Elastic ripening and inhibition of liquid-liquid phase separation
Kathryn A. Rosowski, Tianqi Sai, Estefania Vidal-Henriquez, David, Zwicker, Robert W. Style, Eric R. Dufresne

TL;DR
This paper demonstrates how elastic stresses within polymer networks can suppress phase separation, stabilize mixtures, and induce a novel, faster ripening process driven by stiffness gradients, with implications for cellular organization.
Contribution
It introduces the concept of elastic ripening and shows how network stresses influence phase separation and droplet dynamics in synthetic systems.
Findings
Compressive stresses suppress phase separation.
Network stresses stabilize mixtures beyond phase boundary.
Stiffness gradients drive rapid elastic ripening.
Abstract
Phase separation has recently emerged as an important organizational principle in the dense and heterogeneous environment within living cells. Here, we use a synthetic system to show that compressive stresses in a polymer network suppress phase separation of the solvent that swells it. These stresses create a barrier to droplet nucleation that leads to robust, stabilized mixtures well beyond the liquid-liquid phase separation boundary. Network stresses not only alter the stability of mixtures, but they also have a dramatic effect on the ripening of droplets. Gradients in network stresses can drive a new form of ripening, where solute is transported down stiffness gradients. This elastic ripening can be much faster than conventional surface tension driven Ostwald ripening.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1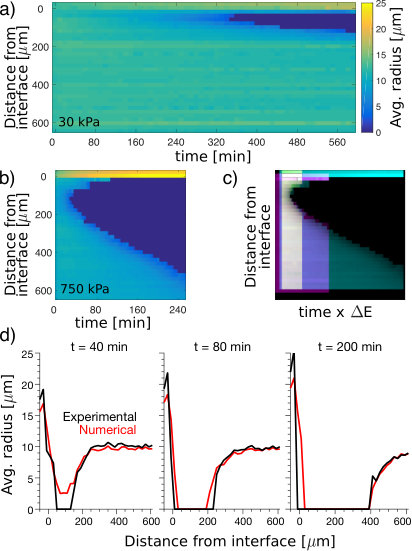 Figure 2
Figure 2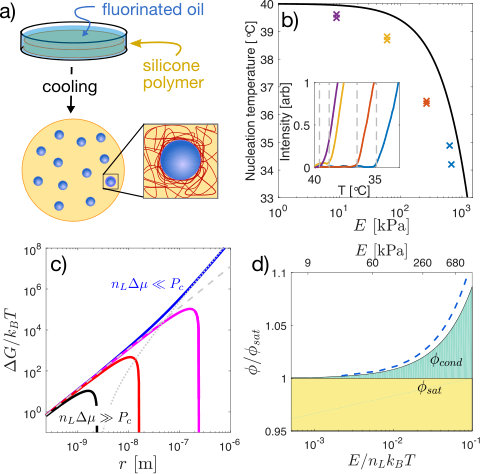 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
††thanks: KAR and TS contributed equally††thanks: KAR and TS contributed equally
Elastic ripening and inhibition of liquid-liquid phase separation
Kathryn A. Rosowski
Department of Materials, ETH Zürich, 8093 Zürich, Switzerland.
Tianqi Sai
Department of Materials, ETH Zürich, 8093 Zürich, Switzerland.
Estefania Vidal-Henriquez
Max Planck Institute for Dynamics and Self-Organization, 37077, Göttingen, Germany
David Zwicker
Max Planck Institute for Dynamics and Self-Organization, 37077, Göttingen, Germany
Robert W. Style
Department of Materials, ETH Zürich, 8093 Zürich, Switzerland.
Eric R. Dufresne
Department of Materials, ETH Zürich, 8093 Zürich, Switzerland.
Abstract
Phase separation has recently emerged as an important organizational principle in the dense and heterogeneous environment within living cells. Here, we use a synthetic system to show that compressive stresses in a polymer network suppress phase separation of the solvent that swells it. These stresses create a barrier to droplet nucleation that leads to robust, stabilized mixtures well beyond the liquid-liquid phase separation boundary. Network stresses not only alter the stability of mixtures, but they also have a dramatic effect on the ripening of droplets. Gradients in network stresses can drive a new form of ripening, where solute is transported down stiffness gradients. This elastic ripening can be much faster than conventional surface tension driven Ostwald ripening.
condensation, phase separation, cavitation, gels, elastomers
††preprint: APS/123-QED
Phase separation has recently emerged as an important route to compartmentalization within living cells. It is widely assumed to be governed by the physics of liquid-liquid phase separation Brangwynne et al. (2009); Hyman et al. (2014); Patel et al. (2015); Shin and Brangwynne (2017). However, the cell interior is not a simple liquid, but a crowded macromolecular stew including polymer networks that impart significant elasticity Janmey and McCulloch (2007).
Recent experiments have shown that network elasticity can dramatically impact liquid-liquid phase separation in swollen synthetic polymer networks. In a homogeneous network, droplets grow to a fixed size, controlled by the network stiffness Style et al. (2018). When the network has an anisotropic state of stress, droplets grow with scale-independent ellipsoidal shapes Kim et al. (2018).
In the nucleus of living cells, artificial phase separating domains formed preferentially in chromatin-poor regions Shin et al. (2018). After drops were triggered to grow in chromatin-rich regions, they migrated toward chromatin-poor regions. Theory and simulation suggest that these observations could be driven by gradients in network stiffness Style et al. (2018); Shin et al. (2018). However, these exciting observations are not sufficient to establish the role of network elasticity in droplet nucleation and migration. There are two main challenges. First, the heterogeneous mechanical properties of the nucleus remain unquantified. Second, the nucleoplasm is a multicomponent mixture, and the chemical solubility of one component (i.e. chromatin) can dramatically effect the solubility of other components (see e.g. the phase diagrams of three-component mixtures Veatch and Keller (2003); Vitale and Katz (2003)).
Here, we reveal the impact of network mechanics on droplet nucleation and ripening with experiments in a synthetic polymer system, where mechanical properties and chemical solubility can be tuned independently Style et al. (2018). We find that network elasticity can fully suppress droplet nucleation deep inside the thermodynamically immiscible region, thus mechanically stabilizing supersaturated mixtures. The long-term stability of droplets is strongly affected by network elasticity. While we observe no ripening in homogenous networks, in a mechanically heterogeneous network, solute moves from stiff to soft regions by diffusive transport through the dilute phase. Superficially resembling surface tension driven Ostwald ripening, this elastic ripening can be much faster and is driven by gradients of network elasticity.
To investigate the impact of network elasticity on nucleation, we drove phase separation in silicone gels (Gelest). First, we saturated them in a bath of fluorinated oil (Fluorinert FC-770) at Style et al. (2018). After several hours, the volume fraction of oil saturated at , independent of Young’s modulus, , as shown in Figures S3,S4. Then, we quenched samples in a temperature controlled microscope stage (TSA12Gi, Instec), while recording bright-field images with a 20X NA 0.5 objective. With sufficient undercooling, droplets nucleate and grow, shown schematically in Figure 1a. Nucleation events were identified by quantifying the average pixel-wise intensity difference at each temperature from a reference image, which grows rapidly at the point of nucleation, as shown in the inset panel of Figure 1b. At quench rates below , the nucleation temperature, , has no significant dependence on the quench rate (Fig. S6). Thus we used a quench rate of to measure the droplet nucleation temperature as a function of network stiffness from 9 kPa to 680 kPa. The nucleation temperature depends strongly on the network stiffness. In flexible networks, kPa, droplets nucleated at only below . In stiff networks, kPa, droplets did not appear until the sample was cooled about below (Fig. 1b).
These results suggest that network elasticity may constrain droplet nucleation. When the radius of the droplet exceeds the network mesh size, Rubinstein and Colby (2003), the network can squeeze the growing droplet. However, as long as the pressure within a droplet exceeds a critical value, , it can overcome elastic stresses and continue to grow Gent and Wang (1991); Zimberlin et al. (2007); Kim et al. (2018). We assume here that , a classic result for nonlinear-elastic solids Gent and Wang (1991), which is a good approximation for many polymer networks Zimberlin et al. (2007). This mechanical resistance is readily incorporated into classical nucleation theory, which considers the free energy, , of a droplet as a function of its radius, ,
[TABLE]
Here, as in simple condensation, nucleation is driven by the chemical potential difference between the dilute and condensed phases, , and resisted by interfacial energy, . The number density of molecules in the condensed phase is , and is the Heaviside step function. The product is equivalent to the stall pressure, , the maximum pressure a growing droplet can exert against its surroundings Style et al. (2018). The last term is a simple approximation of the mechanical work required to expand the cavity beyond the scale of the network mesh, Gent and Wang (1991). It can have a dramatic impact on the free energy landscape governing droplet growth. Figure 1c shows the predicted size dependence of the droplet free energy for four different supersaturation levels in a 300 kPa sample. At the highest supersaturations ( , black curve), elasticity has no impact on the free energy landscape and droplets can nucleate and grow normally. At lowest supersaturation ( , blue curve), the droplet free energy monotonically increases with radius, completely forbidding droplet growth.
Droplet nucleation is thus allowed only when . Assuming the ideal form of the chemical potential of the dilute phase, , we identify a minimal volume fraction of solute needed for condensation,
[TABLE]
The ratio thus plays a central role. As it increases beyond one, the supersaturation required for droplet nucleation diverges exponentially. In these experiments, , and reaches about 1.05, as shown in Figure 1d.
We identify three regimes of stability. When (shaded yellow in Figure 1d), the mixture is stable, independent of elasticity. When (shaded white), the mixture is unstable. When (shaded green) the supersaturated system is stable against nucleation and growth of droplets, but can demix by expelling solute beyond the boundaries of the polymer network. In this regime, compositions that would be unstable in the absence of elasticity are completely stable in the limit of infinite system size (i.e the thermodynamic limit).
To compare these predictions to our nucleation data, we determined , the temperature where Eq. 2 is satisfied, i.e. . Using independently measured values of (Fig. S4), this yields the solid line in Figure 1b. As expected, the stiffness dependence of follows the measured trend of , but at a consistently higher temperature, likely reflecting kinetic limitations to nucleation. To further test our hypothesis that the supersaturated state is stable to nucleation of droplets, we cooled a 700 kPa sample to , where . At this undercooling, softer samples nucleate and grow droplets in a matter of seconds. However, we fixed the temperature at this value for minutes, and found no evidence of nucleation in the stiffer sample.
Thus, elastic forces hinder nucleation and stabilize the supersaturated mixed state. Similarly, we expect the dilute phase to remain supersaturated even after droplet growth is completed. Consider a system quenched to , driving droplet nucleation. As droplets grow, they deplete solute from the dilute phase, and decreases. When , the free energy liberated by condensation is just sufficient to overcome the mechanical work needed to deform the polymer network, and droplet growth is arrested. At this point, , and the concentration in the dilute phase is stably supersaturated.
Since increases with network stiffness, stiffness gradients should result in gradients of supersaturation, and transport of solute from stiff to soft regions Weber et al. (2017). This simple physical picture rationalizes transport down stiffness gradients observed in simulations Shin et al. (2018).
To test this hypothesis, we nucleated and grew droplets in samples with stiffness gradients, shown schematically in Figure 2a. Samples were saturated with oil at to establish a uniform concentration , and quenched quickly so that both sides nucleated and grew droplets. Experimental data with a side-by-side step change in stiffness from 10 kPa to 700 kPa are shown in Figure 2b,c. As expected Style et al. (2018), we found larger droplets on the soft side than the stiff side. While droplets far from the interface were stable (Fig. S9 and Movies S1,S2), droplets on the stiff side, adjacent to the interface, started to dissolve soon after growth. Over time, more droplets dissolved on the stiff side, leaving a growing band of clear gel adjacent to the interface (Fig. 2b, Movie S3). Simultaneously, the droplets on the soft side, immediately adjacent to the interface, grew (Fig. 2c). This experiment reveals significant solute transport down the stiffness gradient, supporting our hypothesis that the solute is still supersaturated when droplet growth is arrested.
This phenomenon resembles conventional surface tension driven Ostwald ripening de Gennes et al. (2004), as small droplets shrink to feed the growth of larger droplets. To rule this out, we performed two experiments. First, we tested if Ostwald ripening occurs between droplets in a homogeneous silicone network. We produced a bi-dispersed droplet size distribution in a homogeneous 80kPa sample by making a sudden change in the quench rate during droplet growth, described in the Supplement. This method allowed us to produce droplets with radii of about and , with a typical spacing of about . We monitored the size of the droplets for 170 minutes after the end of growth, and found no change in size (Fig. 2d, Movie S4). Compare this to the 140 kPa gradient sample in Figure S10, with droplets of and juxtaposed across the interface. In this case, smaller droplets more than from the larger ones are dissolved completely within 100 minutes.
Second, we repeated the experiment with a side-by-side stiffness gradient, as in Figure 2 b, but with slow and controlled cooling, at . As expected from our data on nucleation in homogeneous samples (Fig. 1), droplets appeared at higher temperatures on the soft side than on the stiff side (Fig. 2e). Additionally, droplets never appeared near the stiff side of the interface. This indicates that stiffness-driven gradients of supersaturation depleted solute from the stiff side even before droplets could form there. While this is consistent with our proposed mechanism, it is not consistent with surface tension driven Ostwald ripening, which requires droplets of different sizes.
Having ruled out surface tension as a driving force for ripening, we now quantify the effect of stiffness gradients. We measured the average droplet size as a function of time and distance from the soft-stiff interface, visualized as a kymograph in Figure 3a,b. Keeping the stiffness at fixed at 7 kPa, we varied the stiffness for from 30 to 750 kPa. Results for the extremes are shown in Figure 3a,b, the rest in the Supplement. In all cases, droplets on the stiff side disappear near the interface after a delay, and a dissolution front invades the stiff side. The dissolution front appears sooner, and moves faster, in stronger stiffness gradients. In Figure 3c, we stretch these kymographs by an amount proportional to the stiffness difference, , and superimpose them. Intriguingly, with a small shift in starting time and starting position, the shapes of the dissolution fronts match (Fig. 3c). The rate of advance of the dissolution front therefore increases with .
A simple model of solute transport captures the essential features of these results. It is based on simple diffusion in the dilute phase, where the concentration is pinned to at the surface of the droplets (see Supplement). This simple theory, shows quantitative agreement for large , as shown in Fig. 3d (and Movie S5). It overestimates the delay before front formation for smaller stiffness differences, as shown in Fig. S10.
In general, both elasticity and surface tension can drive ripening. In Ostwald ripening, the driving pressure is given by the difference in Laplace pressure. In elastic ripening, the driving pressure emerges from differences in pressure applied to the droplets by the polymer network, of order . The relative strength of the driving forces for Ostwald and elastic ripening are therefore captured by the elastocapillary number, , Style et al. (2015). When this is much smaller than one, elastic forces dominate. In these experiments, . But could elastic ripening occur in living cells? For membraneless organelles, reported values of vary from N/m Brangwynne et al. (2009, 2011); Feric et al. (2016); Taylor et al. (2016), and from Brangwynne et al. (2009); Feric et al. (2016). The cytoskeleton can readily reach stresses on the order of kPa Wang et al. (2002); Rotsch and Radmacher (2000); Gardel et al. (2006). Thus, the elastocapillary number of membraneless organelles could readily reach , where elastic ripening dominates.
We have shown that a host polymer network can dramatically alter the nucleation and ripening of phase-separated liquid droplets. In classic liquid-liquid phase separation, these processes are controlled by interfacial energy. Here, the structure of the polymer network introduces a new term into the free energy, reflecting that droplets can grow only when their internal pressure exceeds a critical pressure, of order Young’s modulus. This splits the conventional phase boundary in two. One curve, , determines the saturation equilibrium of a system in contact with a reservoir of solute. Another, , determines the limit of stability of the mixture against droplet nucleation and growth.
These phenomena further suggest that living cells could regulate the localization of membraneless organelles through gradients of their mechanical properties. Since cells are well-known to regulate their heterogeneous mechanical properties Hoffman et al. (2006); Janmey and McCulloch (2007), this physical hypothesis is biologically reasonable. By contrast, recent physical modelling of the localization of phase-separated domains has focused on the role of activity gradients, Brangwynne et al. (2009); Zwicker et al. (2014, 2017); Weber et al. (2019). We should note that the current work is limited to static networks with relatively simple rheology. New phenomena are anticipated in living cells where the timescale of phase separation can be comparable to the viscoelastic relaxation timescale Tanaka (2000) or the remodelling timescale of active networks Blanchoin et al. (2014); Needleman and Dogic (2017).
We acknowledge the SNF National Centre of Competence in Research ‘Bio-Inspired Materials’ for funding, as well as Larry Wilen, Sanat Kumar, and Tal Cohen for helpful discussions.
The datasets generated and analysed during the current study are available from the corresponding authors on reasonable request.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Brangwynne et al. (2009) C. P. Brangwynne, C. R. Eckmann, D. S. Courson, A. Rybarska, C. Hoege, J. Gharakhani, F. Jülicher, and A. A. Hyman, Science 324 , 1729 (2009).
- 2Hyman et al. (2014) A. A. Hyman, C. A. Weber, and F. Jülicher, Ann. Rev. Cell Dev. Bio. 30 , 39 (2014).
- 3Patel et al. (2015) A. Patel, H. O. Lee, L. Jawerth, S. Maharana, M. Jahnel, M. Y. Hein, S. Stoynov, J. Mahamid, S. Saha, and T. M. e. a. Franzmann, Cell 162 , 1066 (2015).
- 4Shin and Brangwynne (2017) Y. Shin and C. P. Brangwynne, Science 357 , eaaf 4382 (2017).
- 5Janmey and Mc Culloch (2007) P. A. Janmey and C. A. Mc Culloch, Annu. Rev. Biomed. Eng. 9 , 1 (2007).
- 6Style et al. (2018) R. W. Style, T. Sai, N. Fanelli, M. Ijavi, K. Smith-Mannschott, Q. Xu, L. A. Wilen, and E. R. Dufresne, Phys. Rev. X 8 , 011028 (2018).
- 7Kim et al. (2018) J. Y. Kim, Z. Liu, B. M. Weon, C.-Y. Hui, E. R. Dufresne, and R. W. Style, ar Xiv preprint ar Xiv:1811.00841 (2018).
- 8Shin et al. (2018) Y. Shin, Y.-C. Chang, D. S. W. Lee, J. Berry, D. W. Sanders, P. Ronceray, N. S. Wingreen, M. Haataja, and C. P. Brangwynne, Cell 175 , 1481 (2018).