A Case Report of Aggressive Post-Infectious Hemophagocytic Lymphohistiocytosis in an Immunocompetent Adult
Nyein Wint Yee Theik

TL;DR
This case report highlights a rare aggressive immune disorder in an adult, emphasizing the need for early diagnosis and treatment to improve outcomes.
Contribution
The case underscores the importance of early suspicion and treatment of HLH in adults using updated diagnostic criteria.
Findings
HLH can present aggressively in immunocompetent adults with non-specific symptoms.
Timely treatment with etoposide and steroids is critical for survival, even without bone marrow confirmation.
The HScore criteria offer a broader diagnostic scope compared to older pediatric-focused criteria.
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is an acute inflammatory syndrome triggered by immune events such as infections, inflammation, autoimmune diseases, and malignancies. Initial presentations can range from vague symptoms to infectious features such as fever. Given its aggressive nature, timely diagnosis and immediate treatment are crucial to achieving optimal patient outcomes. Recently, the HLH score (HScore) criteria have been applied as diagnostic criteria, offering a broader scope compared to the previous HLH-2004 score, which was primarily based on pediatric populations. The standard treatment for decades has involved the combination of etoposide and high-dose steroids, and it is recommended to initiate treatment as soon as possible, even in the absence of a bone marrow test or when there is suspicion of the diagnosis. In this case presentation, we aim to underscore the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
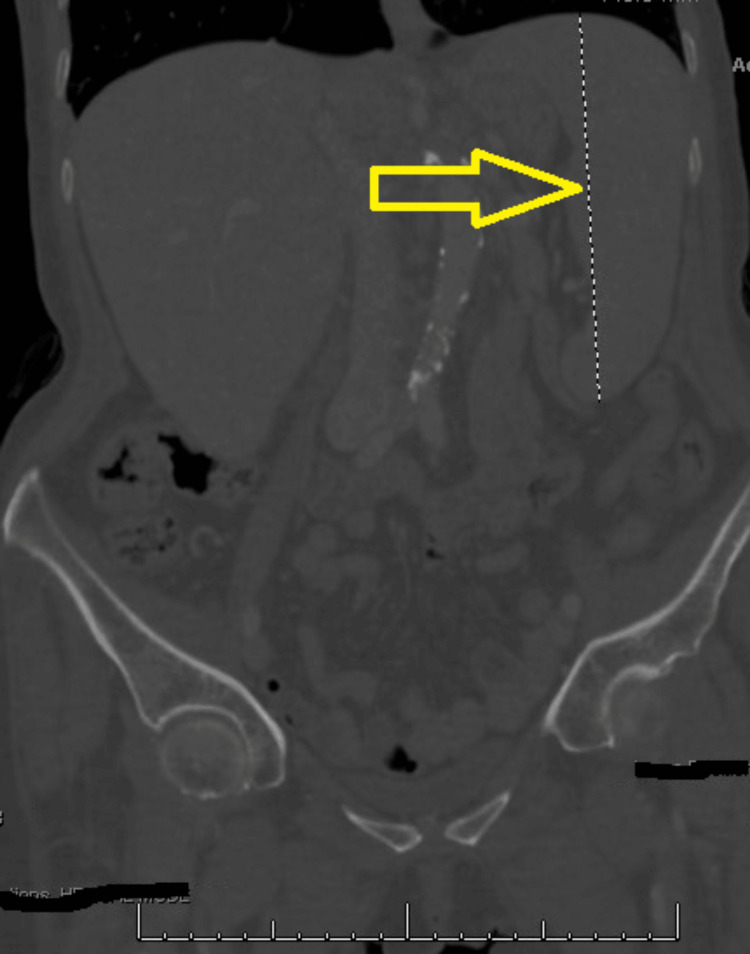 Figure 1
Figure 1| Metrics | Results | Scores | Patient results on Day 1 | Patient results on Day 5 |
| Known immunosuppression | No | +0 | Present | Present |
| Yes | +18 | |||
| Temperature (°C) | <38.4 | +0 | ||
| 38.4-39.4 | +33 | |||
| >102.9 | +49 | Present | Present | |
| Organomegaly | No | +0 | ||
| Hepatomegaly or splenomegaly | +23 | |||
| Hepatomegaly and splenomegaly | +38 | Present | Present | |
| Number of cytopenias | 1 lineage | +0 | ||
| 2 lineages | +24 | Present | ||
| 3 lineages | +34 | Present | ||
| Ferritin (ng/mL) | <2,000 | +0 | Present | |
| 2,000-6,000 | +35 | |||
| >6,000 | +50 | Present | ||
| Triglyceride (mg/dL) | <132.7 | +0 | ||
| 132.7-354 | +44 | |||
| >354 | +64 | Present | Present | |
| Fibrinogen (mg/dL) | >250 | +0 | ||
| <250 | +30 | Present | Present | |
| AST (units/L) | <30 | +0 | ||
| >30 | +19 | Present | Present | |
| Hemophagocytosis features on BM aspirate | No | +0 | ||
| Yes | +35 | Not performed | Not performed | |
| Total Score | 224 | 284 | ||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research · Parvovirus B19 Infection Studies · Immune Cell Function and Interaction
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is an acute inflammatory syndrome resulting from uncontrolled immune activation, primarily due to severe macrophage activation and the release of many inflammatory cytokines [1]. It is diagnosed less frequently than its actual occurrences, especially in adult populations [2]. A late diagnosis can occasionally lead to a poor prognosis and life-threatening situations. HLH can manifest either as a primary condition, known as familial, or sporadic, known as a secondary condition, commonly caused by infections, inflammation, malignancies, autoimmune diseases, and medications such as immune checkpoint inhibitors [3]. The most common infectious trigger is viral infections. Clinical features are nonspecific and often mimic infectious symptoms such as fever and multi-organ involvement [3].
The finding of hemophagocytosis in bone marrow biopsies is usually considered the standard label for the diagnosis of HLH [4]. The HLH-2004 diagnostic criteria were created based on the pediatric clinical trial and do not reflect to diagnose adult secondary HLH or distinguish adult HLH from inflammatory conditions such as sepsis and rheumatologic diseases [5]. The HLH score (HScore) is a newly developed risk stratification score in 2014 that can be applied reliably to diagnose HLH in pediatric and adult populations. The scoring system also helps guide further investigation and specific treatment plans [6]. HLH is commonly associated with a very high mortality rate regardless of etiology. Patients without treatment can have a rapid disease progression, leading to fatality within weeks or months from initial diagnosis [7]. Therefore, timely diagnosis is crucial in HLH to initiate specific treatment [8]. In this case report, we would like to discuss the patient with a severe aggressive disease course of idiopathic HLH who unfortunately passed away within a week of diagnosis.
Case presentation
A 77-year-old Hispanic woman presented to the emergency department (ED) with fever, drenching sweats, generalized weakness, and unintentional weight loss for two weeks. The patient had a history of poorly controlled hypertension, hyperlipidemia, and gastroesophageal reflux disease (GERD). Her home medications included pantoprazole for GERD, metoprolol, amlodipine, hydrochlorothiazide, and enalapril for hypertension. A week before presenting to the ED, she visited an urgent care clinic for the same symptoms. Prior to the ED visit, she was diagnosed with a urinary tract infection caused by pan-sensitive Escherichia coli, although she did not exhibit urinary symptoms. She also took amoxicillin-clavulanate before admission, but her symptoms did not improve with antibiotics. The patient had no significant social history, such as smoking, heavy alcohol intake, or recreational drug use. Regarding family history, her father had passed away from coronary artery disease, but there were no other significant family health issues.
Aside from a mild fever with a temperature of 38°C, she had normal blood pressure, a stable heart rate, and adequate oxygen levels in an initial assessment. Physical examination revealed signs of cachexia and dehydration, including dry mucus membranes. The abdominal examination showed hepatosplenomegaly, but other systemic examinations, including the cardiopulmonary assessment, were unremarkable. A complete blood count (CBC) revealed a decreased white cell count (WCC) of 2.1 x 10^3^/µL and a platelet count of 60 x 10^3^/µL. The comprehensive metabolic panel showed elevated liver function parameters, including total bilirubin and alkaline phosphatase. Right upper quadrant abdominal ultrasound indicated hepatomegaly with non-specific lymphadenopathy. Large splenomegaly was noted on abdominal and pelvic computed tomography (CT) scan (Figure 1).
Splenomegaly on CT scan
The patient was initially prescribed broad-spectrum antibiotics to address the possibility of septic shock from the recent infection and fluid replacement. On her second day of admission, her WCC worsened despite antibiotics, and her hemoglobin dropped to 9.8 g/dL from the baseline. A biopsy was impossible due to the small size of the lymph nodes. Anemia workup included an iron panel, ferritin, vitamin B12, folate, and a hemolysis panel. The tests revealed an elevated ferritin level of 1790 ng/mL and signs of hemolysis, such as low haptoglobin and elevated lactate dehydrogenase. Blood leukemia-lymphoma phenotype analysis did not reveal abnormal blasts or aberrant antigenic expression. Fibrinogen levels decreased to 78 mg/dL, and triglycerides from the lipid panel elevated at 388 mg/dL. Autoimmune antibodies, used to rule out autoimmune hemolytic anemia, returned as unfavorable.
Considering the clinical manifestations and laboratory findings, an HScore was calculated to assess the possibility of HLH, resulting in a score of 234, indicating a 98-99% probability. Standard causative viral panels, such as Epstein Barr virus (EBV), cytomegalovirus, and Herpes tests, came back negative, along with sterile blood and urine cultures. On the third day of admission, the patient was treated with dexamethasone while scheduling biopsies, including a bone marrow and liver biopsy, to rule out malignancies as potential etiologies and confirm the diagnosis. The patient's condition was unstable, and intermittent febrile episodes, tachycardia, low blood pressure, and intermittent desaturation complicated her hospital course. Immediate biopsies could not be performed due to unstable vitals, and the patient declined to start etoposide despite it being recommended for immediate treatment initiation. The HScore diagnostic criteria comparison between day 1 and day 5 is shown in Table 1 [6].
Follow-up blood work, such as CBC and comprehensive metabolic panels on the fourth and fifth days of admission, revealed worsening thrombocytopenia, liver function, and bilirubin levels. The patient also became acidotic, with a gradual decrease in bicarbonate levels. Unfortunately, the patient decompensated and died on the fifth admission day, and an autopsy could not be performed.
Discussion
The HScore is a valuable tool for estimating the probability of HLH among suspected individuals. Notably, the probability of HLH is relatively low, less than 1%, for an HScore of less than or equal to 90 [6]. In contrast, an HScore of 250 or more is associated with a high probability of 99% [6]. We found that using a cutoff of 169 had 93% sensitivity and 86% specificity in diagnosing HLH [9]. To establish a diagnosis of HLH, patients must meet at least five out of nine HScore criteria.
Among the elderly population, common causes of HLH include viral infections, such as EBV, bacterial or fungal infections, certain malignancies such as T-cell lymphoma, and autoimmune diseases [10]. In our case, we ruled out active infections through blood culture and viral panels and malignancy through CT imaging. Given the aggressive nature and high mortality rate of HLH, immediate treatment, typically within a week, is recommended to improve patient outcomes [11]. The recommended treatment regimen consists of dexamethasone and etoposide, as treating with dexamethasone alone is considered inadequate [12].
Several predictive factors are associated with poor overall survival indicators in HLH patients. These factors include age over 45 years, low platelet counts, EBV association, and hyperferritinemia [13]. Our patient exhibited three factors: age over 45 years, initial presentation with thrombocytopenia, and hyperferritinemia, which unfortunately led to the patient's demise within five days of the initial diagnosis. The patient's refusal to commence etoposide treatment in combination with steroids may have also contributed to this unfortunate outcome.
Conclusions
This case emphasizes the difficulties of diagnosing HLH and underscores its aggressive nature. Healthcare providers should maintain a high level of suspicion for HLH when they come across patients exhibiting compatible clinical features, even if classic symptoms or risk factors are absent. It is crucial to promptly initiate treatment upon suspicion of the diagnosis without waiting for results such as a bone marrow biopsy, which can be time-consuming and may lead to treatment delays. This proactive approach is vital in improving patient outcomes and preventing the progression of this life-threatening condition.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hemophagocytic syndromes Blood Rev Janka GE 2452532120071759025010.1016/j.blre.2007.05.001 · doi ↗ · pubmed ↗
- 2Nationwide survey of hemophagocytic lymphohistiocytosis in Japan Int J Hematol Ishii E Ohga S Imashuku S 58658620071767526810.1532/IJH 97.07012 · doi ↗ · pubmed ↗
- 3Adult haemophagocytic syndrome Lancet Ramos-Casals M Brito-Zerón P López-Guillermo A Khamashta MA Bosch X 1503151638320142429066110.1016/S 0140-6736(13)61048-X · doi ↗ · pubmed ↗
- 4Novel mutation in perforin gene causing familial hemophagocytic lymphohistiocytosis type 2 in an Egyptian infant: case report Egypt J Med Hum Genet Almalky M Saleh SHA Baz EG Fakhr AE 24212020
- 5HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis Pediatr Blood Cancer Henter JI Horne A AricóM 1241314820071693736010.1002/pbc.21039 · doi ↗ · pubmed ↗
- 6Development and validation of the H Score, a score for the diagnosis of reactive hemophagocytic syndrome Arthritis Rheumatol Fardet L Galicier L Lambotte O 261326206620142478233810.1002/art.38690 · doi ↗ · pubmed ↗
- 7High mortality of HLH in ICU regardless etiology or treatment Front Med (Lausanne) Bichon A Bourenne J Allardet-Servent J 735796820213469272710.3389/fmed.2021.735796 PMC 8526960 · doi ↗ · pubmed ↗
- 8How I treat hemophagocytic lymphohistiocytosis Blood Jordan MB Allen CE Weitzman S Filipovich AH Mc Clain KL 4041405211820112182813910.1182/blood-2011-03-278127 PMC 3204727 · doi ↗ · pubmed ↗