Genome-wide screens connect HD82 loss-of-function to purine analog resistance in African trypanosomes
Anna Trenaman, Michele Tinti, Abdelmadjid Atrih, David Horn

TL;DR
Researchers found that HD82, a protein in African trypanosomes, helps these parasites resist purine analog drugs by managing nucleotide levels, offering new insights into drug resistance.
Contribution
The study identifies HD82 as a novel factor in purine analog resistance in trypanosomes, linking it to nucleotide metabolism.
Findings
HD82 knockdown increases resistance to adenosine analog ara-A in trypanosomes.
HD82 is a dNTP triphosphohydrolase that preferentially breaks down dATP.
Genome-wide screens identified Tb927.6.2800 and HD82 as key players in nucleoside analog resistance.
Abstract
Nucleoside analogs have been used extensively as anti-infective agents, particularly against viral infections, and have long been considered promising anti-parasitic agents. These pro-drugs are metabolized by host-cell, viral, or parasite enzymes prior to incorporation into DNA, thereby inhibiting DNA replication. Here, we report genes that sensitize African trypanosomes to nucleoside analogs, including the guanosine analog, ganciclovir. We applied ganciclovir selective pressure to a trypanosome genome-wide knockdown library, which yielded nucleoside mono- and diphosphate kinases as hits, validating the approach. The two most dominant hits to emerge, however, were Tb927.6.2800 and Tb927.6.2900, which both encode nuclear proteins; the latter of which is HD82, a SAMHD1-related protein and a putative dNTP triphosphohydrolase. We independently confirmed that HD82, which is conserved among…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
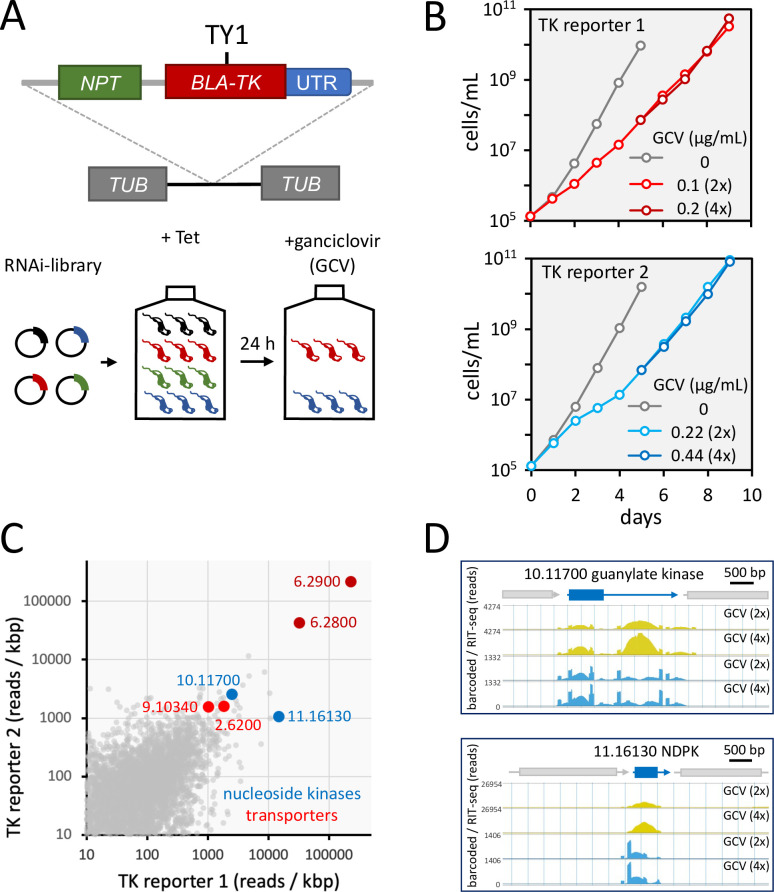 Fig 1
Fig 1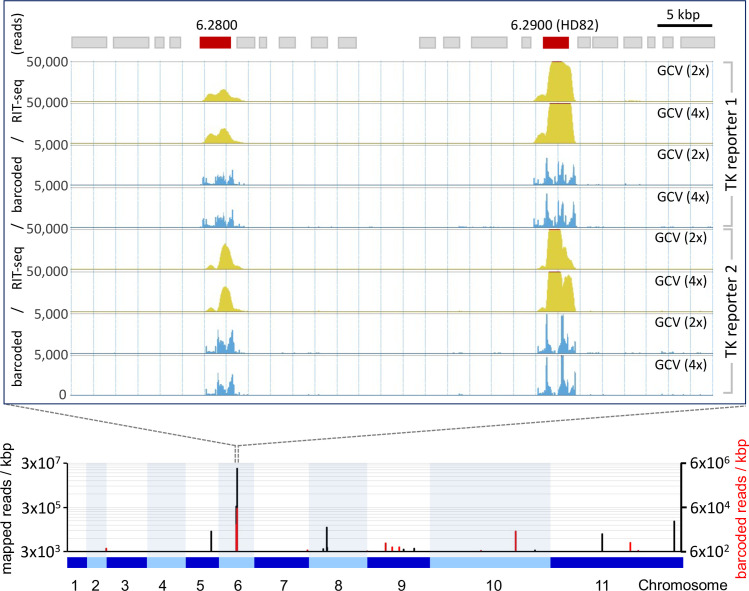 Fig 2
Fig 2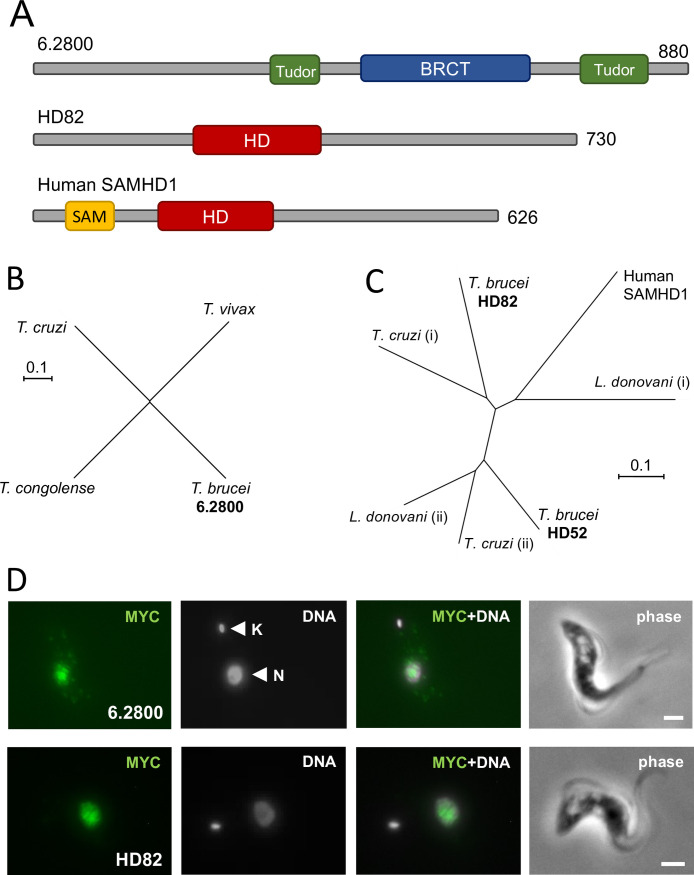 Fig 3
Fig 3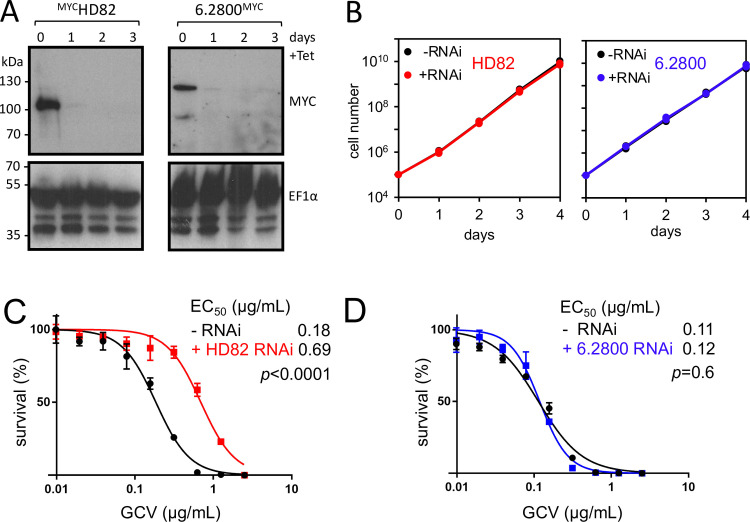 Fig 4
Fig 4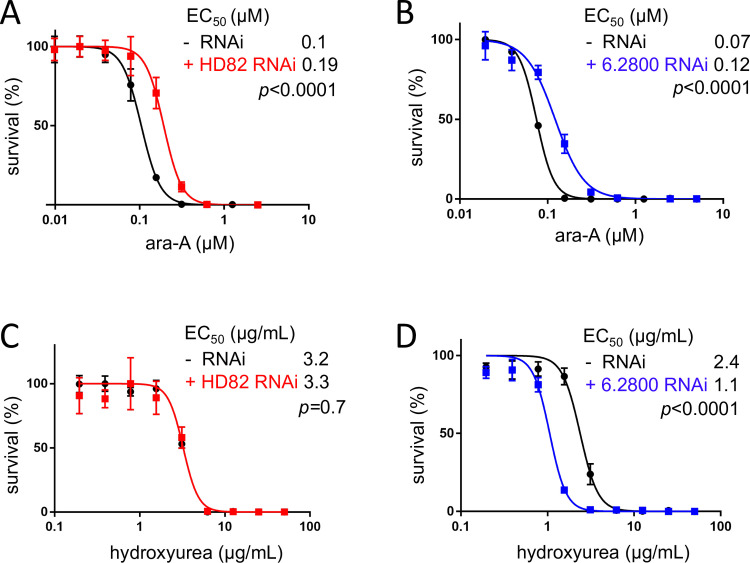 Fig 5
Fig 5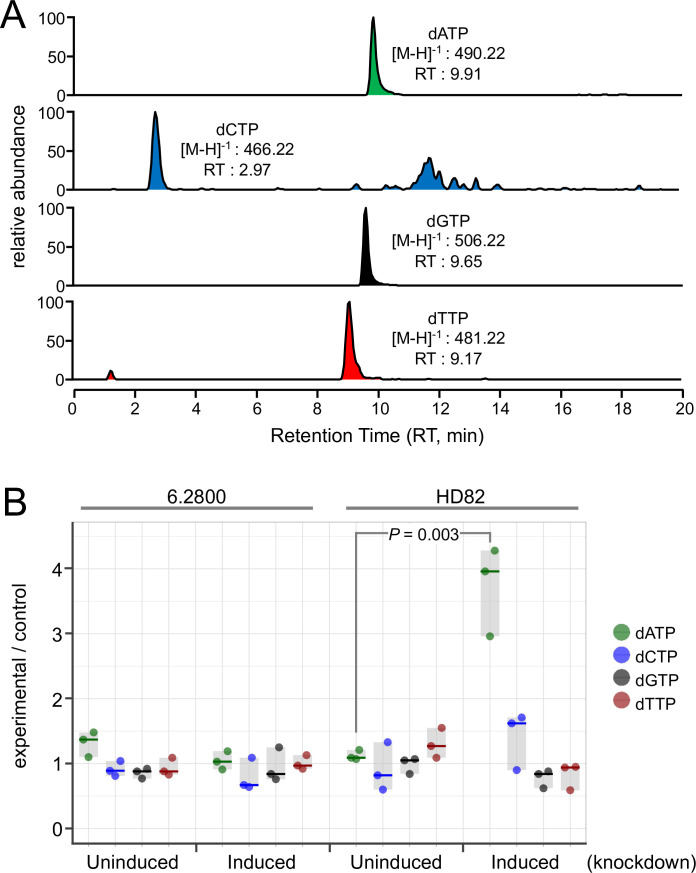 Fig 6
Fig 6- —Wellcome Trust (WT)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTrypanosoma species research and implications · Biochemical and Molecular Research · HIV/AIDS drug development and treatment
INTRODUCTION
The African trypanosome, Trypanosoma brucei, is a protozoan parasite that causes devastating diseases in both humans and animals, while related trypanosomatids cause other devastating and neglected tropical diseases (1). There has been substantial recent interest in developing nucleoside analogs as anti-trypanosomal therapeutics (2–8), specifically because trypanosomatids, unlike their mammalian hosts, lack the capacity for de novo purine biosynthesis (5). Consequently, trypanosomatids are purine auxotrophs that, being incapable of de novo purine synthesis, salvage purines from their hosts, for the biosynthesis of nucleic acids.
Nucleoside analogs have been used extensively as anti-infective agents, particularly against viral infections (9), with examples including aciclovir, for the treatment of herpes simplex virus (HSV) infections, ganciclovir (GCV) for the treatment of cytomegalovirus infections, and the “ProTide” drug remdesivir, for the treatment of COVID infections. For the reasons above and because trypanosomatids have efficient mechanisms for nucleoside uptake (10), such analogs are considered promising anti-trypanosomal agents. Indeed, the purine analog, allopurinol, continues to be widely used to treat canine leishmaniasis (11). Pathogenesis in human leishmaniasis can also be enhanced by a leishmania virus such that purine analogs that target the virus could yield therapeutic benefit (12).
Another application of purine analogs in T. brucei is for negative selection, in combination with expression of herpes simplex virus thymidine kinase (HSV-TK), which effectively uses both pyrimidines and purines as substrates. Such negative selection has been used to investigate mutagenesis (13), glycosome biogenesis (14), and antigenic variation (15) and for Cre-Lox-based conditional null strategies (16). Ganciclovir is typically used as the HSV-TK substrate and pro-drug (17), and HSV-TK substantially increases sensitivity to ganciclovir when expressed in T. brucei (13). T. brucei thymidine kinase cannot be used for negative selection as it is essential for viability and is involved in both salvage and de novo synthesis (18, 19); it also fails to sensitize cells to ganciclovir, possibly due to low-level expression (20).
Here, we use genome-wide RNA interference (RNAi) library screens to identify T. brucei genes that sensitize cells to nucleoside analogs. A primary hit is HD82, a nuclear HD domain protein related to SAMHD1, a human viral restriction factor and triphosphohydrolase. Indeed, T. brucei HD82 also appears to be a triphosphohydrolase with dATP as the preferred substrate.
RESULTS
A genome-wide T. brucei screen for ganciclovir-sensitizing genes
Trypanosoma brucei RNAi target sequencing (RIT-seq) screens have been used to identify several drug resistance mechanisms or to identify regulatory factors, using positive selectable marker genes as reporters (1). No such screen has been reported using negative selection, however, with cells expressing a protein that sensitizes cells to a drug. We established a RIT-seq screening assay using a negative selectable marker to identify mechanisms of resistance to the nucleoside analog, GCV. T. brucei are sensitized to GCV by expression of the HSV-TK. HSV-TK phosphorylates this thymidine analog (21) which, in its tri-phosphorylated state, acts as a competitive inhibitor of deoxyguanosine triphosphate (dGTP), thereby inhibiting DNA replication. Our screens had the potential to identify other genes that sensitize T. brucei to GCV and to other nucleoside analogs. Since we used HSV-TK reporters with 3′ untranslated regions from either aldolase (TK reporter 1) or variant surface glycoprotein (VSG; TK reporter 2) genes (Fig. 1A), our screens also had the potential to identify loss of positive control operating via these distinct 3′ untranslated regions.
A genome-wide RIT-seq screen using an HSV-TK negative selectable marker. (A) Schematic of the reporter construct that was integrated within a T. brucei tubulin gene array. The reporter comprises a blasticidin deaminase gene (BLA) fused to a herpes simplex virus thymidine kinase gene with a centrally located TY1 epitope tag. The neomycin phosphotransferase gene facilitated recombinant clone selection without applying blasticidin selective pressure. A genome-wide RNAi plasmid library was transfected into the reporter strains, and RIT-seq screens were carried out using GCV selection. (B) The cumulative T. brucei library population was monitored over 9 days using reporters with two distinct 3′ untranslated regions: (i) aldolase and (ii) VSG. Tetracycline was added at day 0 to induce knockdown. GCV selection was added at day 1 and the respective effective concentration (EC50) was increased from 2× to 4× to generate additional parallel cultures on day 5. Genomic DNA was harvested on day 9. (C) RIT-seq read mapping data are compared for all T. brucei genes and for both reporters; data from selection with 2× GCV EC50 are shown. Notable hits are highlighted. (D) RIT-seq read mapping is shown for two nucleoside kinase hits and flanking genes. Total reads (yellow) and reads encompassing the barcode sequence from the RNAi library cassette (blue) are shown; these data are from the screen with reporter 1.
We established reporter strains expressing a BLA : HSV-TK fusion protein with 3′ untranslated regions from aldolase or VSG. BLA : HSV-TK expression was confirmed by protein blotting (Fig. S1A), while dose response assays revealed GCV half maximal EC_50_ values of 0.05 and 0.11 µg/mL, respectively (Fig. S1B and C). Both strains were used to assemble T. brucei RNAi libraries (Fig. 1A), as described (22). RNAi-based knockdown was induced in each library using tetracycline for 24 h prior to GCV-selection (Fig. 1A) and at two times the respective EC_50_ values. We observed an initial delay in growth under GCV selection, as expected, followed by an increased growth rate, in both cases (Fig. 1B). After 4 days of GCV selection, cultures were split and subjected to continued GCV selection at two or four times the EC_50_. We observed little difference between cultures selected at two or four times the EC_50_ over a further 4 days (Fig. 1B). Parasites were harvested for genomic DNA extraction from all four GCV-selected cultures at this point. We PCR amplified RNAi target fragments and deep sequenced the amplicons. Sequence reads were mapped to the T. brucei reference genome, and reads were quantified for each gene (Fig. 1C).
GCV must be phosphorylated in a three-step process to generate the active ganciclovir triphosphate (GCV-TP). HSV-TK is responsible for producing the monophosphate, guanylate kinase is responsible for producing the diphosphate, and nucleoside diphosphate kinase is responsible for producing the triphosphate. An analysis of the top hits (Fig. 1C; Data file S1) revealed guanylate kinase (Tb927.10.11700) and nucleoside diphosphate kinase [Tb927.11.16130 (23)], consistent with GCV-activation through multiple phosphorylation steps (Fig. 1D). Notably, genes that failed to register as hits in our screens included hits from prior RNAi screens using adenosine analogs for selection (10, 24): adenosine kinase (Tb927.6.2300), 4E-interacting protein (Tb927.9.11050), and a nucleoside transporter (Tb927.5.286b), all consistent with distinct activation and potentially also transport mechanisms for adenosine and guanosine analogs. Another nucleoside transporter (Tb927.2.6200) and a polyamine transporter (Tb927.9.10340), however, did feature as hits in our screens (Fig. 1C); these transporters may impact intracellular dNTP pools or GCV uptake. Taken together, these hits provided excellent validation for our screening approach.
The top ganciclovir-sensitizing hits are nuclear HD82 and 6.2800
The two most dominant hits to emerge from our screens were “6.2800” and “6.2900” (Fig. 1C; Fig. 2). From the four experiments analyzed, we mapped an average of 2.5 ± 0.6 million RIT-seq reads to genes, with 6% ± 2% and 23% ± 5% of these mapped to 6.2800 and 6.2900, respectively. These two genes are within 28.3 kbp of each other, in the same polycistronic transcription unit on chromosome 6 (Fig. 2), although this does not necessarily reflect clustering of genes with related function; this polycistron incorporates approx. 120 putative protein-coding genes, with genes between the hits encoding an ATPase, a protein kinase, and a kinesin. Figure 2 depicts the relevant portion of the polycistronic unit and shows RIT-seq read mapping profiles from all four GCV-selected cultures. Since “barcoded” reads indicate the termini of RIT-seq fragments, we concluded that several RIT-seq fragments, from independent, multi-genome coverage T. brucei RNAi libraries, specifically and reproducibly identify these top two hits.
Two prominent GCV-sensitizing hits emerge from the RIT-seq screens. RIT-seq read mapping is shown for a 60-kbp region incorporating both the top two hits on chromosome 6. Total reads (yellow) and barcoded reads (blue) are shown; data from both reporter 1 and reporter 2 screens are shown. The lower panel is a genome-wide chromosomal map showing the barcoded reads over the 11 T. brucei megabase chromosomes, incorporating >7,400 genes. Total reads (black) and barcoded reads (red); data from the screen with reporter 1 and selection at 2× GCV EC50.
While 6.2800 is uncharacterized and annotated as a “conserved hypothetical protein,” 6.2900 has been designated as HD82, an HD domain protein with a predicted mass of 82 kDa (25, 26). HD domains, named after the conserved histidine (H) and/or aspartate (D) residues, which are thought to coordinate divalent cations, are typically found in predicted dNTP phosphohydrolases (27). Examples include the human immunodeficiency virus restriction factor, SAMHD1 (28, 29) and T. brucei HD52 (Tb927.7.4810), which is a putative mitochondrial phosphohydrolase that controls pyrimidine homeostasis (26). The domain structures of 6.2800 and HD82, relative to SAMHD1, are depicted in Fig. 3A. The predicted mass of 6.2800 is 97 kDa, with two putative Tudor domains and a BRCT (BRCA1 C Terminus) domain. Tudor domains are often found in histone methylation “readers” (30), while BRCT domains are predominantly found in proteins involved in the DNA damage response (31). No additional domains were identified in HD82, beyond the HD domain. Indeed, HD82 differs from SAMHD1 in that it lacks a predicted SAM (sterile alpha motif), a protein interaction module.
The top two hits HD82 and 6.2800 are nuclear proteins. (A) The schematic maps show protein domains identified in 6.2800, HD82, and human SAMHD1. (B) The unrooted phylogenetic tree shows T. brucei 6.2800-related proteins in the trypanosomes. T. congolense, TcIL3000_0_08110; T. vivax, TvY486_0602280; T. cruzi, TcCLB.511501.10. (C) The unrooted phylogenetic tree shows human SAMHD1 (NP_056289.2) and T. brucei HD82 and HD52 (Tb927.7.4810)-related proteins in the trypanosomatids; T. cruzi, TcCLB.507021.70 (i) and TcCLB.508579.10 (ii); Leishmania donovani, LdBPK_322680.1 (i) and LdBPK_060160.1 (ii). (D) Immunofluorescence microscopy localizes 6.280012MYC or HD8212MYC to the T. brucei nucleus. DNA is stained with DAPI. K, kinetoplast; N, nucleus; scale bars, 2 µm.
A phylogenetic analysis revealed 6.2800 orthologs in other African trypanosomes and in the American trypanosome, Trypanosoma cruzi (Fig. 3B), but only a substantially more divergent protein in the other trypanosomatid, Leishmania (E = 2^−6^). HD82, and HD52, on the other hand, have conserved orthologs in T. cruzi and in Leishmania (Fig. 3C). To determine subcellular protein localization in T. brucei, we fused c-MYC epitope tags to one allele of each of these genes: at the N-terminus for HD82 and at the C-terminus for 6.2800. Both proteins migrated in gels as expected based on their predicted molecular mass (see below), and both proteins presented a punctate nuclear localization by immunofluorescence microscopy (Fig. 3D). Notably, SAMHD1 is also a nuclear-localized protein (32). Neither protein appeared to display major differences in abundance or localization during the cell cycle (Fig. S2).
HD82 and 6.2800 sensitize T. brucei to purine analogs
To investigate GCV sensitization by HD82 and 6.2800 in more detail, we first generated inducible knockdown strains for each gene in a reporter 2 background. We also fused c-MYC epitope tags to one allele of each gene in these respective strains, to facilitate monitoring of knockdown. Efficient and sustained inducible knockdown was demonstrated by protein blotting in both cases (Fig. 4A). Neither knockdown was associated with a growth defect (Fig. 4B), consistent with prior observations (26, 33). Identification of both HD82 and 6.2800 as GCV-sensitizing hits in our RIT-seq screens, using HSV-TK-reporters with distinct 3′ untranslated regions, suggested that both proteins sensitized T. brucei to GCV in a manner that was independent of TK expression control by these 3′ untranslated regions. Consistent with this hypothesis, we observed no discernible change in expression level of the HSV-TK reporter following knockdown of either HD82 or 6.2800 (Fig. S3).
HD82 and 6.2800 knockdown and GCV dose response analysis. (A) The protein blots show effective and sustained knockdown of 6MYCHD82 or 6.280012MYC. EF1α serves as a loading control. The estimated mass of 6MYCHD82 is 91 kDa and that of 6.280012MYC is 114 kDa. (B) The growth curves show exponential and unperturbed growth following either HD82 or 6.2800 knockdown. (C) GCV dose-response analysis before and after knockdown for HD82. (D) GCV dose-response analysis before and after knockdown for 6.2800. (C, D) Error bars, SD; three replicate assays. EC50 values and P-values (F-test) are shown.
GCV selection was applied for 8 days in our RIT-seq screens, and we now sought to validate the HD82 and 6.2800 hits using a conventional dose response assay, conducted over 3 days of GCV selection. These assays confirmed that HD82 knockdown rendered T. brucei 3.8-fold resistant to GCV (Fig. 4C). The assays revealed no significant shift in GCV EC_50_ following 6.2800 knockdown, however (Fig. 4D). We, therefore, assessed GCV sensitivity after Tb927.6.2800 knockdown in a 6-day growth assay, which indicated that 6.2800 knockdown cells did indeed display increased tolerance to GCV selection at two times the EC_50_ (Fig. S4). These results are consistent with our RIT-seq profiles (Fig. 1C; Fig. 2) and with the idea that HD82 knockdown has the greatest impact on GCV resistance.
Since GCV activity depends upon phosphorylation by ectopic expression of HSV-TK, we next tested an alternative purine analog that may be fully phosphorylated by native T. brucei kinase(s). In cells lacking HSV-TK, both HD82 and 6.2800 knockdown significantly increased the EC_50_ for adenine arabinofuranoside (ara-A or Vidarabine), an analog with the D-ribose replaced with D-arabinose (Fig. 5A and B). HD82 and 6.2800 may sensitize T. brucei to GCV and ara-A by limiting dNTP pools. To explore this hypothesis, we carried out dose-response assays with hydroxyurea, which itself reduces dNTP pools by inhibiting ribonucleotide reductase (34). Knockdown of HD82 revealed no significant shift in hydroxyurea EC_50_, while knockdown of 6.2800 did indeed render T. brucei hyper-sensitive to hydroxyurea (Fig. 5C and D).
HD82 and 6.2800 sensitize T. brucei to purine analogs. (A) Dose response analysis for ara-A before and after HD82 knockdown. (B) Dose response analysis for ara-A before and after 6.2800 knockdown. (C) Dose response analysis for hydroxyurea before and after HD82 knockdown. (D) Dose response analysis for hydroxyurea before and after 6.2800 knockdown. Error bars, SD; three replicate assays. EC50 values and P-values (F-test) are shown.
dATP appears to be the preferred substrate of the HD82 triphosphohydrolase
To further probe the mechanisms by which 6.2800 and HD82 sensitize T. brucei to purine analogs, we established an assay to measure nucleotide pools before and after knockdown. We extracted nucleotides from these cells and assessed their abundance in triplicate for each sample using liquid chromatography and mass spectrometry (LC-MS). Heavy dATP was used as a reference in each sample, and the parent T. brucei strain was also analyzed and used as an unperturbed control. All four dNTPs were readily detected using this assay (Fig. 6A). Quantification of dNTPs in 6.2800 knockdown cells revealed no significant changes in dNTP pools (Fig. 6B). In contrast, quantification of dNTPs in HD82 knockdown cells revealed a significant and specific 3.3-fold increase in dATP abundance following knockdown (Fig. 6B). These results are consistent with the view that HD82 is indeed a triphosphohydrolase, with dATP as the preferred substrate; another recent study also found that dATP abundance was substantially increased following knockout of T. brucei hd82 (25). Taken together with the results above, these findings suggest that HD82 sensitizes T. brucei to purine analogs by reducing purine pools, while 6.2800 sensitizes T. brucei to purine analogs by a distinct mechanism.
dATP abundance is specifically increased following HD82 knockdown. (A) The example data set shows the read-out of the LC-MS assay for dNTP abundance. (B) dNTP pools were quantified in T. brucei cell extracts before and after knockdown; n = 3 samples per condition. The horizontal lines indicate median values, and the gray boxes indicate the range of each data set. Heavy dATP was used as a reference in each sample, and values are expressed relative to a control parent T. brucei strain. Significance was assessed using an unpaired, two-tailed t-test.
DISCUSSION
We used genome-wide RNAi library screens to identify T. brucei genes that sensitize cells to nucleoside analogs. HD82 and Tb927.6.2800 emerged as the top two hits, both of which localized to the T. brucei nucleus. HD82 is a HD domain protein related to SAMHD1, a human viral restriction factor and triphosphohydrolase. Additional results suggest that HD82 and 6.2800 impact purine analog incorporation into DNA by distinct mechanisms. Our findings suggest that HD82 is a triphosphohydrolase that sensitizes cells to purine analogs by reducing the native dATP pool. In contrast, we suggest that the product of 6.2800, which incorporates a BRCT domain, sensitizes cells to purine analogs through DNA repair.
HD domains are typically found in predicted dNTP phosphohydrolases (27). Human SAMHD1 controls dNTP/deoxynucleoside homeostasis and thereby enhances nucleoside-analog efficacy against human immunodeficiency virus (35), presumably by depleting dNTPs and increasing the effective availability of nucleoside analog triphosphates. dNTP turnover is thought to dominate outside of S-phase during the cell cycle, when dNTP use is low (29, 36). A second T. brucei HD domain protein was previously found to localize to the mitochondrion and to control dTTP/dCTP pools; specifically, HD52 knockdown increased dCTP and decreased dTTP (26). Notably, HD52 was not a hit in our screens, consistent with specific and competitive inhibition of dGTP by GCV-TP. Additionally, GCV-TP is not a substrate for SAMHD1, while dGTP is (37). HD82 knockdown specifically increases dATP pools, which may explain resistance to the adenosine analog, ara-A, through competition for incorporation into DNA, but this does not readily explain resistance to GCV. Perhaps, unlike SAMHD1, HD82 also metabolizes GCV-TP.
Genome-wide RNAi library screens have been instrumental in revealing novel biology in T. brucei (38). Combining such screens with HSV-TK expression and negative selection presents opportunities for assessing further loss-of-function phenotypes, to identify positive regulators of gene expression, for example. Our findings should facilitate the analysis of such knockdown screens by pointing to those predictable GCV resistance mechanisms identified here. Finally, a prior study on nucleoside metabolism suggested that T. brucei encodes an unidentified 5′-nucleotidase that converts deoxypyrimidine nucleotides to their corresponding nucleosides (18). Tb927.9.10830 emerged as a candidate in that study, a gene that failed to register as a hit in our screens. We speculate that HD82 could fulfil this role. In summary, our results indicate that HD82 reduces nuclear dATP pools and sensitizes T. brucei to purine analogs, while 6.2800 sensitizes T. brucei to purine analogs by a distinct mechanism. These findings provide insights into nucleoside/nucleotide metabolism and nucleoside analog resistance mechanisms in trypanosomatids.
MATERIALS AND METHODS
T. brucei manipulation
Bloodstream-form T. brucei strain Lister 427 wild-type cells, 2T1 cells (39), and derivatives, including 2T1:T7, were used in this study. Cells were grown in HMI-11 media in a humidified incubator at 37°C and 5% CO_2_. Cells expressing an HSV-TK reporter were grown in media lacking additional thymidine, made by supplementing Iscove’s modified Dulbecco’s medium (IMDM; ThermoFisher) with 10% fetal bovine serum (FBS; Sigma), 1 mM hypoxanthine (Sigma), 0.05 mM bathocuproine disulfonic acid (Sigma), 1 mM sodium pyruvate (Sigma), and 1.5 mM L-cysteine (Sigma). Cell density was determined by counting cells on a hemocytometer (Hawksley). Transfection was carried out typically using 3–5 × 10^7^ cells at log phase (approx. 1 × 10^6^ cells/mL) in cytomix buffer using a Nucleofector (Amaxa) set on program X-001. Cells were selected using 2.5 µg/mL G418 (InvivoGen), 2.5 µg/mL hygromycin (Melford), or 10 µg/mL blasticidin (InvivoGen) as appropriate. RIT-seq library transfections were carried out as described previously (22) in 2T1:T7 strains containing an HSV-TK reporter.
RIT-seq analysis
RIT-seq libraries were induced with tetracycline for 24 h prior to the addition of GCV (Sigma). Selected populations were harvested, and genomic DNA extracted using the DNeasy Blood and Tissue Kit (Qiagen). RIT-seq fragments were amplified from genomic DNA by PCR using Lib3F and Lib3R primers and Phusion DNA Polymerase (NEB). PCR amplicons were sequenced at the Beijing Genomics Institute using the DNB-seq platform. The FASTQ files with forward and reverse paired end reads were aligned to the reference genome v46 of T. brucei clone TREU927, reference sorted and indexed, quality evaluated, deduplicated, extracted, and parsed, with genome coverage data extracted, visualized, quantified, and normalized as described (40). The bash script containing the analysis pipeline, a Conda environment specification file for its execution, the python script to extract barcoded reads, and the versions of the software packages used are available in GitHub (https://github.com/mtinti/HD82-loss-of-function) and deposited in Zenodo 10.5281/zenodo.8093907.
Plasmids
The initial reporter cassette, comprising a fusion of a blasticidin deaminase gene, a TY1 epitope tag, and the HSV-TK gene, with a VSG 3′ untranslated region, all flanked by β and α tubulin homology targeting fragments, was synthesized and cloned in a pMA-T vector (GeneArt). An aldolase 5′ untranslated region and neomycin phosphotransferase cassette were then added upstream of the reporter cassette. The VSG untranslated region was then excised using NotI and BglII and replaced with a PCR-amplified aldolase 3′ untranslated region. DNA fragments for transfection were excised from the plasmid by digestion with PmeI. The plasmid sequence can be found in Data file S1. RNAi target fragments were amplified and cloned into pRPa^iSL^ (41). HD82 was tagged with 6xMYC epitopes at the N-terminus using pNAT^6MYCX^ or with 12xMYC epitopes at the C-terminus using pNAT^X12MYC^, and 6.2800 was also tagged with 12xMYC epitopes at the C-terminus (41); these plasmids were linearized with KpnI, NdeI, or HpaI, respectively, prior to transfection. Oligonucleotide sequences can be found in Data file S1.
Protein blotting
Bloodstream-form T. brucei (5 × 10^7^) were harvested and washed once with phosphate buffered saline (PBS) prior to lysis in Laemmli buffer at 1 × 10^6^ cells/µL. Ten microliters of lysate was loaded onto 10% SDS-PAGE gels (Bio-Rad). After protein separation, proteins were transferred onto nitrocellulose membrane (Millipore) using the Trans-blot Turbo (Bio-Rad) 10-min program and Tris/glycine transfer buffer (Bio-Rad). Membranes were blocked in 5% milk powder (Marvel) in PBS, then incubated with primary α-MYC at 1:5,000 (clone 9B11, Cell Signaling), washed with PBS 0.001% Tween 20 (Sigma), and incubated with secondary α-mouse HRP at 1:10,000 (Bio-Rad), followed by further washing. Signals were detected by incubation with ECL Prime (Amersham) and exposure to X-ray film (Amersham) before developing. Blots were stripped with western blot restore stripping buffer (ThermoFisher) before re-blocking and probing with α-eF1α (clone CBP-KK1, Millipore) as detailed above.
Immunofluorescence microscopy
Bloodstream-form T. brucei (1 × 10^6^) were fixed in 3% paraformaldehyde in PBS (Sigma) for 10 min at RT before washing twice with PBS and settling onto poly-L-lysine slides (VWR) for 1 h at RT. Cells were permeabilized with 0.5% Triton X-100 (Sigma) in PBS for 15 min and blocked with 50% FBS (Sigma) in PBS for 15 min, followed by incubation with primary α-MYC (clone 9B11, Cell Signalling) at 1:5,000 in 3% FBS in PBS for 1 h at RT. Finally, slides were incubated with α-mouse Alexa Fluor-488-coupled secondary antibody (ThermoFisher) at 1:2,000 for 1 h at RT before VECTASHIELD (Vector Laboratories) with DAPI (4′,6-diamidino-2-phenylindole) was applied and a coverslip was used to seal on top. Z-stacks were captured using a Zeiss Axioscope Microscope and merged using the ImageJ Maximum Intensity Projection algorithm.
Dose response assays
Cells were seeded in triplicate at 1 × 10^3^/ mL in a twofold serial dilution of either GCV (Sigma), hydroxyurea (Sigma), or ara-A (Sigma). The ara-A plate also contained 1 µM EHNA (Sigma) to prevent deoxyadenosine deamination (42). After 72-h incubation at 37°C, 20 µL resazurin sodium salt (alamarBlue, Sigma) was added at 0.5 mM in PBS to each well and incubated for a further 6 h. The fluorescence signal was determined using an Infinite 200 pro plate reader (Tecan) using an excitation wavelength of 540 nm and an emission wavelength of 590 nm. Data were analyzed, to derive EC_50_ values and statistics using Prism (GraphPad). RNAi knockdown strains were induced with 1 µg/mL tetracycline 24 h prior to plating.
dNTP quantification by LC-MS
dNTPs were extracted as described (43). Briefly, 8 × 10^7^ T. brucei cells were washed in PBS and then lysed in 200 µL 0.5 M perchloric acid. One hundred nanograms of ^13^C and ^15^N labelled dATP (CK Isotopes) was spiked into each sample and used as an internal standard. Samples were centrifuged for 3 min at 17,600 × g at 4°C, and 100 µL supernatant was neutralized by the addition of 25 µL ice-cold 2.3 M potassium bicarbonate. Samples were centrifuged, and the supernatant was stored at −80°C. dNTP (dATP, dCTP, dGTP, dTTP, and heavy dATP) standards were used to optimize the LC-MS-based detection and quantification. dNTPs were measured in MRM mode with optimized collision energies and radio frequencies determined by infusing pure compounds. The TSQ Quantiva mass spectrometer was interfaced with an ultimate 3000 Liquid Chromatography System (Thermo Scientific). A PGC column (HyperCarb 30 × 1 ID 3µ Thermo Scientific) that was maintained at a constant temperature of 42°C was used for the separation. Mobile phase buffer A consisted of 0.3% (vol/vol) formic acid adjusted to pH 9.17 with ammonia prior to a 1/10 dilution. Buffer B was 80% (vol/vol) acetonitrile. The column was initially equilibrated with 3% buffer B for 11 min at a constant flow rate of 0.045 mL/min. Aliquots of 4 µL of each sample were loaded onto the column, and compounds were eluted with a linear gradient of 3%–5% buffer B in 2 min, 5%–80% buffer B in 2 min, and 80%–100% buffer B in 4 min, and the column was then equilibrated in 3% buffer B for 11 min. Eluents were sprayed into the TSQ Quantiva using an Ion Max NG ion source with the ion transfer tube with the temperature set at 350°C and vaporizer temperature at 30°C. The mass spectrometer was operated in negative mode spray voltage of 3,500, Sheath gas 40, Aux gas 10, and Sweep gas 2. One blank was run between each sample.
Protein domain searches
Amino acid sequences of Human SAMHD1 and HD82 were searched for protein domains using InterPro (https://www.ebi.ac.uk/interpro/) (44). This failed to identify domains in 6.2800 so the Protein Databank file was downloaded from AlphaFold (https://alphafold.ebi.ac.uk [45, 46]) and uploaded into the Vector Alignment Search Tool (VAST) (https://www.ncbi.nlm.nih.gov/Structure/VAST/vastsearch.html) to identify protein domains.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Horn D. 2022. A profile of research on the parasitic trypanosomatids and the diseases they cause. P Lo S Negl Trop Dis 16:e 0010040. doi:10.1371/journal.pntd.001004035025891 PMC 8758061 · doi ↗ · pubmed ↗
- 2Barnadas-Carceller B, Martinez-Peinado N, Gómez LC, Ros-Lucas A, Gabaldón-Figueira JC, Diaz-Mochon JJ, Gascon J, Molina IJ, Pineda de Las Infantas Y Villatoro MJ, Alonso-Padilla J. 2022. Identification of compounds with activity against Trypanosoma cruzi within a collection of synthetic nucleoside analogs. Front Cell Infect Microbiol 12:1067461. doi:10.3389/fcimb.2022.106746136710960 PMC 9880260 · doi ↗ · pubmed ↗
- 3Bouton J, Furquim d’Almeida A, Maes L, Caljon G, Van Calenbergh S, Hulpia F. 2021. Synthesis and evaluation of 3'-fluorinated 7-deazapurine nucleosides as antikinetoplastid agents. Eur J Med Chem 216:113290. doi:10.1016/j.ejmech.2021.11329033667845 · doi ↗ · pubmed ↗
- 4Bouton J, Maes L, Karalic I, Caljon G, Van Calenbergh S. 2021. Synthesis and evaluation of a collection of purine-like C-nucleosides as antikinetoplastid agents. Eur J Med Chem 212:113101. doi:10.1016/j.ejmech.2020.11310133385837 · doi ↗ · pubmed ↗
- 5Hofer A. 2023. Targeting the nucleotide metabolism of Trypanosoma brucei and other trypanosomatids. FEMS Microbiol Rev 47:fuad 020. doi:10.1093/femsre/fuad 02037156497 PMC 10208901 · doi ↗ · pubmed ↗
- 6Hulpia F, Bouton J, Campagnaro GD, Alfayez IA, Mabille D, Maes L, de Koning HP, Caljon G, Van Calenbergh S. 2020. C 6-O-alkylated 7-deazainosine nucleoside analogues: discovery of potent and selective anti-sleeping sickness agents. Eur J Med Chem 188:112018. doi:10.1016/j.ejmech.2019.11201831931339 · doi ↗ · pubmed ↗
- 7Hulpia F, Campagnaro GD, Alzahrani KJ, Alfayez IA, Ungogo MA, Mabille D, Maes L, de Koning HP, Caljon G, Van Calenbergh S. 2020. Structure-activity relationship exploration of 3’-deoxy-7-deazapurine nucleoside analogues as anti-Trypanosoma brucei agents. ACS Infect Dis 6:2045–2056. doi:10.1021/acsinfecdis.0c 0010532568511 · doi ↗ · pubmed ↗
- 8Lin C, Hulpia F, Karalic I, De Schepper L, Maes L, Caljon G, Van Calenbergh S. 2021. 6-Methyl-7-deazapurine nucleoside analogues as broad-spectrum antikinetoplastid agents. Int J Parasitol Drugs Drug Resist 17:57–66. doi:10.1016/j.ijpddr.2021.08.00134375904 PMC 8358123 · doi ↗ · pubmed ↗