).
- 5Beijing Genomics Institute. 2012. Whole genome sequencing of rock pigeon. NCBI Sequence Read Archive. SRS 346866.
- 6Beijing Genomics Institute . 2013 a. Pterocles gutturalis genome assembly. Gen Bank. GCA_000699245.1.
- 7Beijing Genomics Institute . 2013 b. Tauraco erythrolophus genome assembly. Gen Bank. GCA_000709365.1.
- 8Benson G . 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2):573–580. doi:10.1093/nar/27.2.573.9862982 PMC 148217 · doi ↗ · pubmed ↗