Constitutional Chromothripsis on Chromosome 2: A Rare Case with Severe Presentation
Afia Hasnain, Laura L. Thompson, Nicole L. Hoppman, Karine Hovanes, Jing Liu, Bita Hashemi

TL;DR
A rare case of chromosomal abnormalities in a child led to severe birth defects and early death, likely due to complex genetic changes on chromosome 2.
Contribution
This paper reports a rare and severe case of constitutional chromothripsis on chromosome 2 with a novel combination of genomic rearrangements.
Findings
The patient had duplications and deletions on chromosome 2, leading to severe congenital abnormalities.
Literature review showed overlapping features but no prior case with such a severe outcome.
Breakpoints analysis found no candidate gene involvement, suggesting structural and dosage effects caused the severity.
Abstract
Chromothripsis is characterized by shattering and subsequent reassembly of chromosomes by DNA repair processes, which can give rise to a variety of congenital abnormalities and cancer. Constitutional chromothripsis is a rare occurrence, reported in children presenting with a wide range of birth defects. We present a case of a female child born with multiple major congenital abnormalities including severe microcephaly, ocular dysgenesis, heart defect, and imperforate anus. Chromosomal microarray and mate pair sequencing identified a complex chromosomal rearrangement involving the terminal end of the long arm of chromosome 2, with two duplications (located at 2p25.3-p25.1 and 2q35-q37.2 regions) and two deletions (located at 2q37.2-q37.3 and 2q37.3 regions) along with structural changes including inverted segments. A review of the literature for complex rearrangements on chromosome 2…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
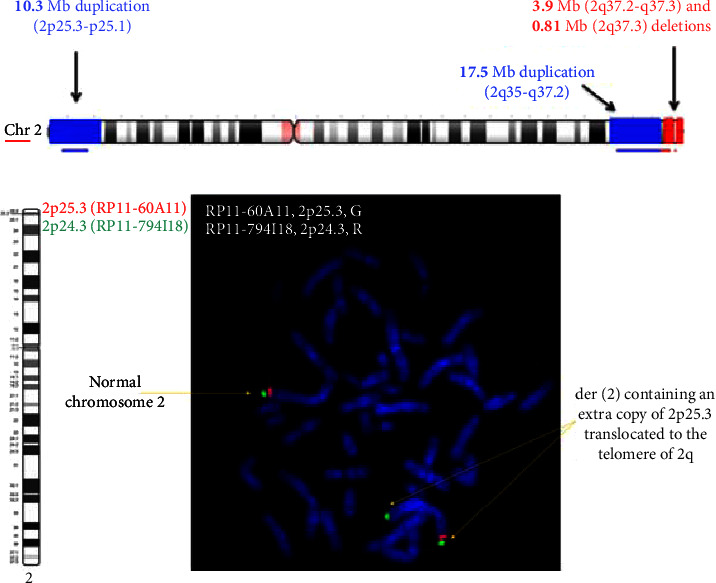 Figure 1
Figure 1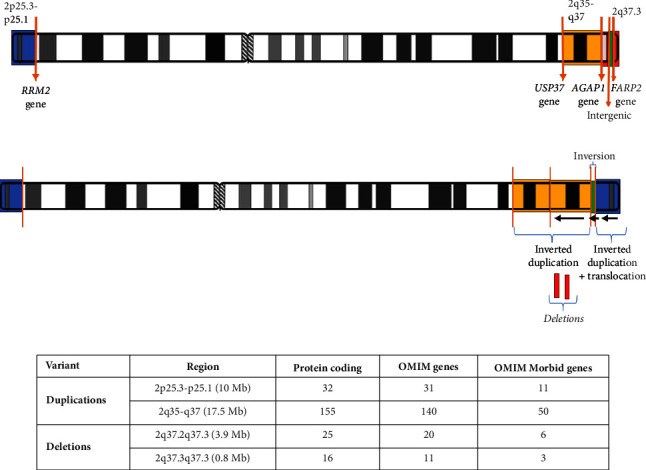 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomic variations and chromosomal abnormalities · Prenatal Screening and Diagnostics · DNA Repair Mechanisms
1. Background
Chromothripsis, a terminology first coined in patients with cancer [1], is defined as chromosomal shattering and random reassembly of fragments through various DNA damage repair mechanisms [2–6]. Soon after, constitutional chromothripsis was reported in several cases with a variety of phenotypes and chromosomal involvement [7–9]. In one case, transmission was reported from a balanced carrier parent [10], whereas one study reported stable segregation of this complex rearrangement in several family members [11]. The underlying mechanism of chromothripsis remains a matter of debate.
Single chromosomal involvement is rare in constitutional chromothripsis with currently only a few cases being reported [7, 12–14].
Terminal deletions and duplications of chromosome 2, as seen in our patient, are rare. Both are usually observed as part of more complex duplication/deletion syndromes. Rarely, terminal deletion of chromosome 2 has been reported in isolation and is known as chromosome 2q37 deletion syndrome. Reported features include hypotonia, brachycephaly, intellectual disability, autism spectrum disorder, short stature, obesity, minor facial dysmorphism, short neck, minor ear anomalies, congenital heart defects, nipple abnormalities, low anterior hairline, and low set ears [15]. To our knowledge, only a few patients have been reported with chromothripsis of chromosome 2, involving the chromosomal region of 2q34q37.2 [16, 17].
Determining the molecular and phenotypic consequences of genomic rearrangements is a major challenge, especially for patients with complex rearrangements. Genomic imbalances, position effects, or potential genes involved at or near breakpoints could be responsible for the clinical phenotype.
The aim of the present study was to characterize a complex rearrangement of chromosome 2, in a patient with severe congenital malformation involving the development of multiple major organs. We used mate pair sequencing in addition to the conventional techniques to explore the breakpoints and structural changes involved.
2. Case Presentation
We report a 2-year-old female born at term to a healthy 33-year-old G1P0 mother via uncomplicated normal vaginal delivery. The patient's family history was unremarkable. She was the first child of a nonconsanguineous couple of Filipino descent. Her maternal aunt passed away due to leukemia at 16 years of age. There was no history of congenital malformation or recurrent miscarriages in the couple or their relatives.
Prenatal history was unremarkable with no reported maternal illnesses or exposure to known teratogens. Prenatal ultrasound showed microcephaly and increased nuchal fold thickness. Maternal serum screening results were categorized as low risk for chromosomal aneuploidies and spina bifida. Parents decided to continue the pregnancy and declined any invasive testing.
At birth, growth parameters included a weight of 3610 g (>50^th^ centile), a length of 54.5 cm (95^th^ centile), and severe microcephaly with a head circumference of 29.5 cm (<2^nd^ centile). Physical examination of the patient at birth identified multiple dysmorphic features including a low anterior hairline, hypertelorism, short nose, depressed nasal bridge, left corneal opacity, right depressed globe, and bilateral simple low-set ears. She also had low-set nipples, bilateral supernumerary nipples in anterior axillary line, and a loud systolic murmur in cardiac examination. The anus was imperforated and anteriorly placed, while examination of external genitalia was typical for a normal female. She had a central and peripheral hypotonia with flexion contractures of elbows and camptodactyly of all fingers and adducted thumbs. She exhibited laryngomalacia.
Due to the patient's multiple congenital malformations, numerous clinical investigations were performed at birth. Her brain MRI revealed a thin corpus callosum with a dysplastic right orbit. Orbital ultrasound confirmed the anophthalmic socket on the right side. On the left side, the lens was contiguous with the cornea and the anterior chamber was flat, explaining the vascularized scar covering most of her left cornea. She failed her newborn hearing screen and was later confirmed to have bilateral moderate to severe sensorineural hearing loss. Her echocardiogram showed a small atrial septal defect with right to left shunt and a mild tricuspid regurgitation. She developed dilated cardiomyopathy with a moderate decrease in left ventricular function around 6 months of age. Abdominal and spinal ultrasounds were unremarkable.
At 3 months of age, she was admitted for a generalized tonic clonic seizure and was initiated on phenobarbital. During this admission, due to her relatively coarse facial features, urine glycosaminoglycan and oligosaccharide testing were performed, and the results came back normal. Her creatine kinase (CK) level was initially high at 5000 U/L, but subsequently normalized.
The patient underwent several corrective repair surgeries for imperforate anus and had a corneal graft in her left eye and right eye prosthesis. With continuing feeding difficulty, she required a surgery for the placement of a gastrostomy tube.
At 1 year of age, she had a severe global developmental delay with absent head control and only occasional cooing. She exhibited progressive microcephaly and developed severe brachycephaly.
The proband passed away at 2 years of age at home due to her multiple comorbidities.
Subsequently, the couple had a second pregnancy, resulting in a healthy daughter with normal development assessed at 6 months of age. The parents have provided consent for the publication of clinical and laboratory data.
3. Investigations
Single nucleotide polymorphism (SNP) array analysis was performed on the proband at birth in 2016 using a custom-designed Illumina SNP array at CombiMatrix/Invitae Corporation (Irvine, CA 92618), and the results were described relative to the reference genome (GRCh37/hg19). FISH analysis was performed at CombiMatrix/Invitae Corporation (Irvine, CA 92618) using probes RP11-60A11 (2p25.3), RP11-94I20 (2q37.1), RP11-526L8 (proximal 2q37.3) and RP11-875C22 (distal 2q37.3). Mate pair sequencing (MPSeq) was performed at the Mayo Clinic (Rochester, MN) as described previously [18, 19]. MPSeq data were analyzed with SVA tools for the detection of structural rearrangements or copy number variants relative to the reference genome (GRCh38/hg38) and visualized using in-house developed software. The breakpoints and junctions were reviewed for gene interruption and/or gene fusions using UCSC Genome Browser (GRCh38/hg38) [20], and CNVs were curated and analyzed for the number of genes, disease association, inheritance pattern, and corresponding triplosensitivity and haploinsufficiency scores using DECIPHER (DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources), ClinVar, OMIM (Online Mendelian Inheritance in Man), DGV (Database of Genomic Variants), and ClinGen Dosage Sensitivity Curation databases [21–25].
4. Results
SNP microarray analysis identified a total of four copy number variants (CNVs) with a total of 5 breakpoints on chromosome 2: the two duplications that encompass the 2p25.3-p25.1 region (10.3 megabases (Mb) in size) and the 2q35-q37.2 (17.5 Mb) region and the two deletions which encompass the 2q37.2-q37.3 (3.9 Mb) region and the 2q37.3 (0.81 Mb) region. There was an intact region of approximately 1.6 Mb in size between the two deletions (Figure 1(a)). Metaphase FISH analysis confirmed these CNVs and showed that the 2p25.3 duplication is translocated to the telomeric region of 2q (Figure 1(b)). MPSeq analysis confirmed this translocation and showed that the duplication in the 2q35-q37.2 region was a tandem inverted duplication. The analysis further identified the intact 2q37.3 region between the two deleted segments as inverted in orientation.
Parental FISH testing and microarray performed on the couple's second, clinically unaffected child was normal, supporting the de novo occurrence of this complex chromosomal abnormality in the proband.
5. Discussion
Constitutional chromothripsis is a rare event and mostly de novo in origin. Most reported cases have two or more chromosomes involved in the complex chromosomal rearrangement. To our knowledge, only a few patients have been reported with chromothripsis implicating a single chromosome.
Chromothriptic events involving chromosome 2 have been reported previously in two cases with fewer breakpoints and smaller CNVs resulting in milder phenotypes including growth retardation and intellectual disability [16, 26].
In this study, we utilized SNP array to detect CNVs and MPSeq to detect the balanced and unbalanced structural rearrangements at a higher breakpoint resolution in our patient with de novo complex rearrangement of chromosome 2.
The five breakpoints on chromosome 2 resulted in two inverted duplications, two deletions, and one inversion (Figure 2(b)). To interpret this complex structural rearrangement in the context of this patient's severe phenotype, we closely examined and reviewed each breakpoint and CNV in DECIPHER, ClinVar, OMIM, DGV, and ClinGen databases.
The two duplicated regions harbored 187 protein coding genes in total, 171 of which were OMIM genes and 61 were OMIM Morbid genes. None of the duplicated genes in these regions were found to be associated with triplosensitivity, in which an additional copy of a gene produces a phenotype. The two deleted regions contained 41 protein coding genes in total, 31 of which were OMIM genes and 9 were OMIM Morbid genes. None of these genes have established haploinsufficiency, in which loss of one copy of a gene could be responsible for the phenotype (Figure 2(c)). One of the deleted regions in our patient involves the chromosome 2q37 deletion syndrome (OMIM # 600430) region.
Four breakpoints interrupted the intronic regions of AGAP1, USP37, FARP2, and RRM2 genes, none of which is associated with a known disease phenotype at the present time (Figure 2(a)). One of the breakpoints fell within the intergenic region between NDUFA10 and LOC150935 long noncoding RNA gene. No gene fusions were detected.
For the phenotypic characterization of our patient, previous reports of cases with complex chromosome 2 rearrangements and associated syndromes within the region were reviewed. A summary of the clinical presentation among patients with 2q37 syndrome (OMIM # 600430), and case reports on complex 2p25 duplication and 2q37 deletion [16, 17, 26] with a comparison to our patient phenotype, is presented in Supplementary Table 1.
While we found a significant overlap of features among patients with a combination of deletion and duplication in the region, our patient had a far more severe phenotype with some unique features like imperforate anus, supernumerary nipples, and severe eye deformities not previously described with chromosome 2-related syndromes. The cumulative effect of this complex chromosomal rearrangement with CNVs, involving over 500 genes including 228 protein-coding genes, is likely responsible for the severe phenotype in this patient.
In cases with complex chromosomal rearrangements, a clear genotype-phenotype correlation is difficult to establish, as there is a potential for alterations involving gene regulation or chromatin structure that can disturb the gene expression and contribute to abnormal development. A future direction for research is to study the role of chromatin structures in gene expression in cases with complex rearrangements.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Stephens P. J. Greenman C. D. Fu B. Massive genomic rearrangement acquired in a single catastrophic event during cancer development Cell 20111441274010.1016/j.cell.2010.11.0552-s 2.0-7865095966321215367 PMC 3065307 · doi ↗ · pubmed ↗
- 2Crasta K. Ganem N. J. Dagher R. DNA breaks and chromosome pulverization from errors in mitosis Nature 20124827383535810.1038/nature 108022-s 2.0-8485642490822258507 PMC 3271137 · doi ↗ · pubmed ↗
- 3Koltsova A. S. Pendina A. A. Efimova O. A. Chiryaeva O. G. Kuznetzova T. V. Baranov V. S. On the complexity of mechanisms and consequences of chromothripsis: an update Frontiers in Genetics 201910 p. 39310.3389/fgene.2019.003932-s 2.0-85067862054 PMC 650315031114609 · doi ↗ · pubmed ↗
- 4Nazaryan-Petersen L. Bjerregaard V. A. Nielsen F. C. Tommerup N. Tümer Z. Chromothripsis and DNA repair disorders Journal of Clinical Medicine 202093 p. 61310.3390/jcm 9030613 PMC 714111732106411 · doi ↗ · pubmed ↗
- 5Shoshani O. Brunner S. F. Yaeger R. Chromothripsis drives the evolution of gene amplification in cancer Nature 2021591784813714110.1038/s 41586-020-03064-z 33361815 PMC 7933129 · doi ↗ · pubmed ↗
- 6Zepeda-Mendoza C. J. Morton C. C. The iceberg under water: unexplored complexity of chromoanagenesis in congenital disorders The American Journal of Human Genetics 2019104456557710.1016/j.ajhg.2019.02.0242-s 2.0-8506311332330951674 PMC 6451730 · doi ↗ · pubmed ↗
- 7Genesio R. Fontana P. Mormile A. Constitutional chromothripsis involving the critical region of 9q 21.13 microdeletion syndrome Molecular Cytogenetics 201581 p. 9610.1186/s 13039-015-0199-32-s 2.0-84950269861 PMC 468385526689541 · doi ↗ · pubmed ↗
- 8Gudipati M. A. Waters E. Greene C. Stable transmission of complex chromosomal rearrangements involving chromosome 1q derived from constitutional chromoanagenesis Molecular Cytogenetics 2019121 p. 4310.1186/s 13039-019-0455-z PMC 682245431695749 · doi ↗ · pubmed ↗