Exposure to Gulf war illness-related chemicals exacerbates alcohol- induced liver damage in rodents
Anca Petrescu, Julie Venter, Dana D Danilenko, Daniela Medina, Stephanie Grant, Su Yeon An, Elaina Williams, Patrick Mireles, Matthew Tjahja, Sharon DeMorrow

TL;DR
Exposure to chemicals linked to Gulf War Illness worsens alcohol-related liver damage in mice, and reducing liver macrophages helps.
Contribution
This study shows that GWI-related chemicals increase susceptibility to alcohol-induced liver injury in rodents.
Findings
PER/PB exposure caused long-term liver damage and inflammation in mice.
Prior PER/PB exposure worsened alcohol-induced liver injury, including steatosis and fibrosis.
Macrophage depletion reduced the exacerbated liver damage in PER/PB-exposed mice.
Abstract
Gulf War Illness (GWI) describes a series of symptoms suffered by veterans of the Gulf war consisting of cognitive, neurological and gastrointestinal dysfunctions. Two chemicals associated with GWI are the insecticide permethrin (PER) and the nerve gas prophylactic pyridostigmine-bromide (PB). In this study we assessed the effects of PER and PB exposure on pathology and subsequent alcohol (EtOH)-induced liver injury, and the influence of a macrophage depletor, PLX3397, on EtOH-induced liver damage in PER/PB- treated mice. Male C57BL/6 mice were injected daily with vehicle or PER/PB for 10 days, followed by 4 months recovery, then treatment with PLX3397 and a chronic-plus-single-binge EtOH challenge for 10 days. PER/PB exposure resulted in the protracted increase in liver transaminases in the serum and induced chronic low-level microvesicular steatosis and inflammation in GWI vs Naïve…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
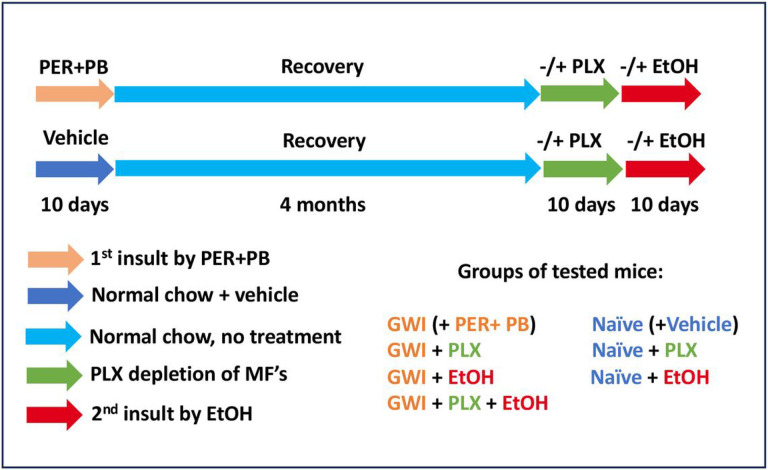 Figure 1
Figure 1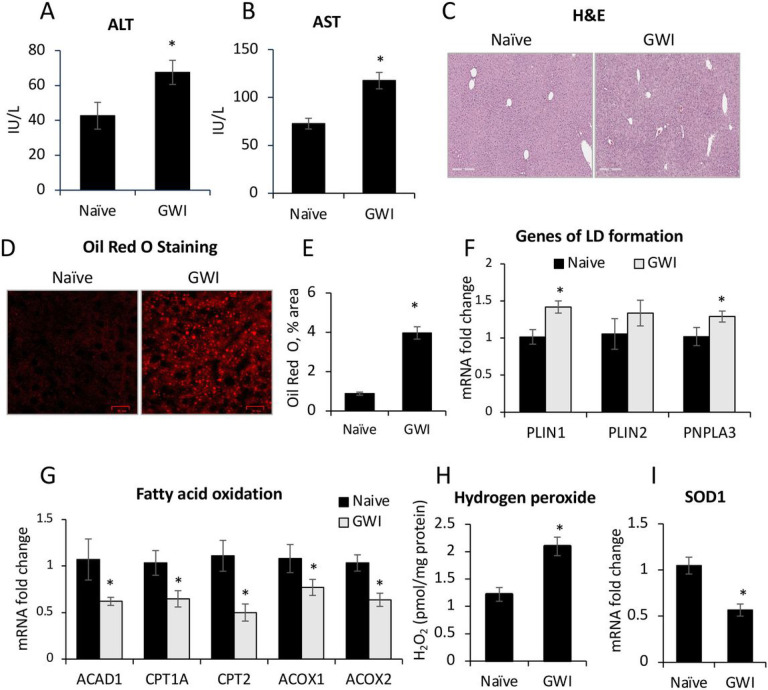 Figure 2
Figure 2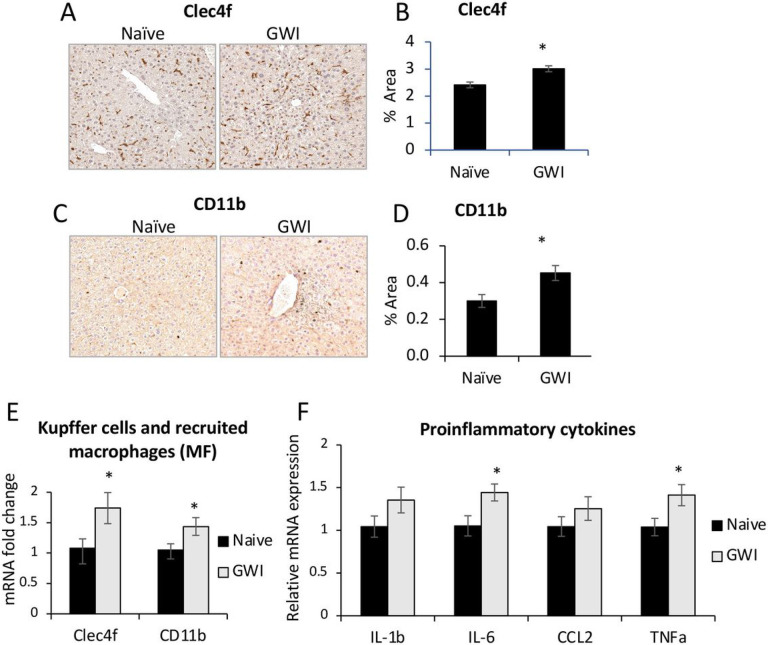 Figure 3
Figure 3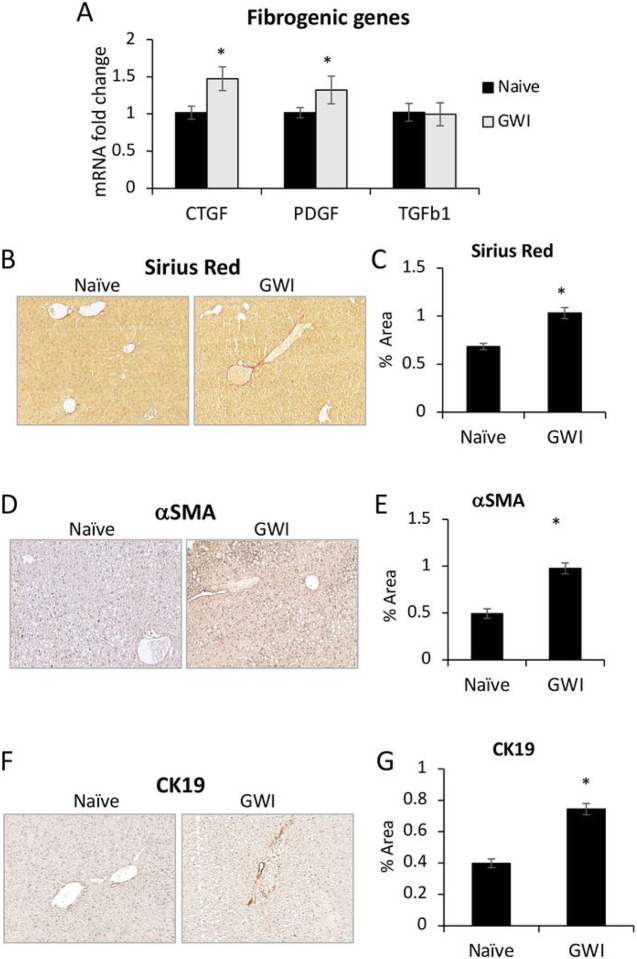 Figure 4
Figure 4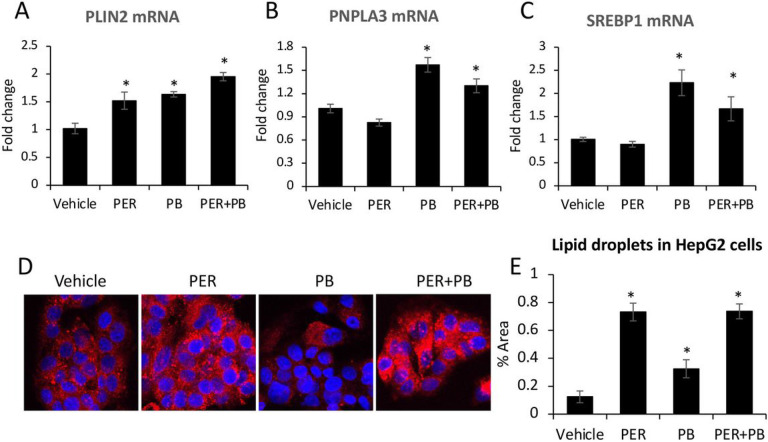 Figure 5
Figure 5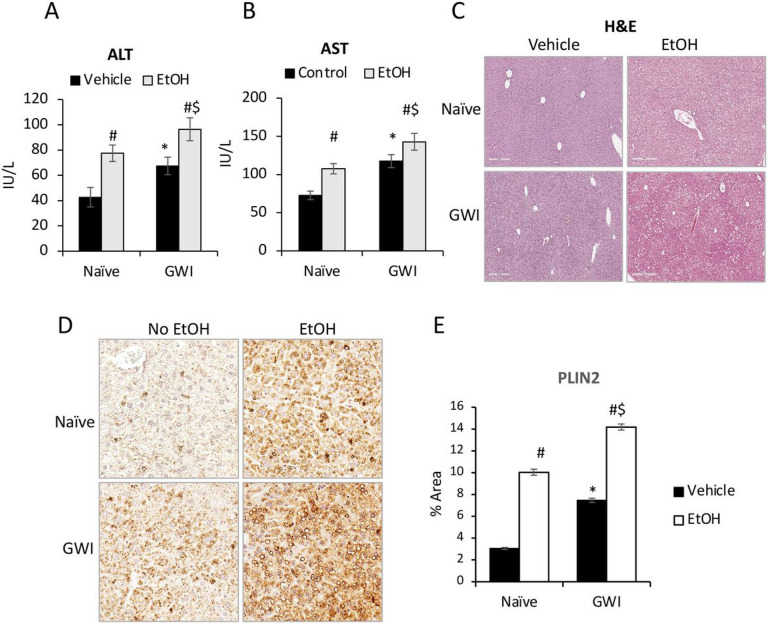 Figure 6
Figure 6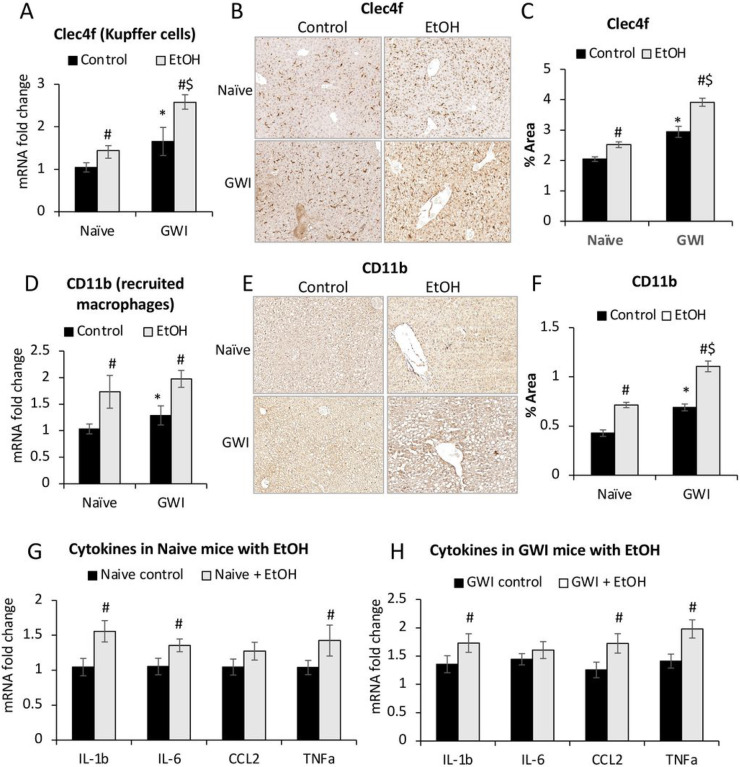 Figure 7
Figure 7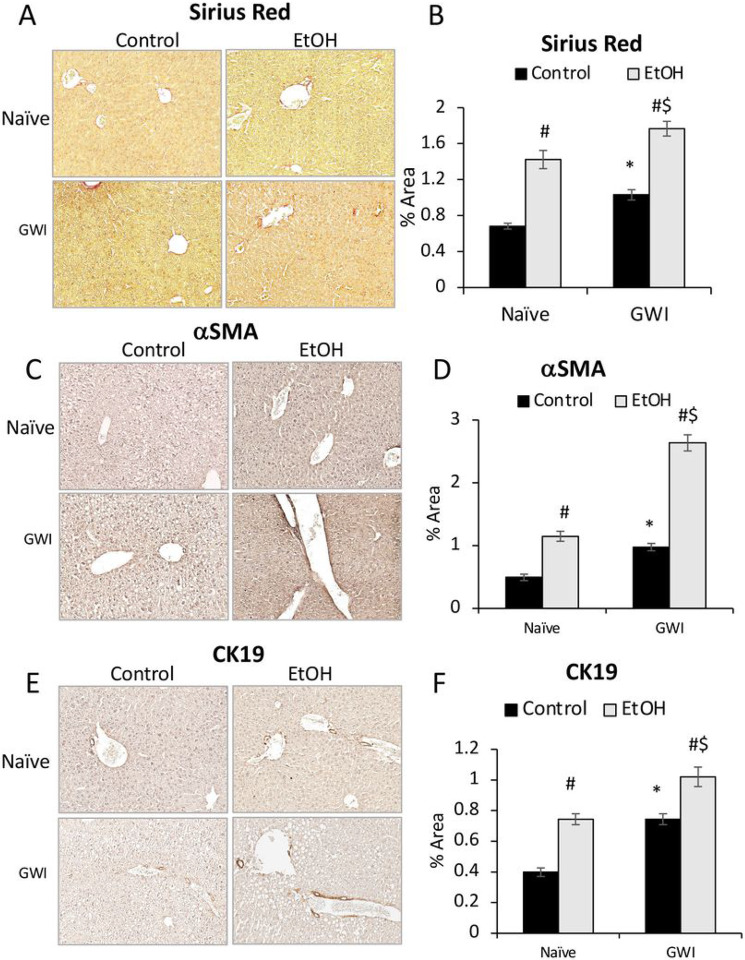 Figure 8
Figure 8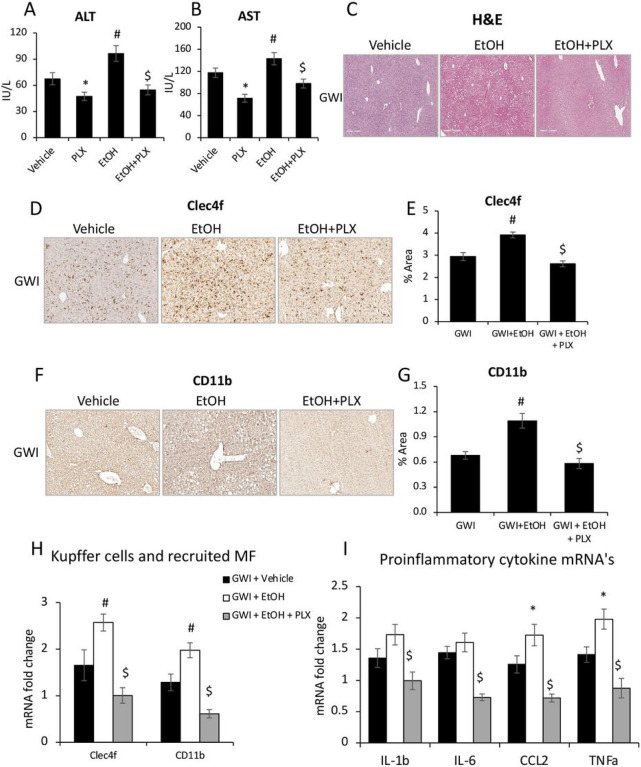 Figure 9
Figure 9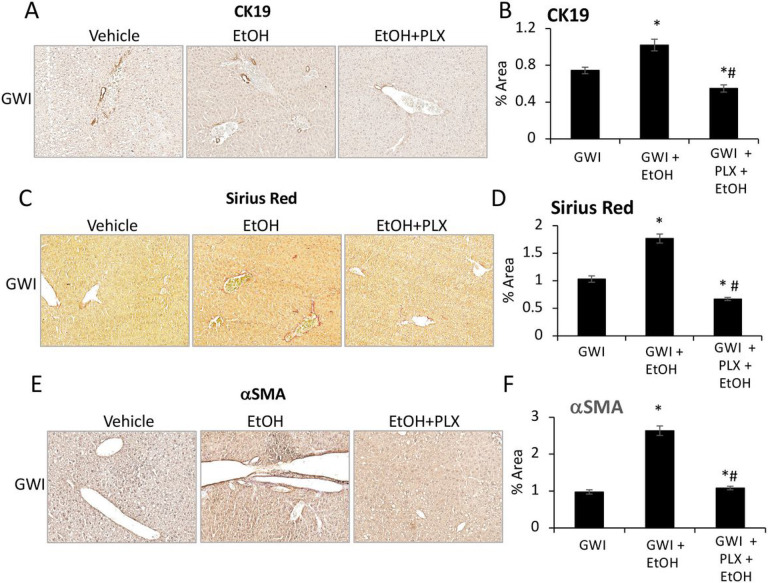 Figure 10
Figure 10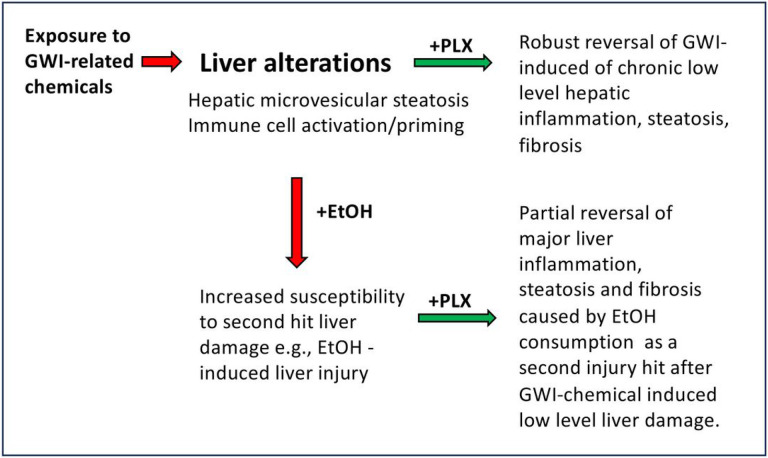 Figure 11
Figure 11Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAlcohol Consumption and Health Effects · Fibromyalgia and Chronic Fatigue Syndrome Research · Drug-Induced Hepatotoxicity and Protection
Introduction
Gulf War Illness (GWI) has been researched extensively not only due to its complexity, but also because more than 25% of the veterans deployed to the Persian Gulf during the 1990–1991 war, were affected by it^1^. GWI is defined as a multi-symptom syndrome characterized by neurological dysfunctions^2,3^, impaired memory and cognition^4^, chronic fatigue^5^, musculoskeletal pain^6,7^, gastrointestinal (GI) conditions^8^ and skin lesions^9^. These symptoms were associated with chronic low-grade increase of serum inflammation markers detected in Gulf War (GW) veterans^10-12^. The symptoms of GWI were characterized as being caused by various factors including dysregulation of the immune system and mitochondrial dysfunction^13^. Thus, increased systemic proinflammatory cytokines such as C-reactive protein and chemokine (C-C motif) ligand 2 (CCL2), were reported by clinical studies^14,15^, and more recently, certain positive acute phase proteins i.e. y-interferon-induced protein and serum amyloid A were associated with worse fatigue or pain in veterans meeting the GWI criteria ^16^. It was also demonstrated that veterans with GWI exhibited greater damage of mitochondrial DNA in peripheral blood mononuclear cells, as compared to healthy controls^13^.
Associated with the development of GWI is exposure to GW-related chemicals including pyridostigmine-bromide (PB), a prophylactic drug against nerve agents like sarin gas, and insecticides such as permethrin (PER) ^1,17^. While a direct association between GWI and a higher incidence of various liver diseases has not yet been suggested in patients, chronic exposure to low level amounts of these chemicals has previously been shown to cause mild toxicity and inflammation in the liver^18^ likely explained as a result of the xenobiotic metabolism of these agents in hepatocytes ^19^. It is conceivable, therefore, that these mild alterations in liver pathology as a result of exposure to GWI-related chemicals may alter the response and susceptibility to subsequent hepatotoxic insults. Indeed, we have previously demonstrated that exposure to GWI-related chemicals primed the hepatic proinflammatory response, and aggravated cholestasis-induced liver damage and fibrosis in rats^20^.
In the current study, we used a mouse model of GWI, to test the long-term effects of exposure to GWI-related chemicals on the susceptibility of the liver to subsequent chronic alcohol exposure. Furthermore, we used PLX3397 (Pexidartinib), an inhibitor of colony stimulating factor 1 receptor (CSF1R), to test the hypothesis that depletion of macrophages in the liver, could mitigate the liver injury caused by alcohol consumption in GWI-mice, as compared to negative controls (Naïve mice).
Results
GW-related chemicals PER and PB, increased serum transaminases and induced hepatic microvesicular steatosis in mice.
The model of GWI used in this study has previously been described and defined^21^ and was obtained by treating the mice with PER and PB for 10 days. In order to dissect the long term consequences of PER/PB treatment on the liver from the acute effects of PER/PB metabolism we allowed the mice to recover from PER/PB treatment for 4-months prior to the subsequent analysis of liver function as described under the Methods section. Mice treated with vehicle (Naive) on the same timeline as GWI-mice (Fig. 1), were used as negative controls. Serum transaminases alanine transaminase (ALT) and aspartate aminotransferase (AST) were slightly increased in GWI-mice as compared to Naïve controls (Fig. 2A, B), while the H&E histopathology test did not show differences between the two groups of mice (Fig. 2C), suggesting minimal overt liver damage due to PB/PER exposure itself. However, staining with Oil Red O demonstrated that livers of GWI-mice exhibited accumulation of fat in very small lipid droplets, a condition known as microvesicular steatosis, and image analysis indicated a significant increase in microvesicular steatosis in GWI-mice compared to Naïve controls (Fig. 2D, E). We assessed the expression of genes involved in lipid metabolism of the liver, and data indicated that the expression of perilipins 1 and 2 (PLIN1,2), which are structural proteins of lipid droplets, as well as the lipase PNPLA3, a marker of lipid droplets, were increased in GWI vs Naïve mice (Fig. 2F). Moreover, the expression of fatty acid oxidation genes encoding for acyl-coenzyme A dehydrogenase 1 (ACAD1), carnitine palmitoyltransferase 1A (CPT1A), (CPT2), acyl-CoA oxidase 1 (ACOX1), (ACOX2) were significantly reduced (Fig. 2G). These data suggested that GWI-related chemicals hindered lipid oxidation and induced lipid droplet formation in the liver. Furthermore, there was increased hydrogen peroxide and decreased superoxide dismutase 1 (SOD1) mRNA expression in the livers of GWI mice as compared to Naïve controls (Fig. 2H, I), suggesting that PER/PB exposure enhanced the level of reactive oxygen species (ROS) which are known to be associated with excess lipid accumulation in the liver^22^.
PER/PB caused a low-grade but long-lasting increase in hepatic inflammation.
We have previously demonstrated an increase in inflammatory cell accumulation in the liver of a rat model of GWI^20^, to determine if this observation holds true in our mouse model of GWI, the density of Kupffer cells and monocyte-derived macrophages in livers of GWI and Naïve mice was assessed by immunolabeling these cells with specific antibodies to Clec4f and CD11b, respectively (Fig. 3). Exposure to PB/PER resulted in a significant increase in both types of macrophages as shown by pictures and quantification data in Fig. 3A, B for Clec4f and in Fig. 3C, D for CD11b. The mRNA levels of Clec4f and CD11b were also augmented in livers of GWI vs Naïve mice (Fig. 3E). Furthermore, several proinflammatory cytokines including IL-1b, IL-6, CCL2, TNFa were assessed, and data indicated similar significant increases in their expression (Fig. 3F).
PER/PB induced low level, long-lasting fibrogenesis and ductular reaction in the liver.
We further investigated the effects of PER/PB on hepatic fibrosis. The expression of genes related to fibrogenesis, i.e., connective tissue growth factor (CTGF), platelet-derived growth factor-beta (PDGFb) and tumor growth factor beta 1 (TGFb1) was measured and found to be increased for CTGF and PDGFb in livers from GWI mice compared to Naïve controls (Fig. 4A). Sirius Red staining of collagen I and III showed a small but significant increase in these components of extracellular matrix (ECM) in GWI mice (Fig. 4B, C). A marker of hepatic stellate cell (HSC) activation is alpha-smooth muscle actin (aSMA) and it was shown to be increased in GWI mice vs Naïve mice by IHC staining of the liver sections (Fig. 4D, E).
Based on reported data^23^, hepatic fibrosis is frequently associated with ductular reaction, i.e., excess proliferation of cholangiocytes. Therefore, we measured the level of a cholangiocyte marker, cytokeratin 19 (CK19) and found that more CK19 was present in liver sections of GWI mice compared to Naïve controls (Fig. 4F, G).
The effects of PER/PB on HepG2 cells in vitro.
To test whether PER and PB exposure interferes with lipid metabolism in hepatocytes in vitro, we treated HepG2 cells with PER only, PB only, or a combination of both, as described under the Methods. The expression of lipid droplets-related genes such as PLIN2 and PNPLA3 at mRNA level, was assessed and the data indicated that: i) PLIN2 was upregulated by each substance individually, and even more by the combination of both PER/PB (Fig. 5A); ii) PNPLA3 was upregulated by PB only either individually or in combination with PER (Fig. 5B). To test the possibility that the lipids accumulated into hepatic lipid droplets were products of de novo synthesis of fatty acids, we measured the changes in mRNA of sterol regulatory element binding protein 1 (SREBP1), a transcription factor that upregulates genes of lipid synthesis^24^. As shown in Fig. 5C, PB only increased SREBP1 mRNA when administered alone or in combination with PER. These data suggested that PB stimulated de novo synthesis of lipids while PER upregulated proteins with role in lipid droplet formation.
The effect of PER, PB and their combination on lipid droplet formation in HepG2 cells was assessed by confocal microscopy imaging of Nile-Red-stained cells as described under the Methods (Fig. 5D, E). While PB had a small stimulatory effect on lipid accumulation into HepG2 cells, PER increased lipid droplets the most when alone or in combination with PB. Taken together, these data suggest that the effects of exposure to GWI related chemicals observed in vivo and likely due to the direct effects of these chemicals on the liver, rather than as an indirect result of their metabolism elsewhere in the body.
Pre-exposed to PER/PB exacerbated subsequent EtOH-induced liver injury.
To determine if these changes in the liver steatosis and inflammation due to exposure to GWI-related chemicals resulted in functional differences and susceptibility to a “second hit”, we treated Naïve and GWI-mice with EtOH or control diets for 10 days after 4 months of recovery from GW-related chemical exposure. The serum transaminases were increased due to EtOH consumption in both groups of Naïve and GWI-mice, however, there was a higher increase in EtOH-consuming GWI-mice compared EtOH-consuming Naïve mice (Fig. 6A, B). H&E staining of liver sections revealed that there was visible steatosis after EtOH treatment in both GWI- and Naïve-mice and (Fig. 6C). To quantify the level of steatosis, PLIN2, a protein associated with lipid droplets, was assessed by IHC in livers from Naïve and GWI mice that had been exposed to EtOH vs no EtOH (Fig. 6D, E). As shown by the IHC images and quantification of the immuno-stained percent area, the level of hepatic PLIN2 was dramatically increased in Naïve and GWI mice that consumed EtOH vs control mice. The amount of PLIN2 was more abundant in GWI mice vs Naïve mice that binged on EtOH.
Further measurements of hepatic inflammation markers were carried out to assess the effect of EtOH consumption on pro-inflammatory macrophages. The expression of mRNA and protein of Clec4f, which is specific to Kupffer cells, was found to be increased by EtOH in both groups of GWI- and Naïve-mice, and the highest level of expression was in GWI-mice that consumed EtOH (Fig. 7A, B, C). The CD11b marker of recruited monocytes-derived macrophages was also increased in EtOH-treated GWI- and Naïve-mice, with higher level in GWI-mice vs Naïve controls (Fig. 7D, E, F).
The effect of EtOH binging on proinflammatory cytokines in livers of Naïve (Fig. 7G) and GWI mice (Fig. 7H) was also tested and indicated that EtOH consumption caused increase in the expression of IL-1b, IL-6, CCL2 or TNFa in both groups of mice. However, some differences in mRNA levels, e.g., for TNFa and IL-1b, were higher in GWI mice compared to Naïve control mice. Moreover, while in the Naïve mice, EtOH binging did not change CCL2 mRNA levels; in GWI mice, there was a significant upregulation of CCL2 gene induced by EtOH consumption.
PER/PB exposure enhanced liver fibrosis and ductular reaction caused by EtOH consumption in mice.
The effects of EtOH on liver fibrosis in GWI-mice vs Naïve controls were assessed using Sirius Red staining of ECM proteins in liver sections and showed that EtOH induced significant increase in these proteins in livers from Naïve and GWI-mice, with a slightly larger expression in GWI-mice vs controls (Fig. 8A, B). The marker for activated HSC, aSMA, was also assessed and indicated that EtOH consumption resulted in upregulation of aSMA in both Naïve and GWI-groups, with a larger increase of aSMA in GWI-mice vs Naïve controls (Fig. 8C, D).
The effect of EtOH on intrahepatic bile duct mass (IBDM) was measured, and all EtOH-consuming mice exhibited increased IBDM as compared to mice that had not been treated with EtOH (Fig. 8E, F). However, the EtOH-treated mice which were pre-exposed to GWI-related chemicals had higher IBDM than Naïve mice.
PLX3397 treatment prior to EtOH consumption attenuated the EtOH-induced liver inflammation in mice pre-exposed to PER/PB.
We investigated the possibility of using PLX3397 (shortly, PLX), a drug that was previously shown to neutralize macrophage activity^25^, to prevent the EtOH-caused pro-inflammatory response in mice that were exposed to GWI-related chemicals. The following groups of GWI-mice were analyzed: i) GWI exposure alone; ii) GWI + EtOH were treated with ETOH in the absence of PLX; iii) GWI + PLX were treated with PLX only; and iv) GWI + EtOH + PLX were treated with PLX prior to EtOH binging as described in the Methods. The level of serum transaminases indicated that PLX had a beneficial effect on the liver of GWI-mice, significantly reducing ALT and AST in GWI mice that were not exposed to EtOH as well as in GWI-mice that consumed EtOH (Fig. 9A, B). The H&E staining showed increased steatosis and hepatocyte distress especially in GWI-mice that consumed EtOH, and this type of damage was not observed in GWI-mice that were treated with PLX prior to EtOH binging (Fig. 9C).
The measurements of Clec4f by IHC, indicated that the EtOH-induced increase in Kupffer cells was prevented in GWI-mice that received PLX prior to EtOH consumption (Fig. 9D, E). Similarly, PLX treatment prevented the EtOH-induced increase in recruited monocyte-derived macrophages (Fig. 9F, G). The expression of Clec4f and CD11b genes at mRNA level also suggested that PLX downregulated the transcription of these genes of liver macrophages (Fig. 9H). Additional data on the influence of PLX on the expression levels of Clec4f, CD68, F4/80 and CD11b mRNA’s in Naïve and GWI-mice treated with vehicle or PLX, are presented in supplementary Fig. 2A, indicating that except for F4/80 marker of macrophages, all the others were increased in GWI-mice and were reversed to normal or even to lower levels, by PLX. Moreover, Clec4f and CD68 mRNA’s were downregulated as result of PLX treatment of Naïve mice as well (Suppl. Figure 1A).
The assessment of mRNA’s encoding for pro-inflammatory cytokines such as IL-1b, IL-6, CCL2 and TNFa indicated that these were upregulated by EtOH in GWI mice and downregulated in livers of GWI mice that were treated with PLX prior to EtOH consumption (Fig. 9I). Additional data on the expression of genes encoding for these cytokines in Naïve mice treated with vehicle or PLX, are shown in supplementary Fig. 1B. These results indicated that PLX did not change the mRNA levels in Naïve mice, while the upregulated levels in livers from GWI mice were reduced to normal in GWI mice that received PLX (Suppl. Figure 1B). The effect of PLX treatment on CD68-expressing macrophages in Naïve and GWI mice, was also measured (Suppl. Figure 1C, D). The data indicated that the increase in CD68 in the liver, which was caused by PER/PB exposure of GWI-mice, was reversed by PLX treatment (Suppl. Figure 1C, D).
PLX3397 attenuated the EtOH-induced biliary hyperplasia and liver fibrosis in mice pre-exposed to PER/PB.
The assessment of IBDM using IHC for CK19 in liver sections, indicated that the increase in IBDM due EtOH, was prevented in GWI mice that were treated with PLX prior to consuming EtOH (Fig. 10A, B). Markers of fibrosis such as Sirius Red staining (Fig. 10C, D) and aSMA (Fig. 10E, F) which were elevated in GWI + EtOH mice vs GWI mice, were found to be lower in GWI + EtOH + PLX mice compared to GWI + EtOH mice. Moreover, Sirius Red staining indicated less ECM proteins in GWI + EtOH + PLX mice vs GWI-mice with no EtOH binging. The expression levels of genes with role in hepatic fibrogenesis, encoding for TGFb1 and CTGF were also determined (Suppl. Figure 2). Thus, Tgfb1 mRNA was not affected by GWI-related chemicals in the absence or presence of PLX, however it was highly upregulated as result of EtOH consumption in both groups of Naïve and GWI mice, with a larger increase in GWI mice that binged on EtOH. CTGF mRNA was upregulated in GWI vs Naïve mice, and ETOH binging enhanced this upregulation (Suppl. Figure 2). Treatment of GWI mice with PLX prior to EtOH, mitigated the effect of EtOH on expression of TGFb1 and CTGF.
Discussion
In this study, we used a mouse model of GWI to investigate several aspects of liver inflammation and injury in GWI vs Naïve mice. Thus, we addressed two main aspects: i) a possible negative influence of PER and PB, on the liver; ii) exposure to GWI substances could prime the liver to over-react to a second insult such as EtOH binging.
Our first hypothesis, that exposure to a combination of PER and PB could result in a chronic low-grade inflammation of the liver, was based on several reports which indicated that in rodent models of GWI as well as in veterans manifesting symptoms of this syndrome, it was found a long lasting, higher than normal level of pro-inflammatory cytokines in the serum^10,11,14^. The primary focus of many publications on GWI studies are on the effects of this systemic low-grade inflammation in the central nervous system (CNS), because the most prevalent symptoms of veterans suffering from GWI were related to neurological diseases^26-28^. However, more recent reports suggest that disorders of CNS, as well as dysregulation of peripheral nervous system that controls the GI organs, were associated with exposure to substances used in the GW^29^.
The hypothesis that exposure to PER and PB could prime the liver to become more vulnerable and suffer increased damage when subjected to a subsequent insult in the form of alcohol-induced liver damage, was based on our prior observations of cholestasis in a GWI model in rats. As previously described^20^, we demonstrated that the exposure of rats to PER and PB followed by a period of recovery had a significant impact, enhancing the level of hepatic inflammation and damage caused by bile duct ligation (BDL)-induced biliary cholestasis. Therefore, in the current study, we tested whether chronic low-grade inflammation caused by PER and PB could affect the process of liver healing after a secondary insult produced by alcohol binging. Indeed, the results showed significant increase in liver damage after alcohol binging in GWI-mice, as compared to Naïve mice, as indicated by more acute inflammation, steatosis, ER stress and fibrosis. When compared to the rat model, the mouse GWI model showed a slightly higher level of pro-inflammatory indicators after PER/PB treatment even without EtOH treatment. Thus, in these mice, pro-inflammatory cytokines IL-1b, IL-6 and TNFa remained upregulated in the livers of GWI-mice even after a long period of recovery post GW-chemical exposure, suggesting a priming of the liver for acute response in case of a subsequent injury. Further investigation of HSC activation, bile duct proliferation and fibrosis concluded that all these symptoms of liver damage, caused by alcohol binging, were more severe in mice that had been exposed to GW substances, compared to Naive mice. It is worth mentioning that alcohol binging by Naïve mice resulted also in steatosis, liver inflammation, ductular reaction and HSC activation, however, the levels of these were lower compared to GWI-mice.
Regarding possible toxic effects of PER on the liver and other organs, a review was published in 1994 (NCBI Bookshelf ID: NBK231573, National Research Council (US) Subcommittee to Review Permethrin Toxicity from Military Uniforms. Health Effects of Permethrin-Impregnated Army Battle-Dress Uniforms. Washington (DC): National Academies Press (US); 1994; https://www.ncbi.nlm.nih.gov/books/NBK231573/?report=printable ). The review found that the only organ affected by PER in animal toxicity trials was the liver. Thus, doses equal or larger than 100 mg/kg PER ingested by rats for 26 weeks resulted in increase in liver weight and hepatocellular hypertrophy, characterized by an enlarged endoplasmic reticulum associated with increased activity of cytochrome-P-450-enzymes. The review concluded that the daily exposure to PER-impregnated uniforms at a level of 6.8x10^−5^ mg/kg/day, the toxicity that might come from wearing PER-impregnated uniforms should not be a concern. However, numerous reports on clinical studies in the following years, pointed out that an increasing number of veterans, up to 25% of those who participated in the GW, were affected by GWI and received treatments for GWI symptoms^30^. Also, additional studies on cytotoxic effects of PER on mouse liver cells reported that PER caused severe damages including proliferation of Kupffer cells, decreased nuclear size and increased vacuoles in hepatocytes^31^. Since PER is the most used insecticide not only in the military but also in agriculture and population disease control, the studies on its safety have been run with considerable scrutiny. Thus, a study that addressed the possibility that PER could affect differently people depending on their age, concluded that at least in animal experiments, exposure to PER at early age had more signs of liver damage such as dysregulated redox system and increased lipid peroxidation, as compared to adult controls^32^. A risk of increased incidence of liver cancer due to long term exposure to PER was also reported, showing that chronic exposure to PER increased the incidents of hepatocellular adenoma in female mice at a concentration levels of 2500 ppm or higher as compared to controls^33^.
Our experiments revealed that a relatively short-term exposure of mice to PER and PB, resulted in hepatic microvesicular steatosis which was persistent even after a long period of recovery. Also, in vitro experiments showed that HepG2 cells exhibited a significant increase in lipid droplets when treated with PER alone or in combination with PB, while PB had no significant influence on fat accumulation in these cells. Interestingly, Yang et al^34^ reported that PER stimulated triglyceride synthesis and lipids droplets while suppressing lipid oxidation in palmitic acid-induced HepG2 cell steatosis model, which is in agreement with our findings.
In conclusion, our data on the effect of PER on the liver, and on HepG2 cells in vitro, are consistent with reports stating that PER-induced toxicity under in vivo and in vitro studies, is explained through PER metabolism in the liver, that generates oxidative stress and steatohepatitis^35^.
To test if the exacerbated response to alcohol-induced liver injury in mice previously exposed to GWI-related chemicals was due to the low level protracted inflammation observed in the liver, we used PLX3397 to deplete the GWI-associated build up of macrophages prior to exposure to alcohol. PLX3397 is an oral tyrosine kinase inhibitor of CSF1R in macrophages, that was able to attenuate the negative effects of inflammation in several chronic conditions including fatty liver, obesity, diabetes and even cancer in preclinical studies^36^. PLX3397 has been approved to be used in clinical trials for various types of cancer that required depletion of macrophages with role in tumorigenesis^37^. Moreover, previous studies found that PLX3397 was also an inhibitor of PDGFb receptor, acting effectively to stop cell proliferation^38^. Since the liver injury involves persistent inflammation and activation of HSC that express PDGFb receptors with role in fibrogenesis, we tested the efficacy of PLX3397 in reducing the alcohol-induced liver damage in GWI mice. As expected, PLX3397 lowered the density of Kupffer cells and monocyte-derived macrophages, as well as pro-inflammatory cytokines in liver samples from GWI-mice treated with vehicle or EtOH. Furthermore, the marker of activated HSC, aSMA, was reduced, which resulted in less fibrosis of the liver in GWI-mice when treated with PLX3397 during EtOH binging. One of the molecular mechanisms involved in liver damage is the ER stress, which can lead to cell death, via apoptosis and autophagy in hepatocytes^39^. Remarkably, the presence of lipid droplets in livers of GWI mice treated with PLX3397 prior to EtOH, was also reduced as compared to GWI mice that received the alcohol without PLX3397. These results suggest that macrophage-derived cytokines could contribute to changes in lipid metabolism and storage in hepatocytes under alcohol-induced stress and damage.
To test the possibility that PER, PB or the combination of both substances could have a direct effect on lipid metabolism in hepatocytes, we did experiments in vitro, using HepG2 cells. The results indicated that the combination of PER and PB was more effective than each of the two components alone, favoring lipid storage inside hepatocytes, via upregulation of SREBP1 gene of de novo synthesis of lipids, as well as structural (e.g., PLIN2) and functional (e.g., PNPLA3) proteins of lipid droplets.
In conclusion, as illustrated in Fig. 11, we have demonstrated that exposure of mice to PER and PB, was conducive to liver alterations such as microvesicular steatosis and low-grade chronic inflammation which primed the liver to react to a subsequent challenge caused by alcohol binging, that resulted in abnormal inflammation, biliary hyperplasia and fibrosis. Our study also showed that using PLX3397 to deplete macrophages during alcohol binging, mitigated the hepatic inflammation and decreased alcohol-induced liver damage. Taken together, while increased incidence of overt liver disease has not yet been associated with GWI, significant changes to liver physiology may be evident resulting in an increased susceptibility of Veterans with GWI to second insults to the liver such as alcohol-induced liver injury.
Methods
Chemicals, kits, tissue culture media, antibodies.
PER was purchased from Chem-Service Inc (West Chester, PA). PB came from Sigma-Aldrich, part of Millipore-Sigma in US (Burlington, MA; www.sigmaaldrige.com). Vectastain, DAB Substrate/peroxidase kits, hematoxylin QS were from Vector Lab (Burlingame, CA). AST, ALT Catalyst kits from IDEXX (Westbrook, ME). Oil Red O and Sirius Red staining kits were from Nova Ultra, IHC World (Ellicott City, MD). PLX3397 was from Thermo Fisher Scientific (Waltham, MA). Antigen-unmasking solution, Cytoseal XYL mounting reagent, Prolong Gold Anti Fade with DAPI mounting solution, cell culture media and solutions including high glucose-DMEM, fetal bovine serum (FBS) and penicillin/ streptomycin were from Gibco BRL purchased through Thermo Fisher Scientific (Waltham, MA). Primary antibodies were obtained from Abcam (Cambridge, MA) unless specified otherwise. The antibody to mouse C-type lectin in F4/80-positive cells (Clec4f) was from Invitrogen by Thermo Fisher (Rockford, IL). All primers used in RT-qPCR were ordered from Qiagen (Germantown, MD). All the other chemicals were purchased from Millipore-Sigma (Burlington, MA) unless otherwise stated, and were of the highest grade available.
Animal experiments
Adult, 8-week-old male C57BL6/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained in a temperature-controlled environment at 20-22°C with a light-dark cycle of 12:12 hour, having free access to food and drinking water. All animal procedures were performed in accord with protocols approved by the Institutional Animal Care and Use Committee of University of Texas at Austin (Austin, TX). The mice were randomly divided into two groups: Naïve mice, injected daily with vehicle (DMSO), and GWI-mice that were administered a mix of 100 mg/kg PER and 0.7 mg/kg PB for 10 days. This model has been established as a mouse model of GWI in other laboratories^21^. After 4 months of recovery, the two initial groups of mice were divided into the following groups: GWI and Naïve mice that were treated with 50 mg/kg PLX3397^40^ by gavage, every second day for 10 days; GWI and Naïve mice treated with vehicle as controls for PLX3397. The GWI and Naïve mice that received either PLX3397 or vehicle, were further treated with a chronic-plus-single-binge EtOH for 10 days, as described^41^. The mice were euthanized by intraperitoneal injection of at least 150 mg/Kg Euthasol (pentobarbital sodium plus phenytoin sodium euthanasia solution, purchased from Animal Health International, Loveland CO, USA) followed by a procedure of thoracotomy. Serum and liver samples were collected from all groups of mice. The timeline and experiment design are schematically shown in Fig. 1.
Assessment of mRNA expression by RT-qPCR
Total RNA was isolated from frozen sample of liver tissue using RNeasy kit from Qiagen (Germantown, MD), followed by cDNA synthesis with iScript kit from Bio-Rad Life Sciences (Hercules, CA), and RT-qPCR using iTaq Universal SYBR-Green Supermix from the same company. RT^2^ qPCR Primer Assays and primers for all our RT-qPCR assays were ordered online from https://geneglobe.qiagen.com/us. The RT-qPCR assays were run using CFX96 C1000 Touch Thermal cycler and Bio-Rad CFX Maestro software from Bio-Rad (Hercules, CA). The data was analyzed as previously described^20^. Fold changes in gene expression were normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Serum biochemistry
Aspartate- and alanine-transaminase (AST, ALT) assays were performed using Catalyst One Analyzer from IDEXX Laboratories (Westbrook, ME).
Immunohistochemistry (IHC)
For IHC, liver paraffin sections of 4 μm were immunolabeled with primary antibodies specific to proteins of interest, and stained using VectaStain kits and counterstained with hematoxylin as previously described^23^. The IHC slides were scanned with a Leica SCN400 scanner at 20X magnification, followed by screenshots at 10X magnification. The percent stained area was measured by image analysis which was carried out using the 1.52a version of ImageJ software, as downloaded from the NIH website (http://imageJ.nih.gov/ij ).
Fluorescence labeling and confocal microscopy of lipid droplets and apoptosis marker
Lipid droplets (LD) were stained with Oil Red O in frozen liver sections from mice. Thus, frozen liver tissue embedded in OCT medium was sectioned at 8μm using Leica Cryostat CM1850 (Leica Biosystems, www2.leicabiosystems.com), and the sections were stained using the Oil Red O kit from IHC World (Ellicott City, MD). Images of Oil-Red-stained LD were taken using a confocal laser scanning system from Leica Microsystems Inc. (Buffalo Grove, IL). The percent stained area was measured by image analysis as described for IHC. In HepG2 cells in vitro, LD were stained with Nile Red as described^42^ and quantified using the ImageJ software. Additionally, specific staining for apoptosis, was performed on HepG2 cells, using the Apoptosis/Necrosis assay kit from Abcam (Cambridge, MA).
Liver histopathology, ductular reaction and fibrosis
Hematoxylin and eosin (H&E) staining was performed on 4 μm sections of paraffin-embedded livers, as previously described^20^. Expression LD-associated genes encoding for perilipin 2 (PLIN2) and patatin-like phospholipase domain-containing 3 (PNPLA3) were measured by RT-qPCR. Biliary hyperplasia was assessed measuring expression of cytokeratin 19 (CK19) marker of cholangiocytes at mRNA and protein level, using RT-qPCR and immunohistochemistry (IHC) staining, respectively. The expression of fibrosis-related genes such as connective tissue growth factor (CTGF), platelet-derived growth factor beta (PDGF b) and transforming growth factor beta-1 (TGF-b1) was assessed by RT-qPCR of liver samples. Collagen 1A1 (Col1A1) and other extracellular matrix proteins associated with fibrosis were measured using Sirius Red staining. All these methods were applied according to the manufacturers’ instructions unless otherwise specified.
Assessment of hepatic inflammation
Changes in density of Kupffer cells and monocyte-derived macrophages recruited to the liver were detected by RT-qPCR and/or IHC for Clec4f and CD11b, respectively. Hepatic inflammatory cytokines interleukin-1 beta (IL-1b), interleukin-6 (IL-6), C-C motif chemokine ligand 2 (CCL2), tumor necrosis factor-alpha (TNF-a) were assayed by RT-qPCR and ELISA.
Statistics
Quantifications of data from RT-qPCR, ELISA or image analyses were done calculating the average and standard error of the mean (SEM) of three replicates for each group of tested animals. For in vivo experiments, the number of animals (N) in each treatment group was 4, or otherwise specified in the Results section for each experiment. The statistical differences between two groups, was calculated by Student’s T-test, and were marked as significant when the p value was less than 0.05.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Elhaj R. & Reynolds J. M. Chemical exposures and suspected impact on Gulf War Veterans. Mil Med Res 10, 11, doi:10.1186/s 40779-023-00449-9 (2023).36882803 PMC 9993698 · doi ↗ · pubmed ↗
- 2Engdahl B. E. Brain Function in Gulf War Illness (GWI) and Associated Mental Health Comorbidities. J Neurol Neuromedicine 3, 24–34 (2018).30882065 PMC 6417922 · pubmed ↗
- 3Engdahl B. E. A Magnetoencephalographic (MEG) Study of Gulf War Illness (GWI). E Bio Medicine 12, 127–132, doi:10.1016/j.ebiom.2016.08.030 (2016).27592598 PMC 5078573 · doi ↗ · pubmed ↗
- 4Toomey R. Neuropsychological functioning of U.S. Gulf War veterans 10 years after the war. J Int Neuropsychol Soc 15, 717–729, doi:10.1017/S 1355617709990294 (2009).19640317 · doi ↗ · pubmed ↗
- 5Kang H. K., Li B., Mahan C. M., Eisen S. A. & Engel C. C. Health of US veterans of 1991 Gulf War: a follow-up survey in 10 years. J Occup Environ Med 51, 401–410, doi:10.1097/JOM.0b 013e 3181 a 2feeb (2009).19322107 · doi ↗ · pubmed ↗
- 6Cook D. B., Stegner A. J. & Ellingson L. D. Exercise alters pain sensitivity in Gulf War veterans with chronic musculoskeletal pain. J Pain 11, 764–772, doi:10.1016/j.jpain.2009.11.010 (2010).20338824 · doi ↗ · pubmed ↗
- 7Lindheimer J. B. Influence of pain anticipation on brain activity and pain perception in Gulf War Veterans with chronic musculoskeletal pain. Psychophysiology 56, e 13452, doi:10.1111/psyp.13452 (2019).31429944 PMC 6803046 · doi ↗ · pubmed ↗
- 8Zhou Q., Verne M. L., Zhang B. & Verne G. N. Evidence for Somatic Hypersensitivity in Veterans With Gulf War Illness and Gastrointestinal Symptoms. Clin J Pain 34, 944–949, doi:10.1097/AJP.0000000000000611 (2018).29570102 PMC 6110965 · doi ↗ · pubmed ↗