Temperature-Dependent Lifetimes of Low-Frequency Adsorbate Modes from Non-Equilibrium Molecular Dynamics Simulations
Francesco Nattino, J\"org Meyer

TL;DR
This study uses non-equilibrium molecular dynamics to calculate temperature-dependent vibrational lifetimes of low-frequency adsorbate modes on metal surfaces, revealing new insights into damping mechanisms.
Contribution
First to extract vibrational lifetimes from non-equilibrium MD simulations considering both phononic and electronic damping channels.
Findings
Damping mechanisms are additive, contrary to textbook predictions.
A simple harmonic-anharmonic model semi-quantitatively matches temperature dependence.
Electronic and phononic contributions to damping are quantitatively separated.
Abstract
We present calculations on the damping of a low-frequency adsorbate mode on a metal surface, namely the frustrated translation of Na on Cu(100). For the first time, vibrational lifetimes of excited adlayers are extracted from non-equilibrium molecular dynamics calculations accounting for both the phononic and the electronic dissipation channels. The relative contributions of the two damping mechanisms, which we show to be additive, are found to disagree with textbook predictions. A simple model based on separable harmonic and anharmonic contributions is able to semi-quantitatively reproduce the temperature dependence of the computed lifetimes.
Click any figure to enlarge with its caption.
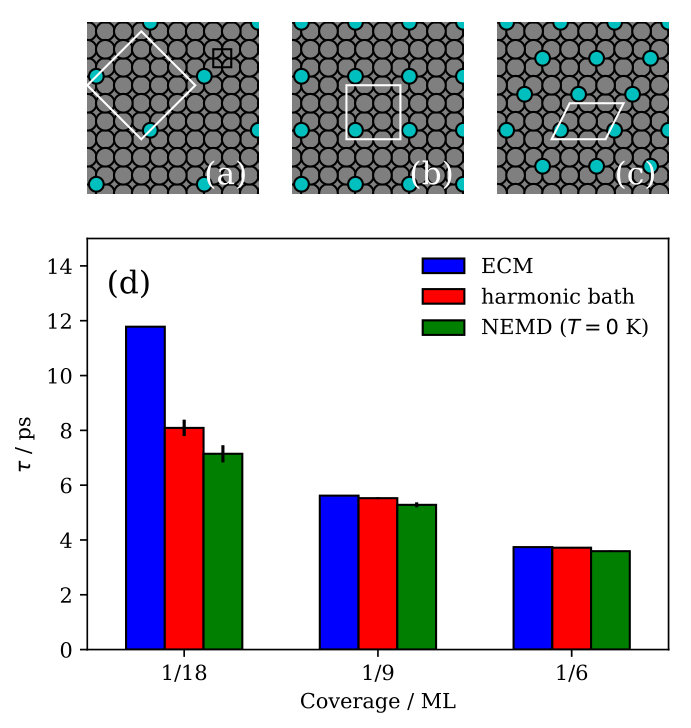 Figure 1
Figure 1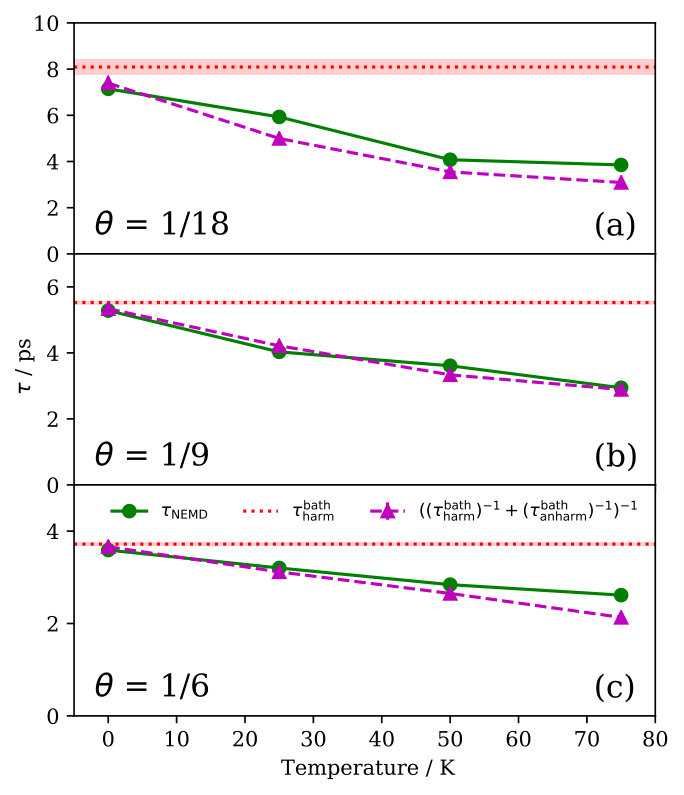 Figure 2
Figure 2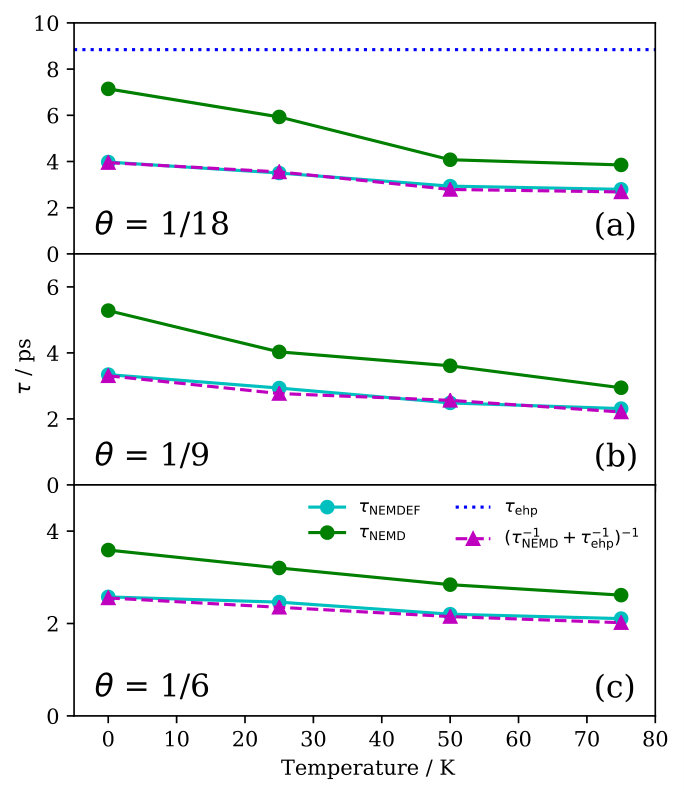 Figure 3
Figure 3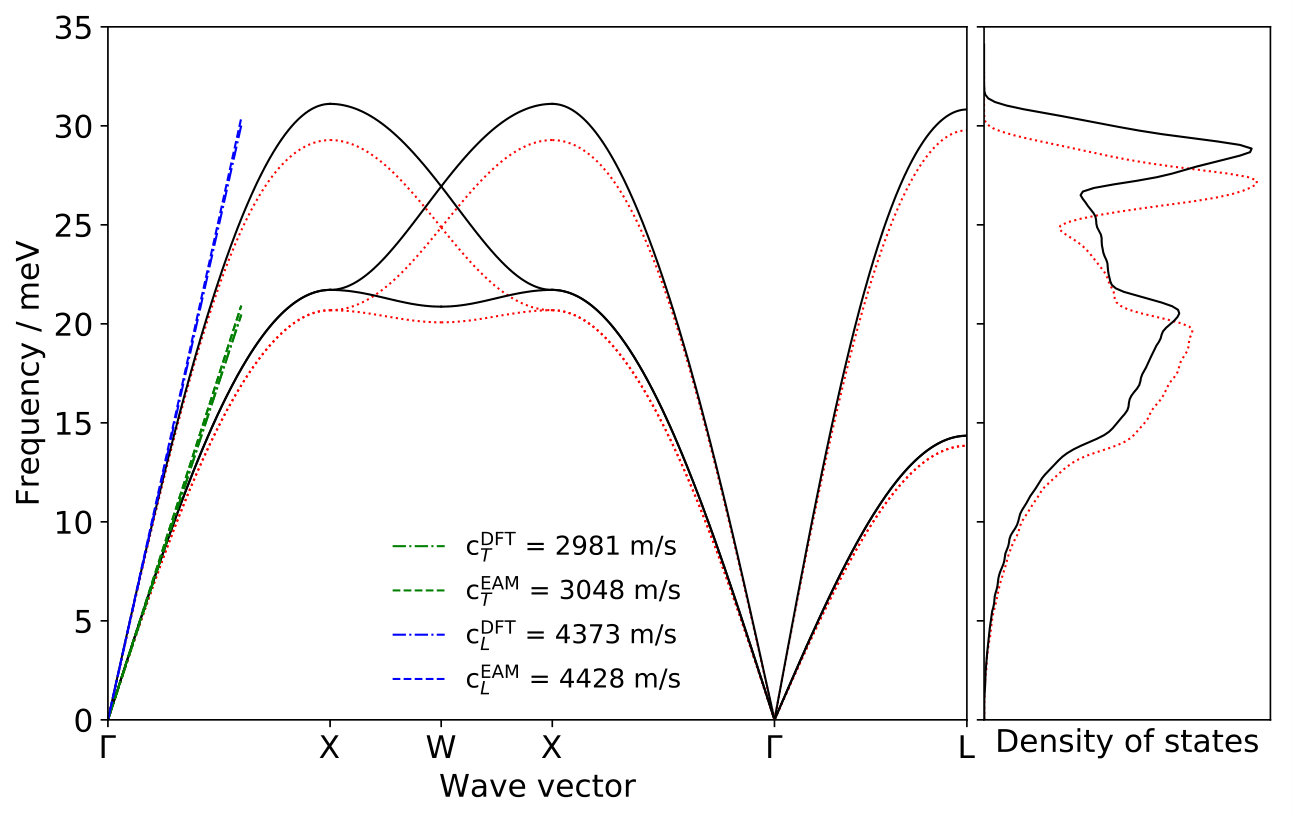 Figure 4
Figure 4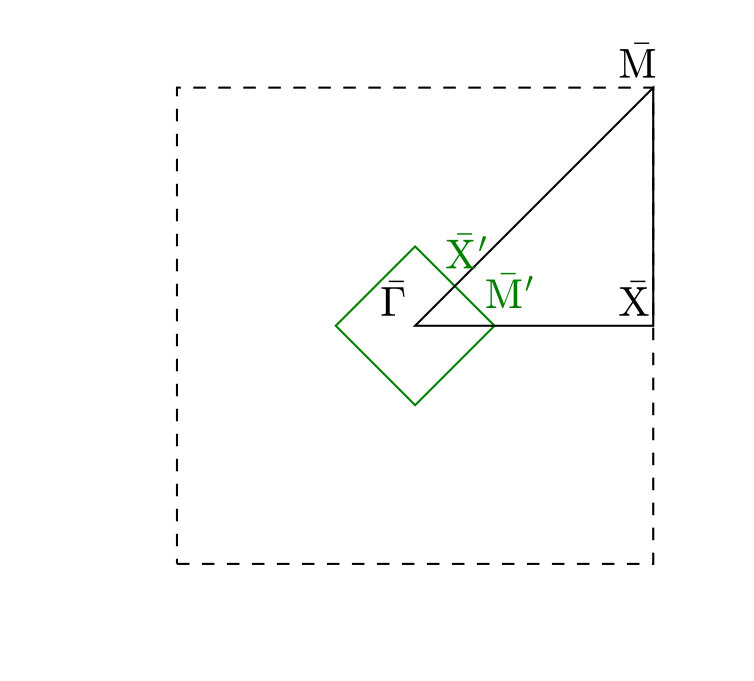 Figure 5
Figure 5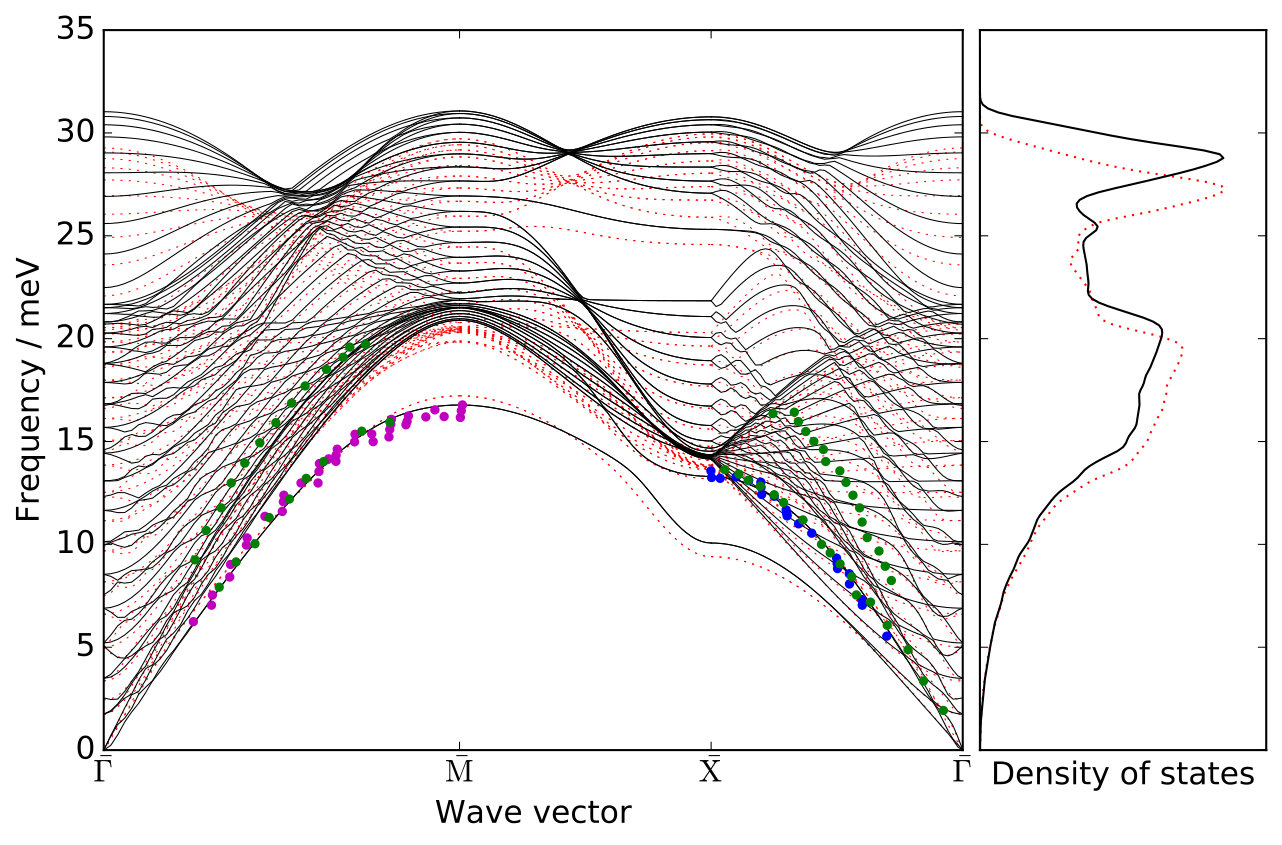 Figure 6
Figure 6 Figure 7
Figure 7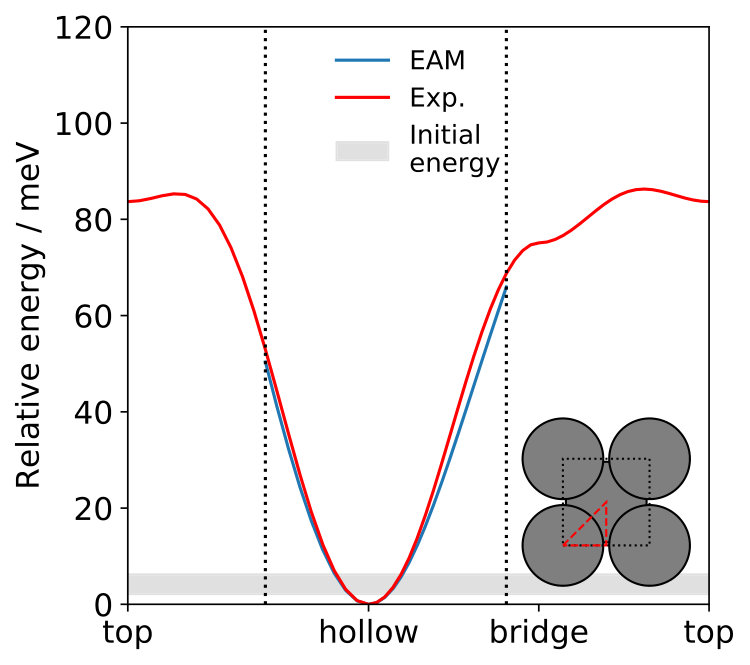 Figure 8
Figure 8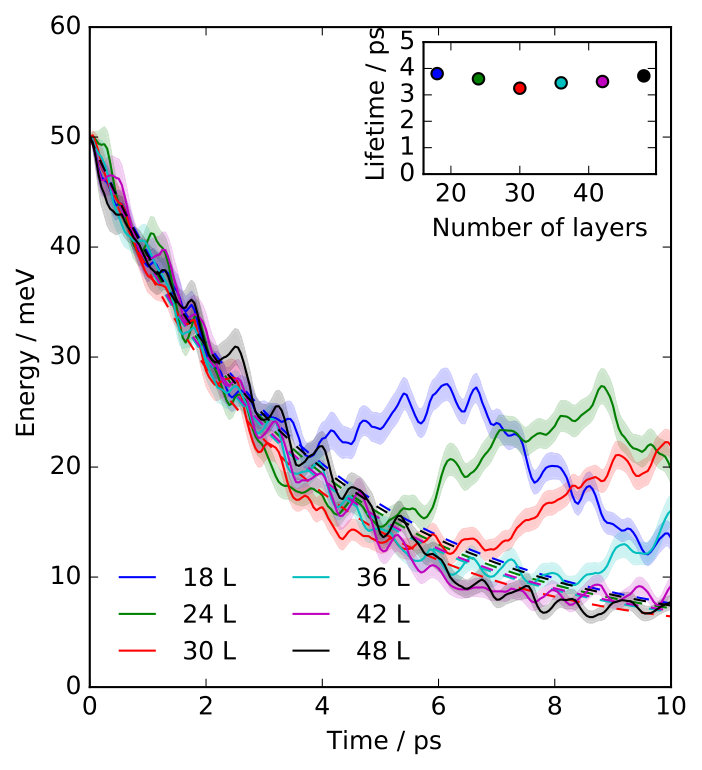 Figure 9
Figure 9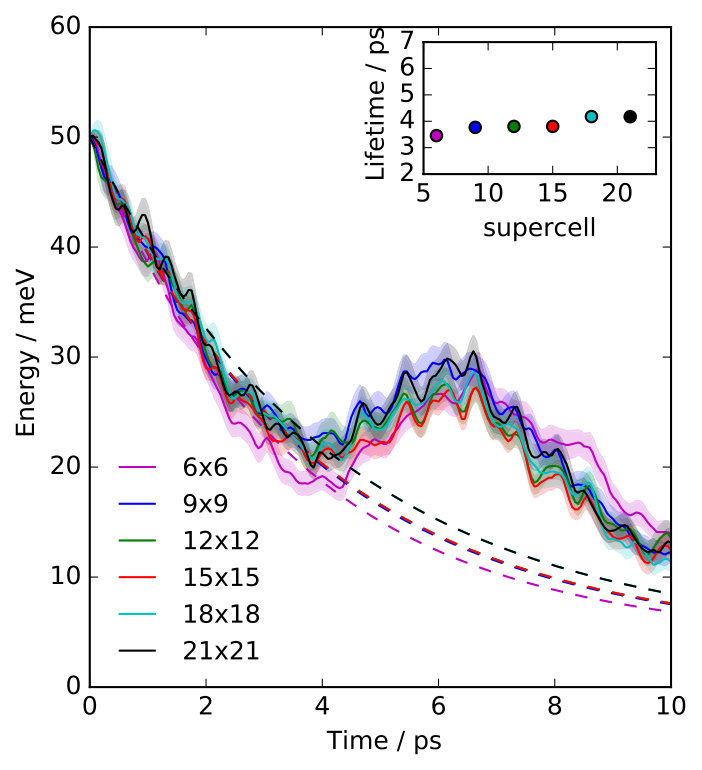 Figure 10
Figure 10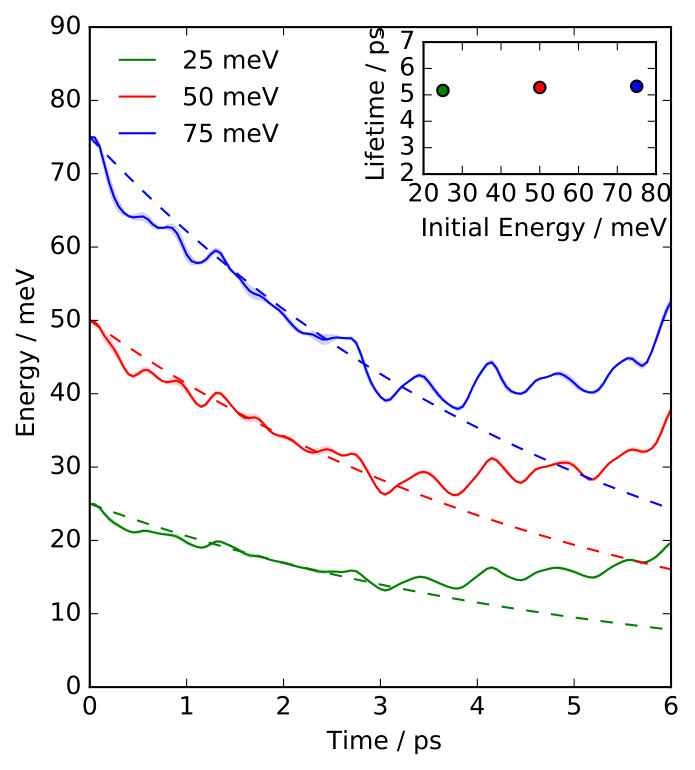 Figure 11
Figure 11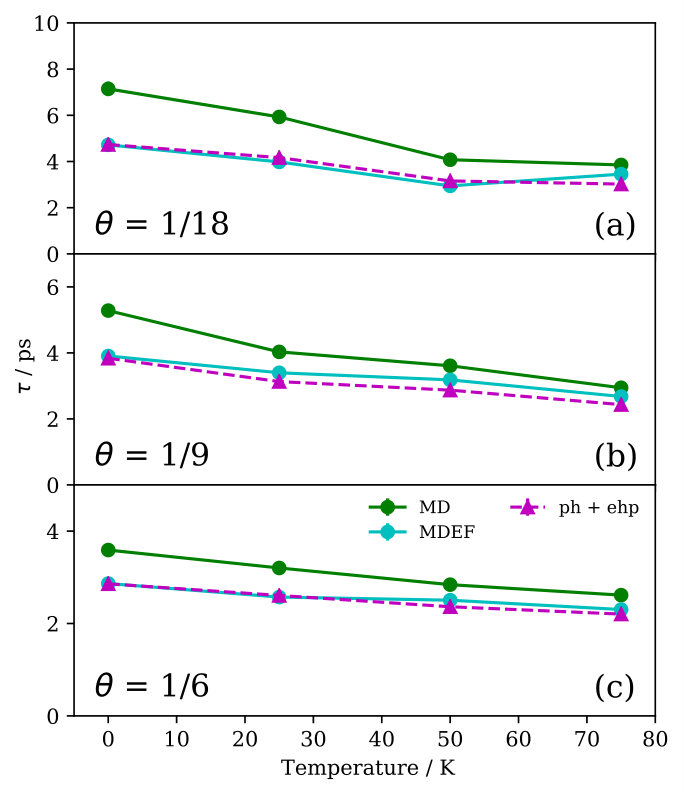 Figure 12
Figure 12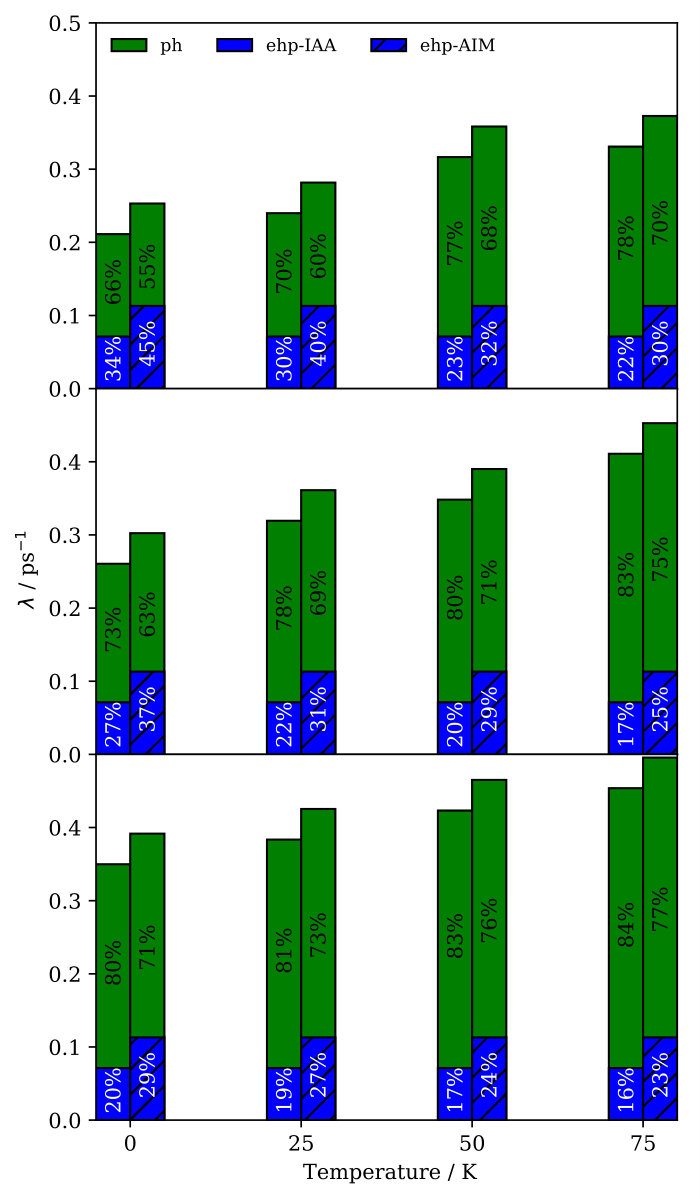 Figure 13
Figure 13Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSpectroscopy and Quantum Chemical Studies · Acoustic Wave Resonator Technologies · Advanced Chemical Physics Studies
Present address: ]Theory and Simulations of Materials (THEOS) and National Centre for Computational Design and Discovery of Novel Materials (MARVEL), École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland
Temperature-Dependent Lifetimes of Low-Frequency Adsorbate Modes from Non-Equilibrium Molecular Dynamics Simulations
Francesco Nattino
[
Leiden Institute of Chemistry, Leiden University, Gorlaeus Laboratories, P.O. Box 9502, 2300 RA Leiden, The Netherlands
Jörg Meyer
Leiden Institute of Chemistry, Leiden University, Gorlaeus Laboratories, P.O. Box 9502, 2300 RA Leiden, The Netherlands
Abstract
We present calculations on the damping of a low-frequency adsorbate mode on a metal surface, namely the frustrated translation of Na on Cu(100). For the first time, vibrational lifetimes of excited adlayers are extracted from non-equilibrium molecular dynamics calculations accounting for both the phononic and the electronic dissipation channels. The relative contributions of the two damping mechanisms, which we show to be additive, are found to disagree with textbook predictions. A simple model based on separable harmonic and anharmonic contributions is able to semi-quantitatively reproduce the temperature dependence of the computed lifetimes.
pacs:
68.35.Ja, 68.43.Pq, 63.22.Np, 34.35.+a,
Metal-based catalysts are widely employed in the chemical and energetic sectors Somorjai and Li (2011). Their role consists in providing a substrate for adsorption, diffusion and transformation of the reactants into the products, which eventually desorb from the catalyst surface Nilsson et al. (2008). The efficiency of the catalyst is crucially affected by the energy exchange between the reaction intermediates and the substrate, which on metal surfaces is ruled by the competition between phonon emission and electron-hole pair excitation. Disentangling the contribution of these two damping mechanisms is therefore extremely relevant for the fundamental understanding of dissipation processes at the atomic scale.
Small adsorbates on single-crystal metal surfaces represent ideal systems to investigate the energy exchange with the underlying substrate, as their vibrational damping can be accurately characterized e.g. through helium scattering experiments Graham et al. (1997a); Graham (2003). For adsorbate modes whose frequency falls well within the substrate phonon spectrum ( meV), textbook knowledge predicts vibrational lifetimes to be dominated by phonon excitations Graham (2003); Politano et al. (2013). According to this picture, the electronic contribution to the lifetime of the frustrated translation (FT) mode for alkali metal adatoms on transition metal surfaces is expected to be negligible Graham (2003); Politano et al. (2013). This expectation stands in clear contrast with the results of a recent study that focused on sodium atoms diffusing on Cu(111) Rittmeyer et al. (2016). In fact, calculations that followed theoretical developments Juaristi et al. (2008); Blanco-Rey et al. (2014); Rittmeyer et al. (2015) of the dynamics beyond the Born-Oppenheimer approximation Tully et al. (1993); Tully (1980); Head-Gordon and Tully (1995) have estimated the electronic dissipation mechanism to be as relevant as of the experimentally-determined friction coefficient Rittmeyer et al. (2016). This is despite the dispersionless FT-mode of Na adatoms on low-index Cu surfaces should very efficiently couple to bulk modes due to the large frequency overlap with the long-wavelength region of the substrate phonon continuum ( meV for Na on Cu(100) Graham et al. (1997a); Politano et al. (2013)).
Unfortunately, the phononic counterpart to the electronic channel in the vibrational damping of adsorbate modes is not straightforward to evaluate in a quantitative way Chen et al. (2018). On the one hand, perturbative approaches relies on the evaluation of the system’s third-order force constants Maradudin and Fein (1962), which typically involves a significant computational effort. On the other hand, molecular dynamics (MD) techniques require large simulation cells in order to represent bulk-projected phonons with a sufficient resolution over the surface Brillouin zone (SBZ) Meyer (2011); Meyer and Reuter (2014). In this scenario, the state-of-the-art method for estimating phonon-dominated vibrational lifetimes remains the elastic continuum model (ECM) initially proposed by Persson and Ryberg Persson and Ryberg (1985) and further developed for periodic adlayers by Rappe et al. Lewis et al. (1998); Pykhtin et al. (1998). This model is based on a continuum description of the substrate and it allows to estimate adsorbate mode lifetimes from a very limited set of system-specific parameters. Despite the simplicity of the ECM, Rappe and coworkers showed that its periodic-adlayer generalization could semi-quantitatively reproduce measured coverage-dependent lifetimes for the carbon monoxide FT-mode on Cu(100) Lewis et al. (1998); Pykhtin et al. (1998).
In this letter, we present an approach that has allowed us for the first time to directly extract adsorbate-mode lifetimes from non-equilibrium simulations. The low computational cost of classical MD calculations enables the atomistic description of large simulation cells, so that a dense sampling of the substrate SBZ is naturally obtained. At the same time, coverage and temperature effects can be realistically modeled. We have focused here on the damping of the FT-mode of Na adatoms on Cu(100), exploiting this prototypical system, relevant to catalysis Lackey and King (1987), to test the validity of the approximations underlying the ECM. We have found a pronounced temperature dependence for the Na FT-mode lifetime, in agreement with experimental trends Graham et al. (1997a); Politano et al. (2013). Moreover, by accounting for electron-phonon coupling via Langevin electronic friction dynamics Tully (1980); Tully et al. (1993); Head-Gordon and Tully (1995); Juaristi et al. (2008) we have found a large electronic contribution to the lifetime, in agreement with the recent simulations from Rittmeyer et al. Rittmeyer et al. (2016). Most importantly, our study produces valuable insight into the harmonic and anharmonic contributions to the phonon-mediated adsorbate damping and to the additivity of the electronic and the phononic dissipation channels.
We base our calculations on a specifically-constructed potential of the embedded atom method (EAM) form Daw and Baskes (1983), which very accurately reproduces the experimentally measured surface phonon band structure for Na on Cu(100) Senet et al. (1999); Graham (2003); SM . Furthermore, it also yields excellent agreement with the Na-Cu interaction potential obtained from helium atom scattering experiments Graham et al. (1997a, b); SM . Based on this potential we perform classical molecular dynamics (MD) simulations with the LAMMPS code Plimpton (1995) for different Na-adlayer structures (vide infra) and including temperature by pre-equilibrating the entire system West and Estreicher (2007); SM . In order to ensure a proper theory-theory comparison, we use the formulation of the ECM for periodic adlayers by Rappe et al. Lewis et al. (1998); Pykhtin et al. (1998) and obtain all parameters required (bulk lattice constant, density, transversal as well as longitudinal speeds of sound and adsorbate frequency) directly from bulk and surface calculations with our EAM potential SM .
Consistent with the previous work on the ECM and representative for experimental conditions encountered in optical pump-probe experiments Watanabe et al. (2005); Fuyuki et al. (2007), we initially excite a FT by depositing an amount of energy larger than the thermal background into the translational degrees of freedom of the Na adatoms , such that all of them initially vibrate in phase along the same crystallographic direction ( excitation). Constant energy MD calculations for these non-equilibrium initial conditions (NEMD) combined with a phonon mode projection scheme McGaughey and Kaviany (2004); Meyer (2011) allow us to follow the equilibration of the small amount of excess energy within the phonon bath. Inspired by the work of Estreicher et al. on the vibrational lifetimes of bulk defects West and Estreicher (2006), we can accurately quantify the lifetime by fitting an exponential decay function to the energy in the projected phonon mode corresponding to the FT after averaging over 100 trajectories SM . Compared to the pioneering work of Tully Tully (1980); Tully et al. (1993), which has relied on time correlation functions of a harmonic system at equilibrium, our approach is much more direct and more accurate.
We have simulated three overlayer structures, illustrated in Fig. 1 (a)-(c): the experimentally observed \left(\begin{array}[]{cc}3&0\\ 1&2\end{array}\right) adsorption structure Graham and Toennies (1997) that corresponds to a surface coverage of a monolayer (ML) and two representative structures for lower coverages, namely the p and the c structures, corresponding to ML and ML, respectively. For each coverage, Na atoms are adsorbed in a fourfold hollow site. Slabs with 24 layers of copper atoms have been employed, based on a multiple of the primitive Cu(100) surface unit cell with fully-relaxed interlayer distances and a surface lattice constant that is commensurate with all three overlayer structures. During the MD simulations the bottommost layer has conveniently been kept frozen without any consequences for the reported lifetimes, which have all been thoroughly checked for convergence with respect to the lateral cell size of the supercell and the slab thickness SM .
The lifetimes obtained for the three overlayer structures when the surface atoms are initially at rest ( K) are presented in Fig. 1 (d), where we compare them to the lifetimes calculated with the ECM. While the ECM predicts accurate lifetimes at the highest coverages, it considerably overestimates the lifetime for the c structure (). Since the parameters of the ECM have been consistently computed with the EAM potential employed in the NEMD simulations, the observed discrepancy has to arise from the breakdown of some of the approximations on which the ECM is based. On the one hand, the continuum approximation for the substrate does not necessarily apply in the context of damping of atomic vibrations. On the other hand, the harmonic approximation that is inherent to the ECM could be too severe to realistically describe the adsorbate coupling to the substrate or the energy transport within the substrate.
We can assess the validity of this harmonic approximation at K by performing a second type of dynamical calculations. The initial excitation of the FT localized in the Na adlayer is described by a superposition of many phonon modes of the strongly-coupled adlayer-substrate system. Our phonon mode projection scheme directly yields the initial components of this phonon wave packet with their amplitudes and phases. The group velocity of this wave packet characterizes the energy transport away from the adlayer into the surface. Unfortunately, this group velocity cannot be easily obtained from the surface phonon band structure, since the wave packet is a linear combination of different (surface) phonon bands at a single phonon wave vector . Instead, in order to describe a harmonic bath with harmonic coupling to the adsorbate layer, we simply switch off all the phonon-phonon couplings between the components of this wave packet by analytically propagating them in the eigenbasis defined by the phonon modes, using the aforementioned amplitudes and phases as initial conditions SM . Lifetimes calculated from these purely harmonic dynamics, also presented in Fig. 1 (d), are in overall good agreement with the values extracted from the NEMD calculations, demonstrating a negligible role of anharmonicity (i.e. phonon-phonon coupling) at K.
Consequently, we have clearly identified the continuum approximation in the ECM as responsible for the large overestimation of the lifetime at low coverages (cf in Fig. 1). Vibrationally excited periodic adlayers can transfer energy to the underlying surface only through substrate phonons that are commensurate to the overlayer structure Lewis et al. (1998); Pykhtin et al. (1998). The range of accessible wavevectors in the SBZ of the substrate becomes narrower for increasing coverages, so that at the highest coverages the adlayer modes can only couple to the substrate phonons with the longest wavelengths. It is in this limit where the elastic continuum approximation is best justified. On the contrary, the lower the coverage, the more phonon modes are backfolded into the smaller and smaller SBZ of the adlayer and thus become accessible – rationalizing the strong effect of the exact surface phonon band structure as captured by our NEMD simulations.
At finite temperature K the situation is quite different, since we have found strong deviations from the harmonic limit. This is illustrated in Fig. 2, where we present Na FT-mode lifetimes calculated in a temperature range at which Na atom diffusion is still negligible ( K). For all coverages we observe a considerable reduction of at K, which even amounts to about 50% at . This temperature dependence can be rationalized by a simple model that separates a temperature-independent harmonic contribution and temperature-dependent anharmonic contribution to the lifetime. We use as described above for the contribution resulting from the (analytic) propagation of the initially excited phonon wavepacket in the harmonic system defined by the phonon modes. In order to quantify the anharmonic contribution , we change the initial conditions in our NEMD calculations to initially exciting the single phonon eigenmode at that has the largest overlap with the FT localized in the Na adlayer excited otherwise SM . The initial excitation energy in this mode equilibrates because of phonon-phonon coupling in the anharmonic bath with anharmonic coupling to the adsorbate layer. Assuming that the harmonic and anharmonic contributions to the lifetime are separable in this way, the corresponding rates should be additive:
[TABLE]
Using our calculated results for and , Fig. 2 shows that this simple model indeed almost quantitatively reproduce the temperature dependence of for all coverages. Consistent with our results above, this model also confirms the negligible role of anharmonicity at K, i.e. since . For increasing temperatures, rapidly decreases ( ps at 75 K) and we consistently observe increasing deviations from the harmonic limit. As expected, the temperature dependence of is therefore completely given by the anharmonic contribution which quantifies the increasing relevance of phonon-phonon couplings for the lifetime with increasing temperatures.
Finally, we investigate the contribution of electron-phonon coupling for the lifetimes by implementing Langevin dynamics on top of our NEMD calculations. The strength of the electron-phonon coupling is given by a (constant) electronic friction (EF) coefficient Head-Gordon and Tully (1995), for which we choose a value that has been obtained from an atoms-in-jellium model and which is representative for Na on copper surfaces Rittmeyer et al. (2016); SM . Results for the lifetime obtained from these NEMDEF simulations are presented in Fig. 3. As expected, , because an additional energy dissipation channel is being accounted for.
Using the same argument as in Eq. 1, the following equation should hold if the phononic and electronic channels are independent from one antoher:
[TABLE]
We quantify the eletron-hole pair contribution to the lifetime simply by , where is the adsorbate mass. Fig. 3 shows The excellent match between the lifetimes predicted through NEMDEF simulations and the lifetimes calculated from the independent phononic and electronic contributions confirms the additivity of the two damping mechanisms over the whole temperature range considered (Fig. 3). This conclusion does not depend on the value of the friction coefficient applied, as similar results are obtained through NEMDEF calculations based on a different estimate Rittmeyer et al. (2016); SM . Note that our comprehensive model predicts the electronic dissipation contribution to represent between 16% and 23% of the total damping for K and , depending on the model employed to estimate SM . This range is in very good agreement with what calculated by Rimmeyer et al., who have estimated a 16%-26% range for the relative contribution of the electron-hole pair dissipation channel for Na atoms diffusing on Cu(111) under similar conditions of coverage ( = 2.5% ML) and temperature ( K) Rittmeyer et al. (2016).
Overall, our estimate of the Na FT-mode lifetime is not so sensitive on the exact value of the friction coefficient applied, with the largest deviations between the two models being . However, the lifetime strongly depends on the coverage structure simulated. Due to the absence of a detailed knowledge about the Na overlayer structure in the available measurements Graham et al. (1997a); Politano et al. (2013), we cannot perform a straightforward theory-experiment comparison using these data. We note, however, that the predicted temperature dependence is consistent with experimental observations of significant lifetime decrease with increasing temperature Graham et al. (1997a); Politano et al. (2013). Lifetime measurements for well-characterized overlayer structures would be desirable for further testing and developments of theoretical models that aim at describing the energy exchange at interphases.
Acknowledgements.
J.M. is grateful for financial support from the Netherlands Organisation for Scientific Research (NWO) under VIDI Grant No. 723.014.009.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Somorjai and Li (2011) G. A. Somorjai and Y. Li, Proc. Natl. Acad. Sci. U. S. A. 108 , 917 (2011) . · doi ↗
- 2Nilsson et al. (2008) A. Nilsson, L. G. M. Pettersson, and J. K. Nørskov (Eds.), Chemical Bonding at Surfaces and Interfaces (Elsevier, Amsterdam, 2008).
- 3Graham et al. (1997 a) A. P. Graham, F. Hofmann, J. P. Toennies, L. Y. Chen, and S. C. Ying, Phys. Rev. Lett. 78 , 3900 (1997 a) . · doi ↗
- 4Graham (2003) A. P. Graham, Surf. Sci. Rep. 49 , 115 (2003) . · doi ↗
- 5Politano et al. (2013) A. Politano, G. Chiarello, G. Benedek, E. Chulkov, and P. Echenique, Surf. Sci. Rep. 68 , 305 (2013) . · doi ↗
- 6Rittmeyer et al. (2016) S. P. Rittmeyer, D. J. Ward, P. Gütlein, J. Ellis, W. Allison, and K. Reuter, Phys. Rev. Lett. 117 , 196001 (2016) . · doi ↗
- 7Juaristi et al. (2008) J. I. Juaristi, M. Alducin, R. Díez Muiño, H. F. Busnengo, and A. Salin, Phys. Rev. Lett. 100 , 116102 (2008) . · doi ↗
- 8Blanco-Rey et al. (2014) M. Blanco-Rey, J. I. Juaristi, R. Díez Muiño, H. F. Busnengo, G. J. Kroes, and M. Alducin, Phys. Rev. Lett. 112 , 103203 (2014) . · doi ↗