Electron Correlation Effects in Attosecond Photoionization of CO$_{2}$
Andrei Kamalov, Anna L. Wang, Philip H. Bucksbaum, Daniel J. Haxton,, and James P. Cryan

TL;DR
This paper investigates how multi-electron interactions influence attosecond-scale photoionization delays in CO₂, revealing effects of autoionization and shape resonances on electron escape times.
Contribution
It combines experimental measurement techniques with theoretical calculations to elucidate electron correlation effects in molecular photoionization.
Findings
Electron correlation impacts time delays via autoionization of Rydberg states.
Shape resonances accelerate electron escape, affecting measured delays.
Multi-electron dynamics are significant in attosecond photoionization of CO₂.
Abstract
A technique for measuring photoionization time delays with attosecond precision is combined with calculations of photoionization matrix elements to demonstrate how multi-electron dynamics affect photoionization time delays in carbon dioxide. Electron correlation is observed to affect the time delays through two mechanisms: autoionization of molecular Rydberg states and accelerated escape from a continuum shape resonance.
Click any figure to enlarge with its caption.
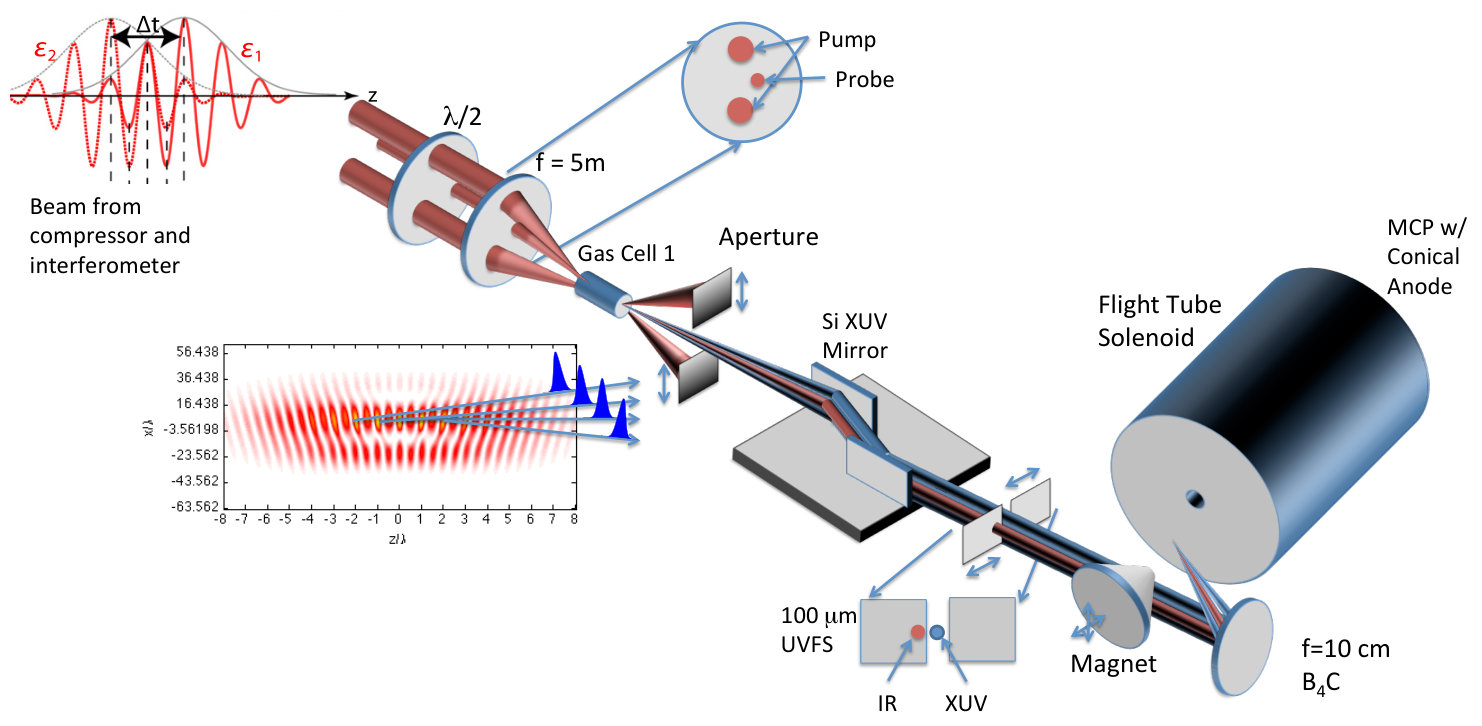 Figure 1
Figure 1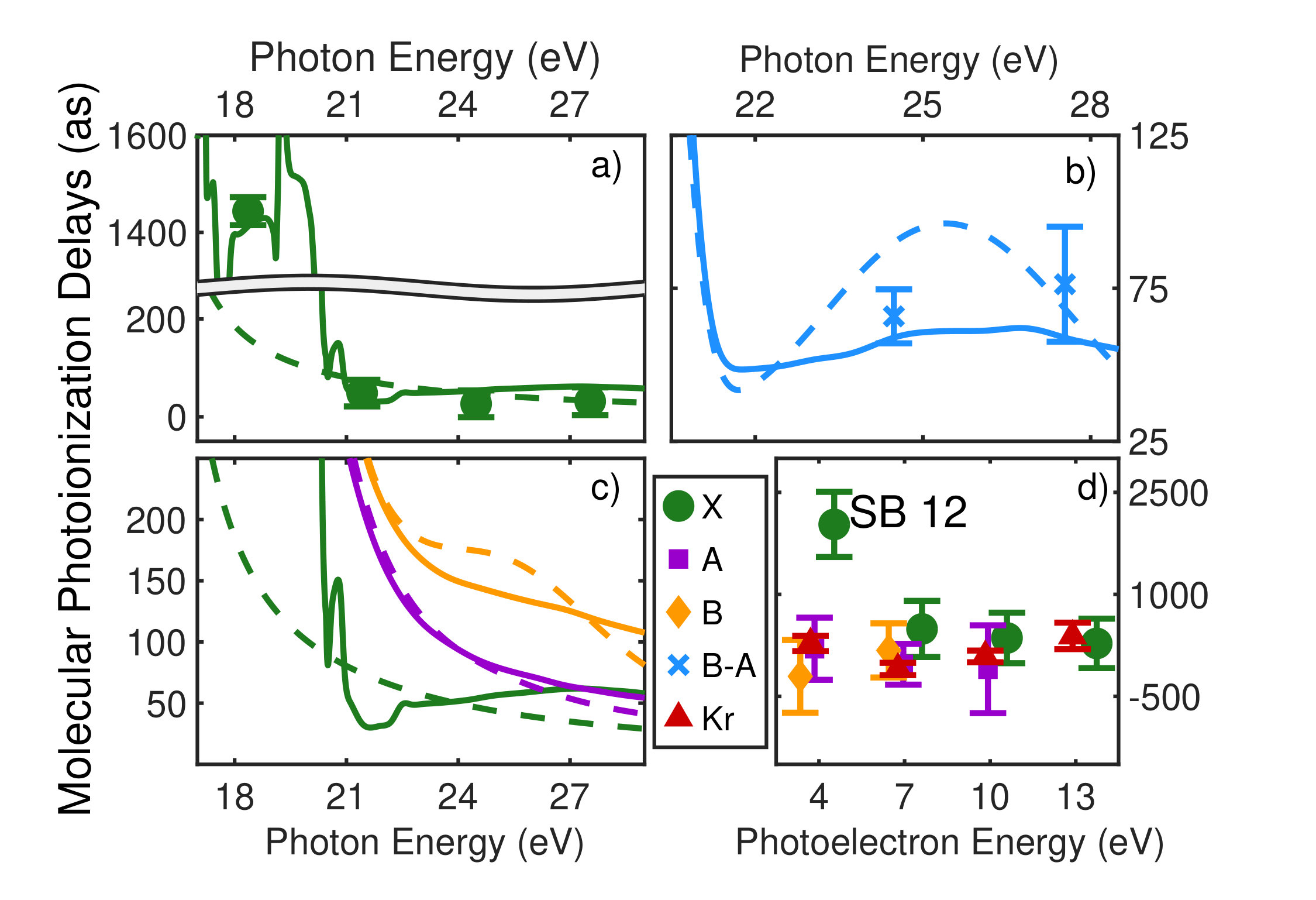 Figure 2
Figure 2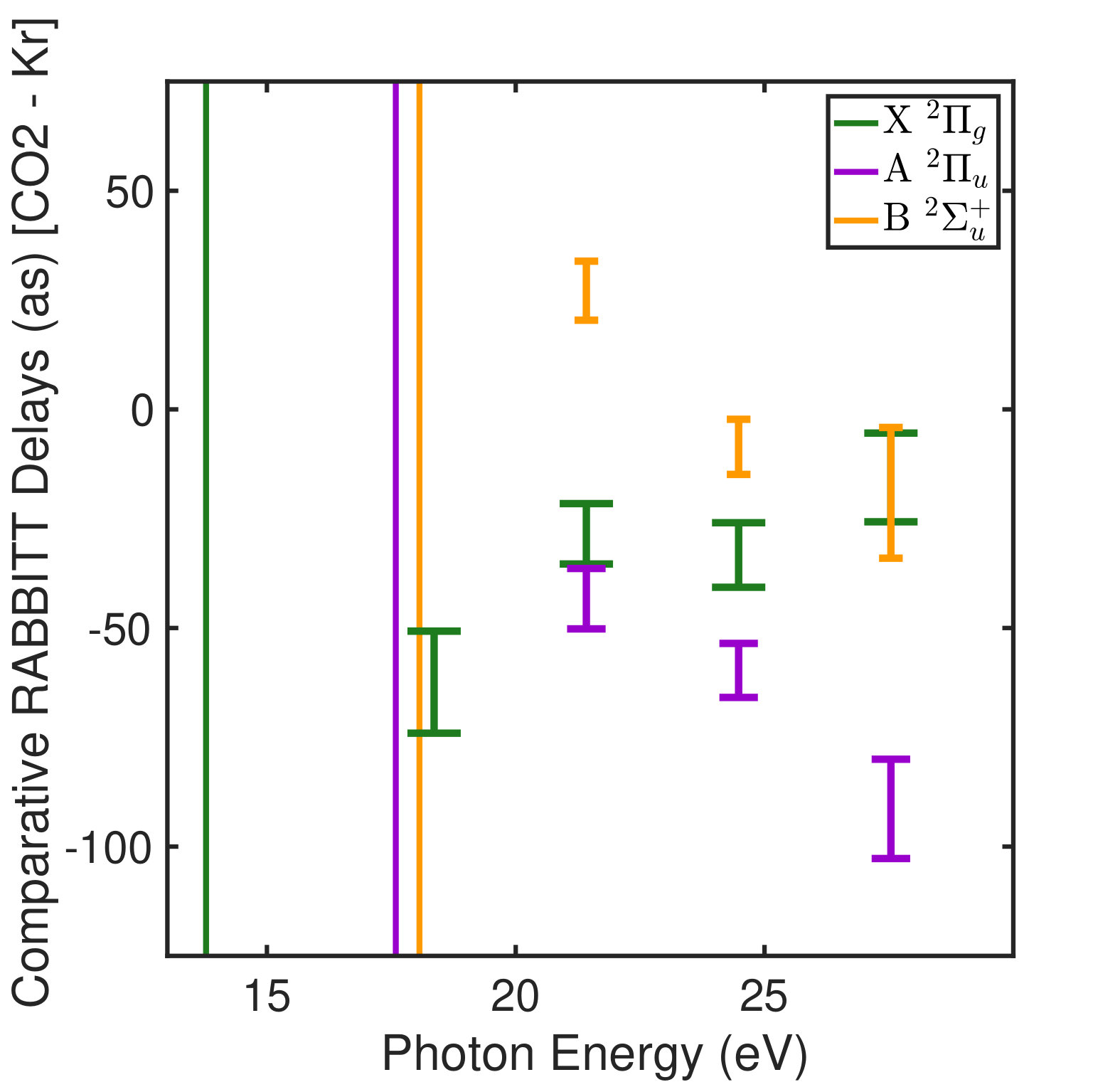 Figure 3
Figure 3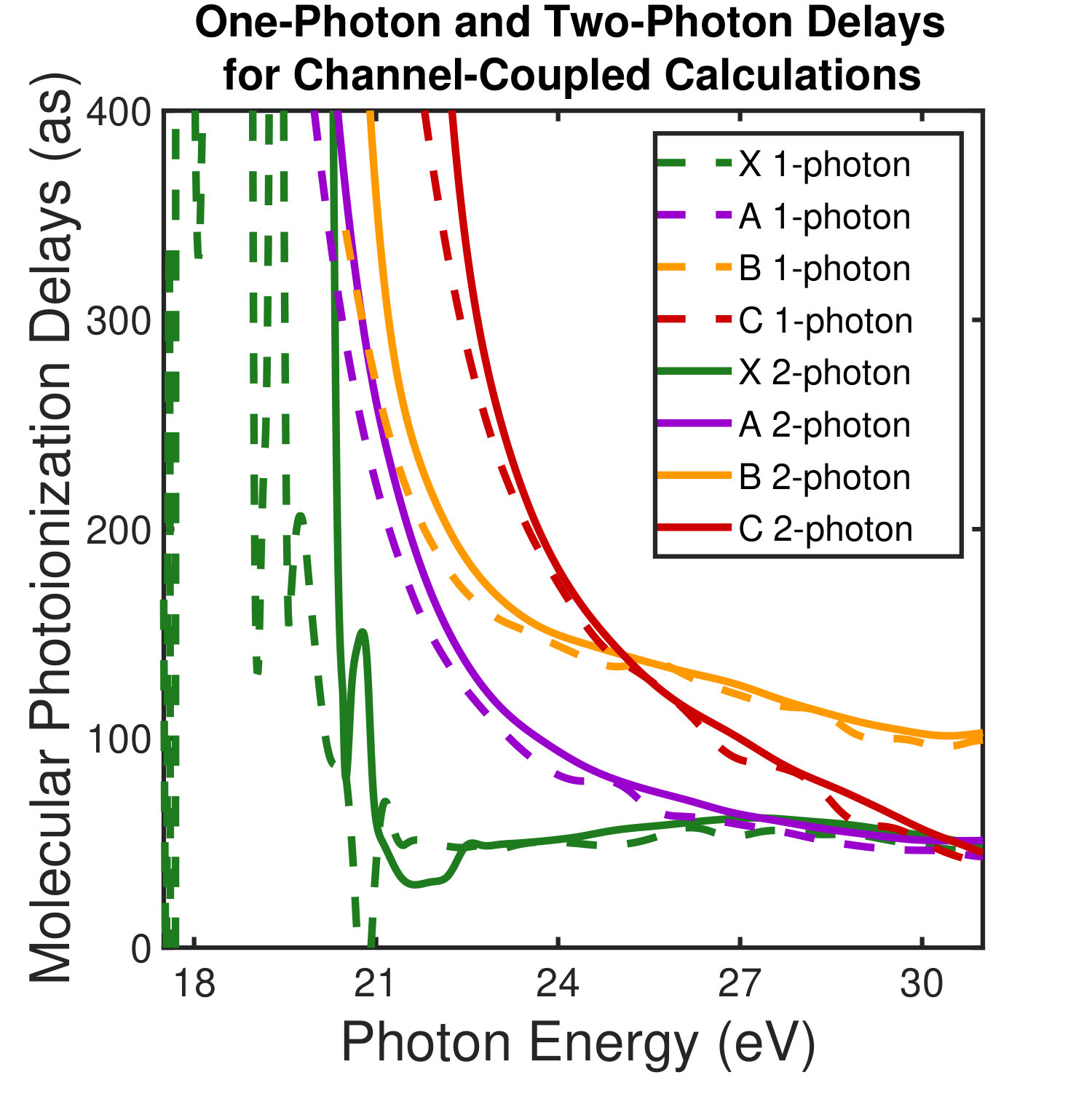 Figure 4
Figure 4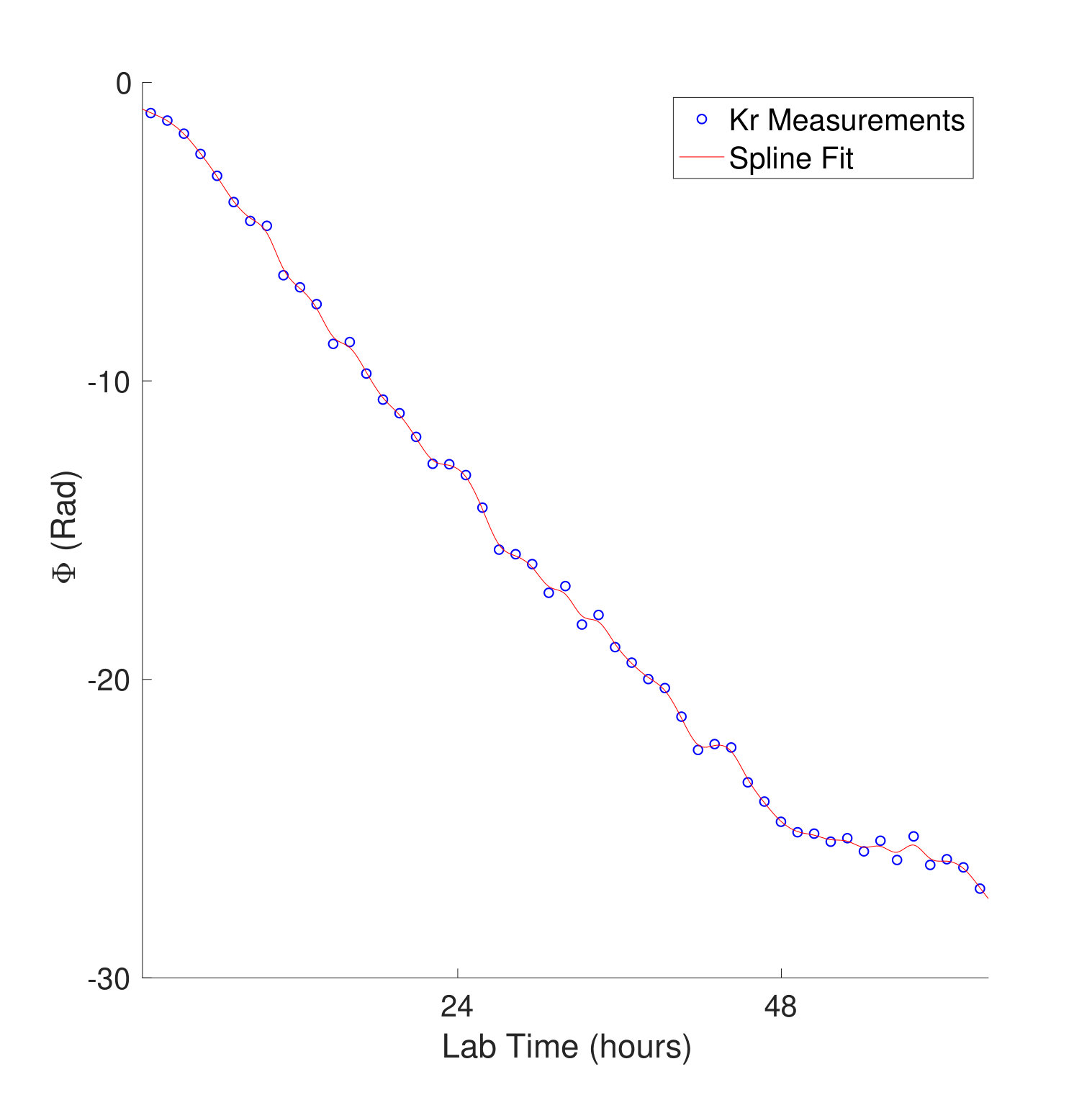 Figure 5
Figure 5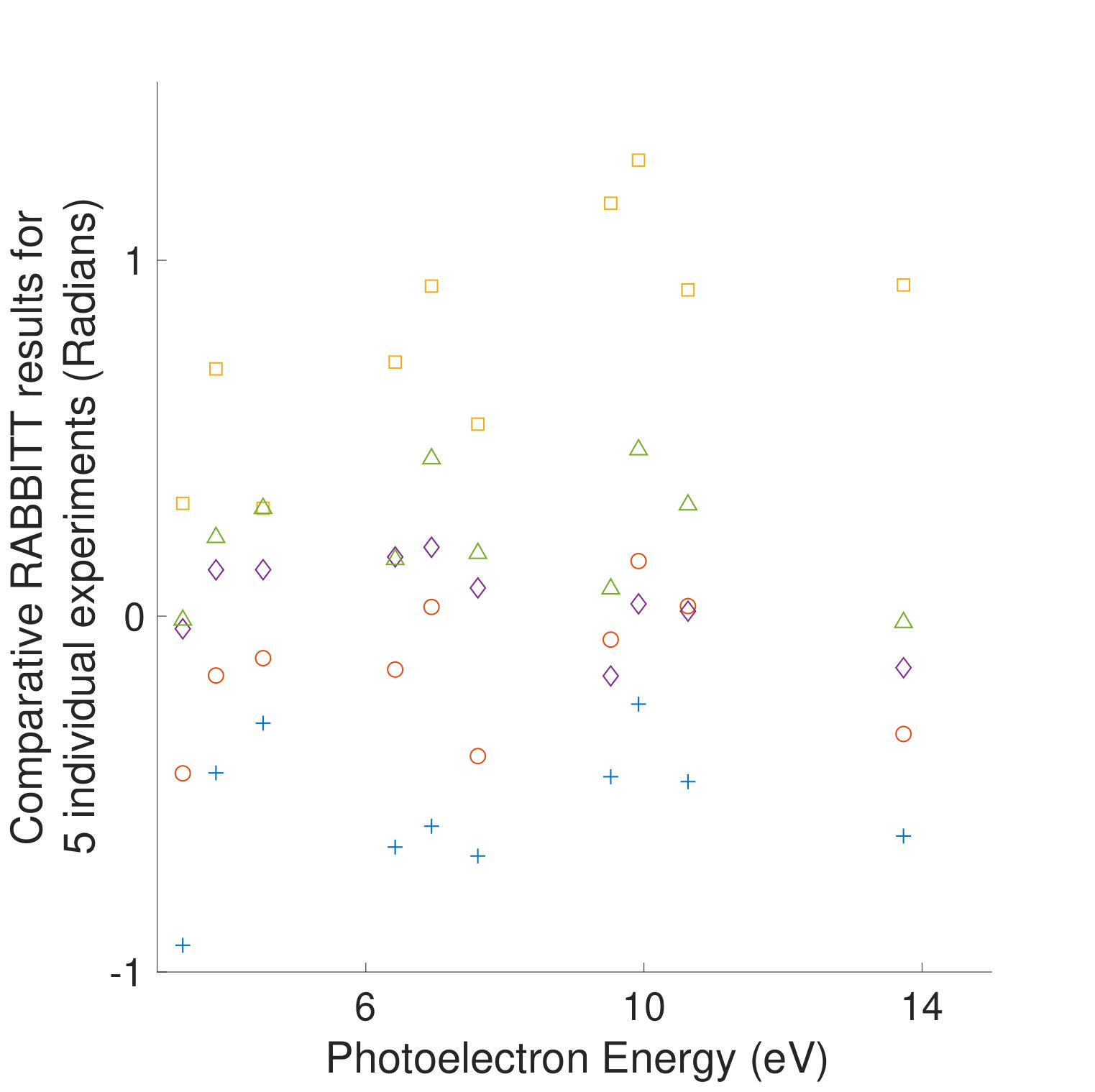 Figure 6
Figure 6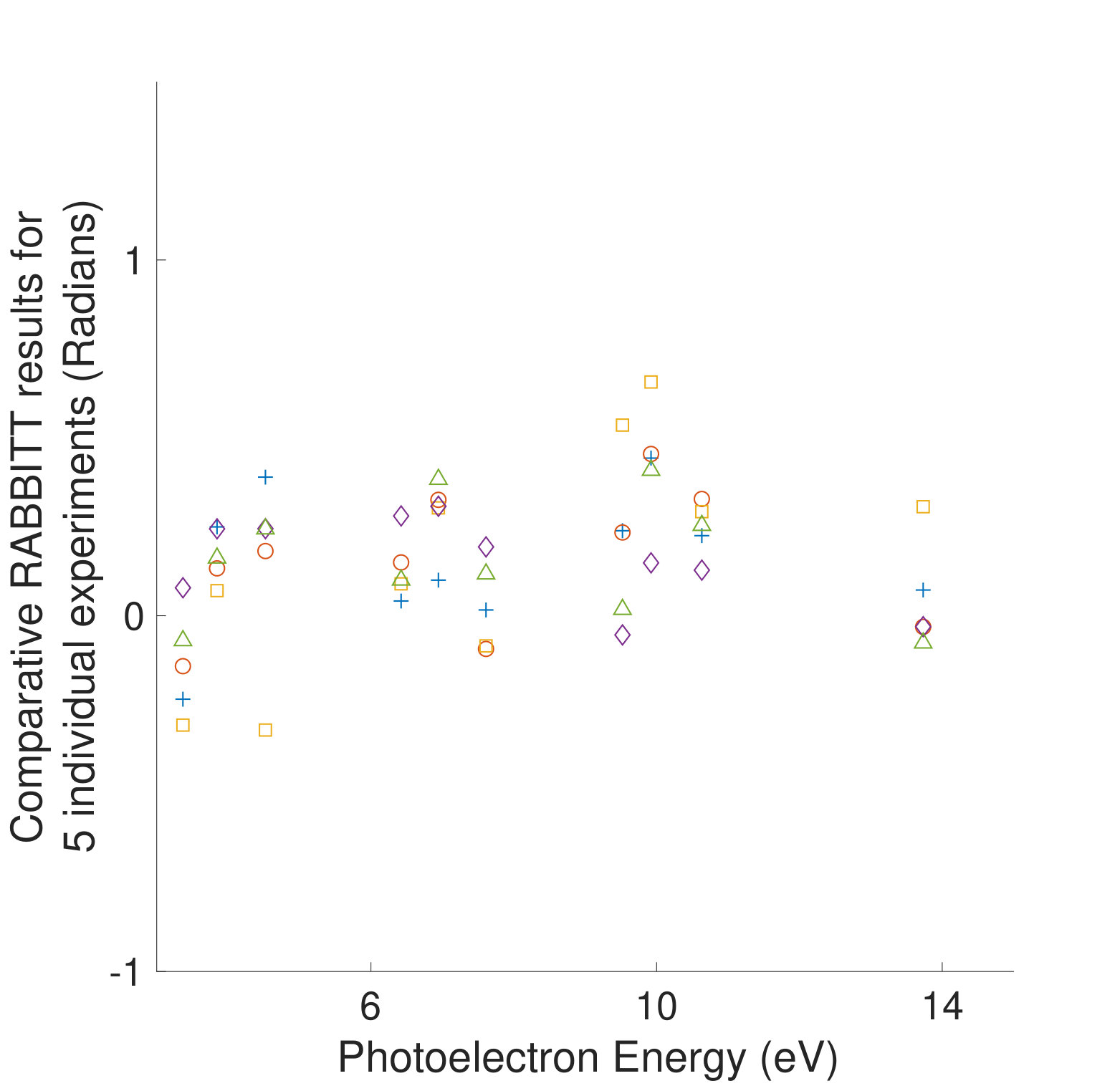 Figure 7
Figure 7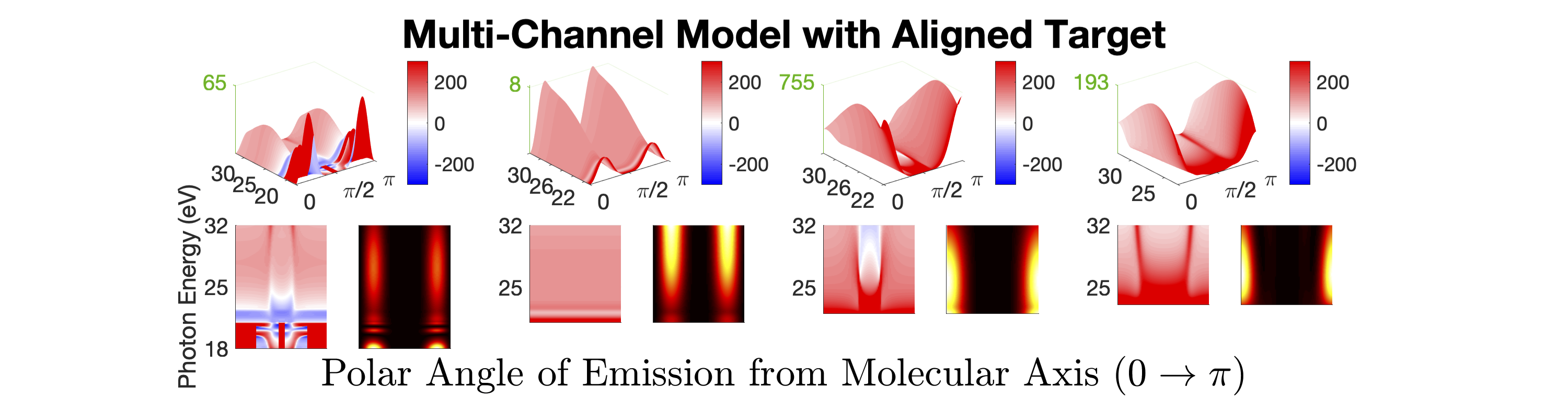 Figure 8
Figure 8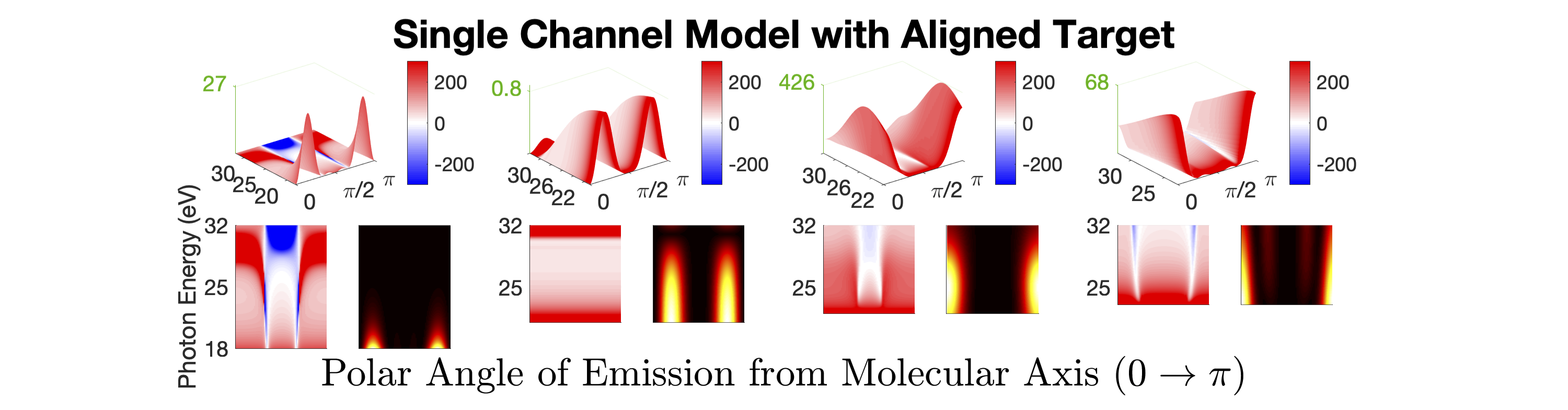 Figure 9
Figure 9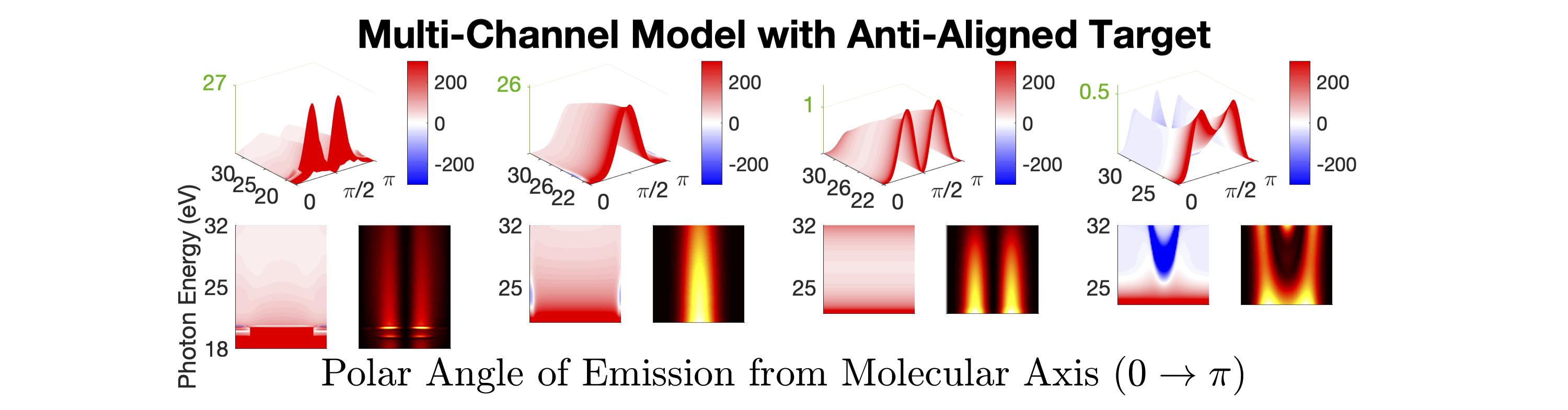 Figure 10
Figure 10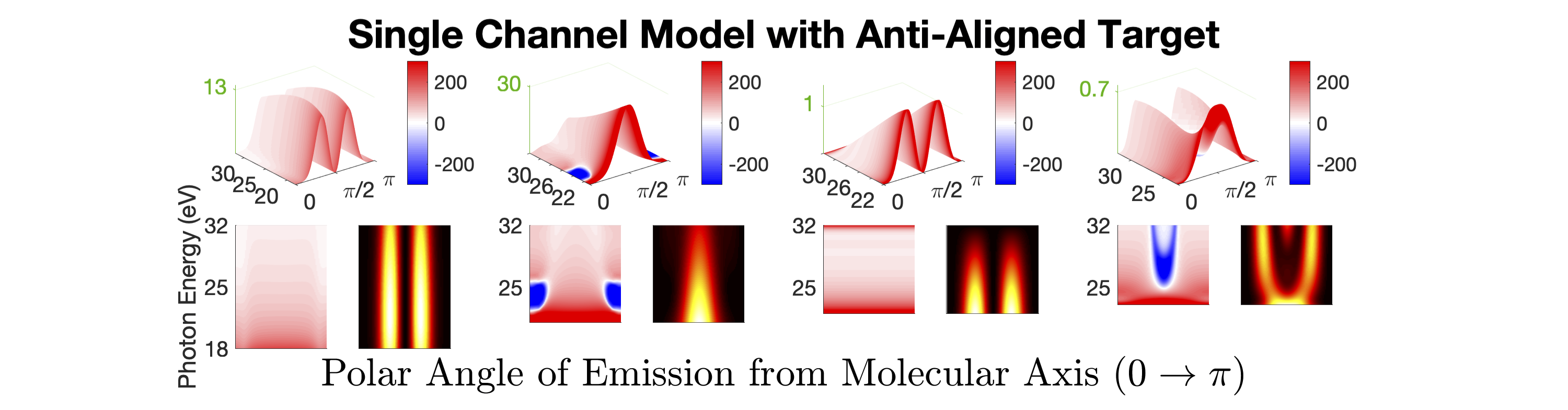 Figure 11
Figure 11 Figure 12
Figure 12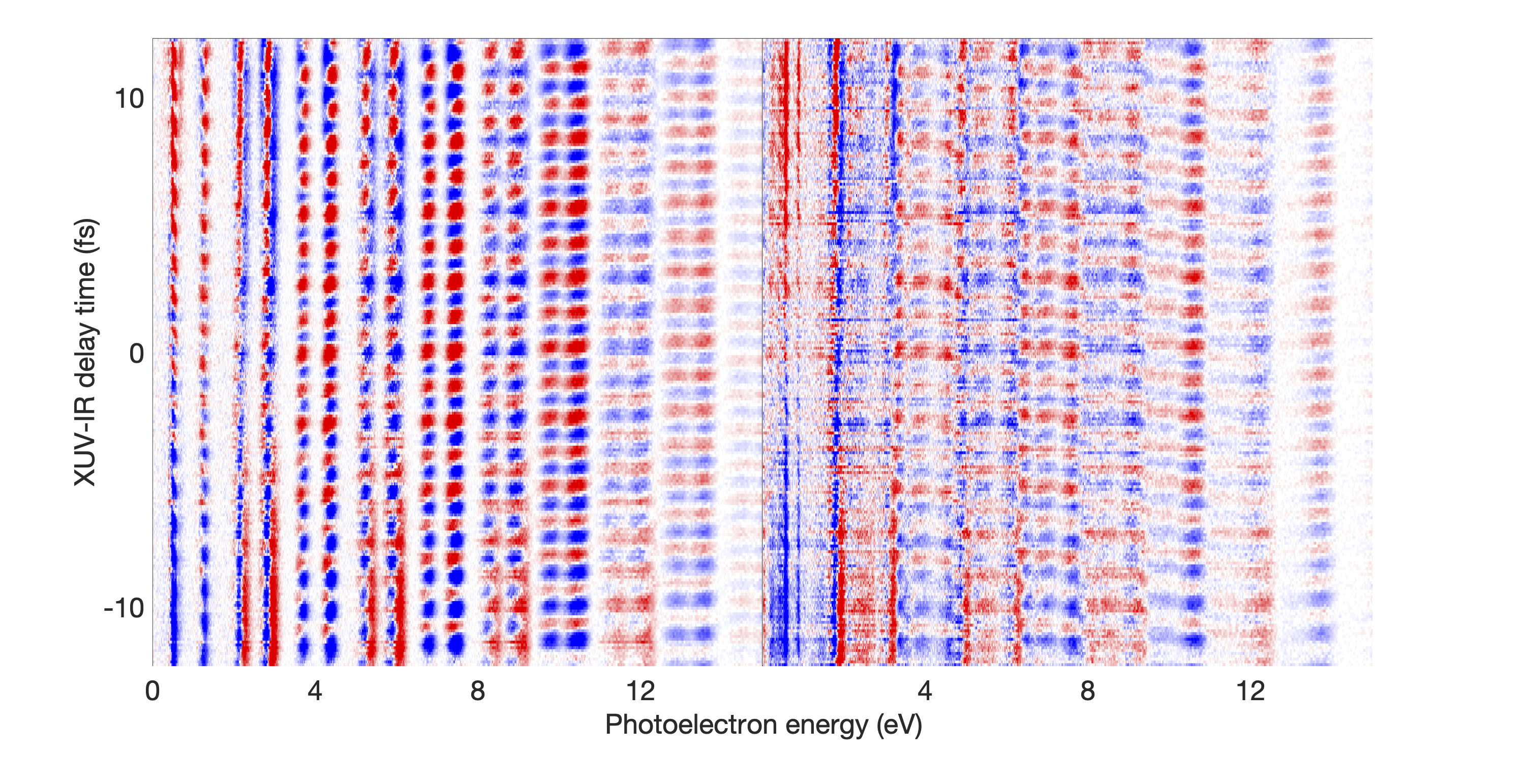 Figure 13
Figure 13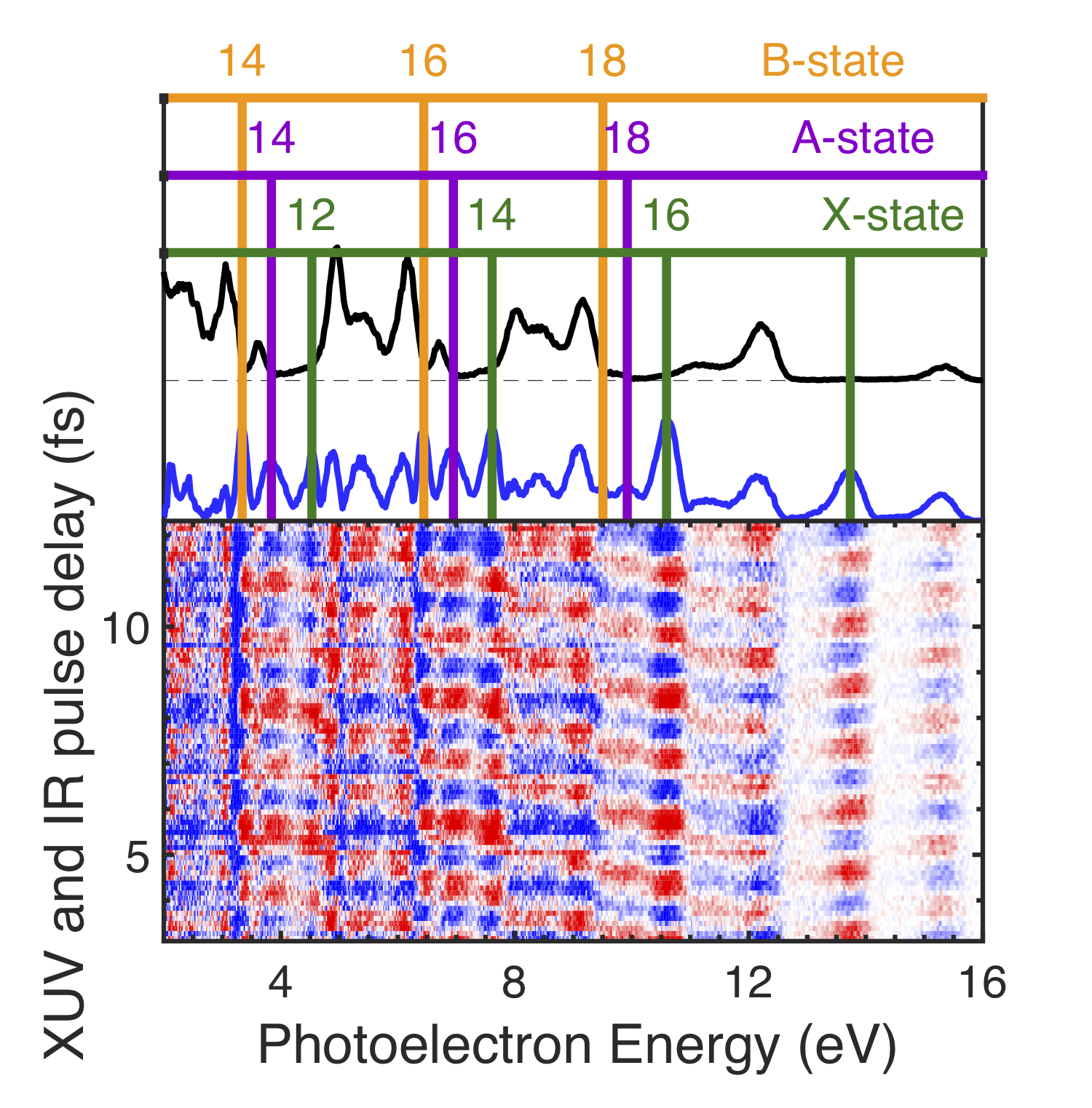 Figure 14
Figure 14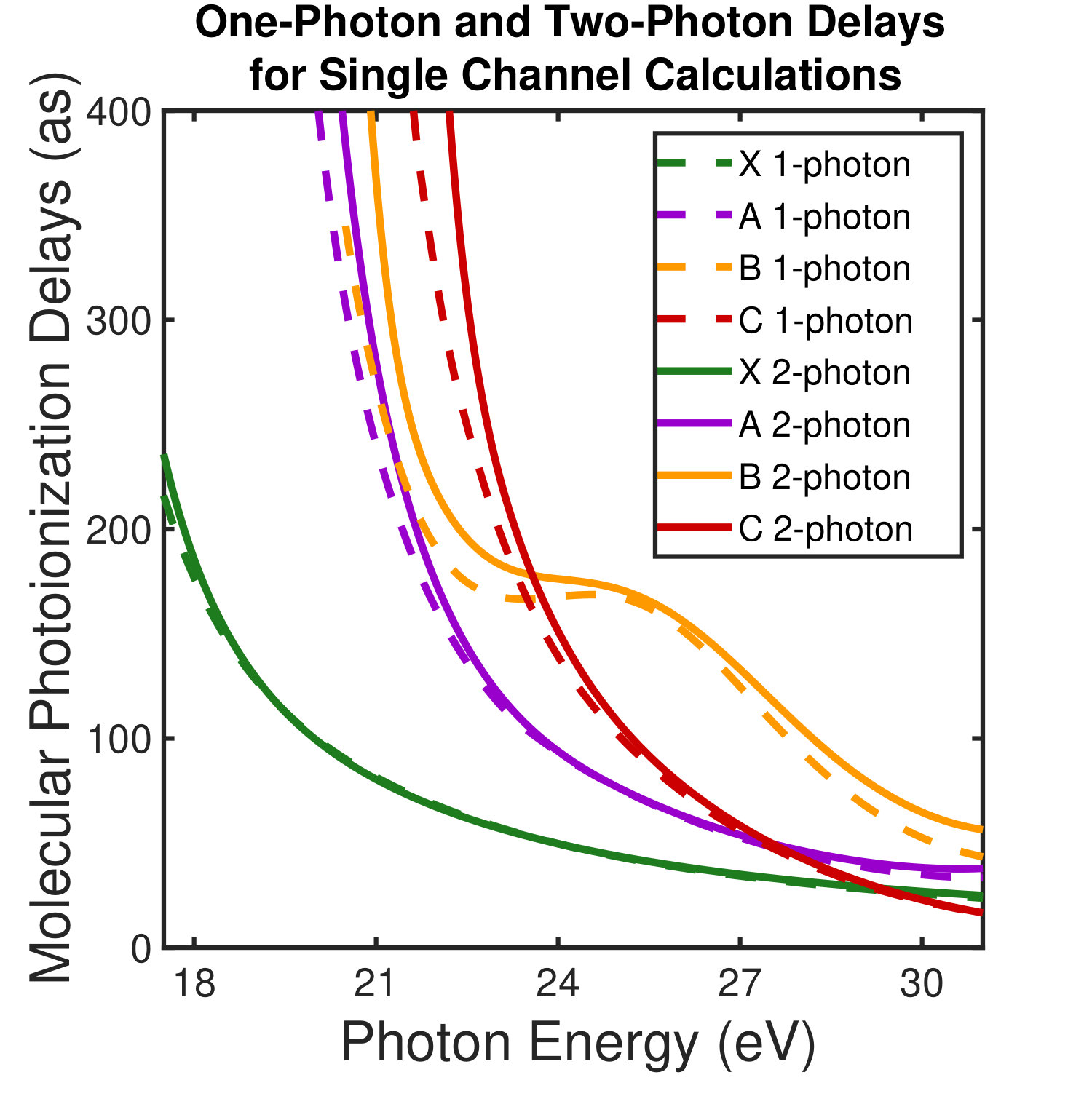 Figure 15
Figure 15Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Electron Correlation Effects in Attosecond Photoionization of CO2
Andrei Kamalov
Stanford PULSE Institute, SLAC National Accelerator Laboratory, Menlo Park, CA, USA
Department of Physics, Stanford University, Stanford, CA, USA
Anna L. Wang
Stanford PULSE Institute, SLAC National Accelerator Laboratory, Menlo Park, CA, USA
Department of Applied Physics, Stanford University, Stanford, CA, USA
Philip H. Bucksbaum
Stanford PULSE Institute, SLAC National Accelerator Laboratory, Menlo Park, CA, USA
Department of Physics, Stanford University, Stanford, CA, USA
Department of Applied Physics, Stanford University, Stanford, CA, USA
Daniel J. Haxton
KLA Corporation, Milpitas, CA, USA
James P. Cryan
Stanford PULSE Institute, SLAC National Accelerator Laboratory, Menlo Park, CA, USA
Linac Coherent Light Source, SLAC National Accelerator Laboratory, Menlo Park, CA, USA
Abstract
A technique for measuring photoionization time delays with attosecond precision is combined with calculations of photoionization matrix elements to demonstrate how multi-electron dynamics affect photoionization time delays in carbon dioxide. Electron correlation is observed to affect the time delays through two mechanisms: autoionization of molecular Rydberg states and accelerated escape from a continuum shape resonance.
Photoionization is a basic quantum scattering process involving the rearrangement of degrees freedom in the total system. In the time domain, this is described by an incoming photon wavepacket that couples to outgoing electron wavepackets (EWPs) in the final-state cation channels. The term “photoionization time delay” refers to the time required for a photoionized EWP to propagate out of the electric potential of the residual cation. It may be defined semi-classically as the extra time required to propagate a photoelectron from its birth location to a detector position, compared to some reference Dahlström et al. (2012); Klünder et al. (2011); Serov et al. (2013). Recent advances in the production of attosecond laser pulses have enabled direct probing of these delays Pazourek et al. (2015). Combining these time delay measurements with theoretical modeling reveals the underlying quantum dynamics of the photoionization process Dahlström et al. (2012); Pazourek et al. (2015); Schultze et al. (2010); Klünder et al. (2011); Dahlström et al. (2012); Guénot et al. (2012); Kheifets (2013); Serov et al. (2013); Guénot et al. (2014); Palatchi et al. (2014); Sabbar et al. (2015); Ossiander et al. (2017); Isinger et al. (2017); Kiesewetter et al. (2017); Busto et al. (2018).
Attosecond electron dynamics of ionization are necessarily violent because additional kinetic energy must be imparted to the bound electron for it to escape the Coulomb potential. The added kinetic energy may be redistributed through Coulomb and exchange scattering with other electrons, dynamically modifying this ionic potential. This is particularly important in molecular systems. Although the asymptotic state of the total system (cation plus ionized electron) is easily understood in a single-electron picture, this picture may break down when the the electron has not yet escaped into the asymptotic region for detection. The modification of the ionic potential is imprinted onto the measured photoionization time delays Pazourek et al. (2015), which provides direct access to the temporal evolution of electron-electron interactions. Previous measurements of photoionization time delays have made use of this effect, which has led to a deeper understanding of electron correlations in shake-up ionization Ossiander et al. (2017) and atomic autoionization Gruson et al. (2016); Kotur et al. (2016); Cirelli et al. (2018); Busto et al. (2018). The present work combines measurements of the photoionization time delays with numerical calculations of photoionization probability amplitudes to demonstrate how multi-electron dynamics affect ionized EWPs in a molecular system.
These dynamics leave a clear signature in the measured photoionization time delay in the vicinity of autoionizing and molecular shape resonances. The enhancement of electron correlation effects near molecular shape resonances was previously considered by Siggel et al.. They found that the photoelectron angular distribution can be sensitive to multi-electron channel coupling phenomena Siggel et al. (1993). The scattering angle is one of two semi-classical scattering observables; the other is time delay. The effect that electron-electron interactions would have on the interpretation of photoionization time delay measurements has yet to be considered in the literature.
Carbon dioxide (CO2) provides a particularly striking example of multi-electron dynamics in molecular photoionization Lucchese and McKoy (1982); Lucchese (1990); Lucchese and McKoy (1981); Harvey et al. (2014a). Straightforward close-coupling expansions require ninety-six individual cation configurations to reproduce the experimental cross section Harvey et al. (2014a), far more states than are energetically available as photoionization channels. The virtual excitations of the closed cationic channels correspond to multiple-electron excitations of the molecular system, which affect the magnitude and phase of the EWPs escaping into the energetically open channels. In the present work we measure the photoionization time delays for CO2 and demonstrate that agreement with calculated time delays is contingent upon including electron correlation effects in the calculation.
Figure 1 shows the measured photoionization time delays for the , , and cationic states of CO2. Details of the measurement procedure are given after a discussion of the results. In order to understand the dynamics captured in the time delay measurements, we compare these data with predicted delays calculated using an implementation Rescigno and Orel (1981); Rescigno and Schneider (1988); Schneider and Rescigno (1988); Orel and Rescigno (1990); Rescigno and Orel (1991); McCurdy and Rescigno (1992); Rescigno et al. (1997); Rescigno and McCurdy (1998); McCurdy et al. (1998) of the complex Kohn variational method Kohn (1948); Nesbet (1968, 1969); Res (1979); McCurdy et al. (1987); Zhang et al. (1988); Rescigno et al. (1995) for photoionization Rescigno et al. (1993); Orel and Rescigno (1997); Miyabe et al. (2009); Sann et al. (2011); Williams et al. (2012); Douguet et al. (2012); Marggi Poullain et al. (2014); Fonseca dos Santos et al. (2015); McCurdy et al. (2017); Champenois et al. (2019) and electron-molecule scattering Hazi et al. (1981); Rescigno et al. (1989); Schneider et al. (1991); Lengsfield and Rescigno (1991); Gil et al. (1993); Rescigno et al. (1999, 2006); Adaniya et al. (2009); Slaughter et al. (2016); Rescigno et al. (2016). The photoionization time delays are calculated in two different levels of approximation and then averaged over molecular orientation and outgoing electron direction, consistent with the measurement scheme used in the experiment. The independent channel method considers the scattering in each continuum channel separately. The coupled-channel method uses fully coupled continuum states, which allows electrons originally produced in one ionization channel to interact with the residual ionic core to produce different final state configurations. More details of the complex-Kohn calculation are given below and in the supplemental material SM .
The independent channel calculations (dashed lines in Figure 1) for the and channels display traditional Coulombic behavior: monotonically increasing photoionization delay with decreasing photoelectron energy Klünder et al. (2011). The channel exhibits an increased photoionization delay time around 25 eV, which is a signature Baykusheva and Wörner (2017); Hockett et al. (2016); Huppert et al. (2016) of a weak shape resonance that has been observed in the CO2 absorption spectrum. The interchannel coupling drastically alters the predicted photoionization time delays (solid line in Figure 1). The photoionization time delays predicted for the fully-coupled continuum become extremely long for low energy ( eV) photoelectrons. This increase is caused by coupling of the continuum to Rydberg states converging to the , and state thresholds, i.e. autoionization. Coupling among the continuum channels also results in a decrease in the photoionization time delay in the vicinity of the shape resonance feature in the -state channel. This decrease is accompanied by an increase in the photoionization time delays in the other channels.
Figures 1 and 1 compare our extracted photoionization delays to the theoretical predictions of both models and show that the measured photoionization delays are consistent with the coupled-channel theory. Moreover, there is strong disagreement with the single-channel predictions in the vicinity of the -state shape resonance. These time-domain measurements show how electron correlation dynamics accelerate the escape of the photoelectron from the molecular potential. Electron interactions cause the EWP in the -state continuum to transition to other available continua while it is trapped in the vicinity of the ionic core. These transitions produce photoelectrons in the , , and continua with increased photoionization time delays. The couplings to the continuum channels act as additional pathways for the electron to escape the shape resonance and thus lower the photoionization delay times for the state.
Figure 1 shows the measured photoionization time delays for the channel along with the single-channel and coupled channel calculations. For photoelectron energies above 20 eV, the measured delays are consistent with both the single-channel and coupled-channel predictions. Below 20 eV, the CO2 absorption spectrum displays a series of sharp peaks associated with two Rydberg series converging to the , ionization thresholds Chan et al. (1993). The observed photoionization time delay confirms the autoionizing nature of the Rydberg states, as shown on the left side of Figure 1.
The measured photoionization time delays displayed in Figure 1 are obtained using a two-color, multi-path interference technique, which overlaps a weak infrared (IR) laser pulse, with photon energy , and an XUV frequency comb with spectral peaks separated by . Such a comb appears in the time domain as an attosecond pulse train (APT), whose pulses provide the temporal resolution needed to measure EWP delays. To produce this pulse arrangement, a titanium-doped:sapphire laser (30 fs, 30 mJ, 100 Hz repetition rate) is split into three co-propagating beams: two temporally overlapped, high energy ( mJ) beams used to drive high harmonic generation (HHG) and produce the XUV APT, and a low energy ( mJ) beam used as an interferometric probe. All three beams are spatially displaced and focused by a common m focusing optic. Near the focus, the three beams intersect in a 10 mm long gas cell filled with torr of argon gas. A temporal advance ( fs) is introduced in the probe beam path so that it does not disrupt the HHG process when passing through the gas cell. The crossed-beam geometry separates the XUV and IR pulses in the far field (See SM Fig. S1) SM . The residual drive laser light is blocked downstream from the gas cell and the probe beam passes through a m fused silica window to temporally overlap with the XUV APT. The temporal delay of the weak IR field is controlled with a piezo-electric driven delay stage. Both the XUV APT and weak IR laser pulse are focused with a B4C coated focusing optic ( cm) into the interaction region of a 1.2 m magnetic bottle spectrometer Mucke et al. (2010). The CO2 target is introduced through a m gas needle near the interaction region. The XUV-only photoelectron spectrum of CO2 is shown in the top panel of Fig. 2, and displays peaks spaced by twice the photon energy of the fundamental drive laser. The introduction of the weak IR field allows for two-photon absorption and produces “sideband” features between adjacent harmonic spectral features. Electron spectra are recorded as a function of XUV/IR delay, resulting in modulation of the sideband features as seen in the bottom panel of Fig. 2. This modulation occurs at twice the IR laser frequency and can be described by
[TABLE]
where is the yield of a sideband peak as a function of , the relative delay between the IR and XUV pulses, and is a phase offset in the sideband modulation that varies with sideband order (). The phase offset, , is recovered from the Fourier transform of the photoelectron spectrogram.The extracted phase offsets are shown in the supplemental material SM .
The sideband modulation is caused by interference between two different ionization pathways formed by single-photon XUV ionization by adjacent harmonics followed by subsequent absorption or emission of an IR photon Paul et al. (2001); Muller (2002); Mairesse and Quéré (2005); Dahlström et al. (2012). The sideband phase offset can be parsed into two contributions:
[TABLE]
where describes the spectral phase difference between consecutive harmonics that contribute to the sideband peak, and describes the phase difference between the two-photon ionization pathways Muller (2002); Dahlström et al. (2012). The reconstruction of attosecond bursts by two-photon transitions (RABBITT) technique was originally developed to characterize APTs by extracting the first term in Eqn. 2 Paul et al. (2001); Muller (2002). Subsequent work has focused on the latter quantity in Eqn. 2 to approximate a delay for the two-photon ionization process ():
[TABLE]
In most cases, this two-photon delay can be separated into a measurement induced (or continuum-continuum) contribution () that simply depends on the energy of the outgoing electron (), and a potential-dependent term () Baykusheva and Wörner (2017); Dahlström et al. (2012); Klünder et al. (2011); Dahlström et al. (2013):
[TABLE]
When the system is spherically symmetric and the ionization process is dominated by a single angular momentum partial wave, the potential dependent term can be shown to approximate the single-photon photoionization time delay Dahlström et al. (2012, 2013). In this limit, the RABBITT technique has been used to investigate photoionization time delays for different continuum channels in atomic targets Klünder et al. (2011); Isinger et al. (2017); Jordan et al. (2017) as well as the relative photoionization time delay between atomic targets Palatchi et al. (2014); Guénot et al. (2014). The RABBITT technique can also be used to observe resonant processes in atomic photoionization Kotur et al. (2016); Gruson et al. (2016); Busto et al. (2018).
The application of this interferometric technique to molecular systems is more challenging. Molecular targets often have several accessible cationic states that lead to substantial overlap of features in the photoelectron spectra (spectral congestion) Jordan and Wörner (2018). Moreover, the partial-wave expansion of outgoing photoelectron wavepackets can contain a large number of coherent contributions, a challenge not typically encountered in atomic targets Fano (1985). Nevertheless, Huppert et al. recently observed the effect of a molecular shape resonance on the measured photoionization time delays in N2O*+* Huppert et al. (2016). Vos et al. were able to study the stereo Wigner time delay in carbon monoxide averaged over a number of dissociative states of the CO+ cation Vos et al. (2018). Due to the excellent kinetic energy resolution afforded by the magnetic bottle spectrometer, we are able to resolve the sideband oscillations for three final state channels in CO2 (middle panel of Figure 2) and compare these results with theory predictions.
The relevant term in Eqn. 4 for theory comparison is the potential-dependent term, . To extract this contribution from the measured phase offsets, we consider the phase differences between a signal and reference channel:
[TABLE]
where and are the kinetic energies of the photoelectrons for the sideband peak in the signal and reference systems, is the difference in the continuum-continuum contribution due to the mismatch in photoelectron energies and is the differential photoionization delay we will compare with calculations. The spectral phase variation of the XUV pulse train, , has canceled out. When the relative difference between and is small, can be calculated very accurately Dahlström et al. (2013), and subtracted from Eqn. Electron Correlation Effects in Attosecond Photoionization of CO2. For the measurements presented in Figure 1 we reference the -state photoionization delay to that of the -state because the two channels have similar ionization potentials ( eV and eV). The -state channel ( eV, Figure 1) is compared with a reference measurement made in krypton gas ( eV). The krypton target is well studied Jordan et al. (2017) and its photoionization delay has been calculated Kheifets (2013), so we remove this contribution in the differential photoionization delay.
We calculate the photoionization time delay starting from the time evolution of the EWP. In the time-domain, the EWP, , is expressed as a coherent superposition of outgoing continuum electron eigenstates () with momentum :
[TABLE]
where describes the final state channel with ionization potential , and is the photoionization probability. Additionally, we define the scattering phase . The form of the outgoing electron wavefunction is described in the complex Kohn formalism and is discussed fully in the supplemental material SM . Initially (at ) the EWP is localized near the ionic core, but it is not stationary. The phase of each eigenstate component,
[TABLE]
evolves with time, and the wavepacket moves and disperses. The spatial location of maximum constructive interference is given by the stationary points of Eqn. 6:
[TABLE]
An observable delay time, , measures the time at which an EWP of energy arrives at a detector some fixed position () from the origin, compared to the case of no scattering Dahlström et al. (2012). Combining Eqn. 7 and 8, this direction-specific value can be shown to be
[TABLE]
Eqn. 9 shows the observed time delay is a direct consequence of the scattering phase, which depends on the charge dynamics that occur during photoionization. is commonly referred to as the single-photon photoionization time delay Dahlström et al. (2012); Baykusheva and Wörner (2017) or simply as the Eisenbud-Wigner-Smith delay, Wigner1955LowerShift . The measurements and calculations presented in figure 1 are related to , but the presence of the dressing laser field, along with the experimental measurement geometry can complicate the relationship Baykusheva and Wörner (2017); Hockett et al. (2016). The delays retrieved from the RABBITT technique, , and become identical in the special case of a spherically symmetric potential, when a single angular momentum channel dominates the photoionization process.
The ionization probability amplitude in Eqn. 6 can be calculated for weak XUV pulses from first order perturbation theory:
[TABLE]
where is the Fourier transform of the incident XUV pulse, is the dipole matrix element between the outgoing and ground state () wavefunctions in the length gauge, and is the polarization direction in the molecular frame. The channel-resolved dipole matrix element, Eqn. 11, is computed using the complex Kohn method for photoionization Rescigno and Orel (1981); Rescigno and Schneider (1988); Schneider and Rescigno (1988); Orel and Rescigno (1990); Rescigno and Orel (1991); McCurdy and Rescigno (1992); Rescigno et al. (1997); Rescigno and McCurdy (1998); McCurdy et al. (1998); Rescigno et al. (1993); Orel and Rescigno (1997); Miyabe et al. (2009); Sann et al. (2011); Williams et al. (2012); Douguet et al. (2012); Marggi Poullain et al. (2014); Fonseca dos Santos et al. (2015); McCurdy et al. (2017); Champenois et al. (2019). The calculations use explicit representations of the initial neutral state and of the final cationic states obtained with one single 11-orbital basis. This basis for the neutral and cationic states was obtained using a state-averaged multiconfiguration self-consistent-field (MCSCF) calculation performed with the COLUMBUS quantum chemistry program Lischka et al. (2009); Shepard et al. (1988); Lischka et al. (2001, 2011, 2012). The orbitals obtained are a compromise between those optimized for the neutral and cationic states. The primitive basis for this MCSCF calculation was Dunning’s aug-cc-pVDZ basis set Dunning (1989), with additional basis functions on the oxygen atom as described in the supplemental material SM . The outgoing wavefunction, , is expanded in a partial-wave representation (), up to , and the dipole matrix element is calculated between these functions and the initial ground state within the approximation of separable exchange Rescigno and Orel (1981); Rescigno and Schneider (1988); McCurdy and Rescigno (1992).
All meaningful partial waves are then coupled by the IR dressing field as described in Ref. Baykusheva and Wörner (2017) to determine the molecular frame (MF) two-photon photoionization time delays. These MF photoionization time delays are then averaged over both the polarization direction and outgoing electron direction to approximate the experimental conditions. This averaged quantity is what we refer to as the laboratory-frame (LF) photoionization delay as defined in Eqn. 4 and plotted in Figure 1. The single photon LF photoionization delays and two-photon MF photoionization delays for all accessible final state channels are shown in the supplemental material SM .
Observing phenomena such as autoionization, which typically have lifetimes longer than the inter-pulse spacing of our XUV pulse train (1.35 fs), requires additional considerations in the analysis. Since each attosecond burst in the train has a coherent relationship to the neighboring pulses, when time-dependent phenomena extend beyond the inter-pulse spacing, the processes induced by a single burst in the train interfere with the signal generated by the adjacent pulses. This interference leads to a burst-dependent photoemission time delay, i.e. the peak emission time is the point where all previous photoemission processes interfere constructively. Such a modulation (burst-to-burst) of the photoemission time delay leads to a variation of the phase across a sideband. This revelation is the motivation behind the development of the Rainbow RABBITT technique Gruson et al. (2016); Busto et al. (2018). The analysis of the slope of across each sideband peak is shown in Figure 1, where each channel is referenced to the krypton measurement for the continuum. The envelope of the IR-drive laser leads to small time shifts in the arrival time of each burst in the APT (referred to as harmonic chirp or femto-chirp Varj√∫ ‚Äñ et al. (2005)) which results in linear phase variation across a sideband peak. This contribution is removed by the krypton referencing because all measurements are made with the same APT. From the residual slope value obtained for the 12 harmonic sideband, there is a clear difference compared to any other sideband peak. Moreover, this difference is consistent with a delay of more than one complete -cycle. Instead of relying directly on the value extracted from Figure 1, we use the slope variation to determine the number of cycles to add to the mean phase extracted from averaging the phase over the sideband peak.
These measurements and calculations demonstrate the effect of electron correlation upon time delays in photoionization of molecules. Photoionization time delays are a direct and easy-to-understand manifestation of the binding interaction that an outgoing electron experiences. We have observed two effects of electron correlation in time-domain measurements of CO2 photoionization, via (1) a shape resonance and (2) autoionizing states, demonstrating that the inclusion of electronic correlation is important when considering resonant features in molecular photoionization with XUV light. While this seems clear for autoionizing states, which are inherently multi-channel phenomena, this result is somewhat surprising for shape resonance features which are typically considered to be single channel phenomena. For a shape resonance, we have shown that electron-electron interactions provide dynamic modifications to the effective potential that can be directly observed in all final state channels. These results highlight the need for including electron correlation when describing time-domain measurements of photoionization of molecular targets, where autoionizing states and shape resonances are omnipresent.
Molecular control (e.g. molecular alignment) techniques can be applied to isolate specific cationic states of CO in future experiments. Continuum-resolved molecular frame measurements will further elucidate the dependence of electron correlation on molecular orientation and electron emission angle. These experiments could be further improved by varying the XUV frequency comb spacing, thus mapping out more energy points in the differential scattering phase.
This work was supported by the AMOS program within the Chemical Sciences, Geosciences, and Biosciences Division of the Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy. Raw and analyzed data, both experimental and theoretical, along with a subset of the analysis code is available at:https://figshare.com/account/home#/projects/63164
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dahlström et al. (2012) J. M. Dahlström, A. L’Huillier, and A. Maquet, Introduction to attosecond delays in photoionization, Journal of Physics B: Atomic, Molecular and Optical Physics 45 , 183001 (2012) . · doi ↗
- 2Klünder et al. (2011) K. Klünder, J. M. Dahlström, M. Gisselbrecht, T. Fordell, M. Swoboda, D. Guénot, P. Johnsson, J. Caillat, J. Mauritsson, A. Maquet, R. Taïeb, and A. L’Huillier, Probing single-photon ionization on the attosecond time scale, Physical Review Letters 106 , 143002 (2011) . · doi ↗
- 3Serov et al. (2013) V. V. Serov, V. L. Derbov, and T. A. Sergeeva, Interpretation of the time delay in the ionization of Coulomb and two-center systems, Physical Review A 87 , 063414 (2013) .
- 4Pazourek et al. (2015) R. Pazourek, S. Nagele, and J. Burgdörfer, Attosecond chronoscopy of photoemission, Reviews of Modern Physics 87 , 765 (2015) . · doi ↗
- 5Schultze et al. (2010) M. Schultze, M. Fieß, N. Karpowicz, J. Gagnon, M. Korbman, M. Hofstetter, S. Neppl, A. L. Cavalieri, Y. Komninos, Th. Mercouris, C. A. Nicolaides, R. Pazourek, S. Nagele, J. Feist, J. Burgdörfer, A. M. Azzeer, R. Ernstorfer, R. Kienberger, U. Kleineberg, E. Goulielmakis, F. Krausz, and V. S. Yakovlev, Delay in photoemission, Science 328 , 1658 (2010) . · doi ↗
- 6Guénot et al. (2012) D. Guénot, K. Klünder, C. L. Arnold, D. Kroon, J. M. Dahlström, M. Miranda, T. Fordell, M. Gisselbrecht, P. Johnsson, J. Mauritsson, E. Lindroth, A. Maquet, R. Taïeb, A. L’Huillier, and A. S. Kheifets, Photoemission-time-delay measurements and calculations close to the 3 s -ionization-cross-section minimum in Ar, Physical Review A 85 , 053424 (2012) . · doi ↗
- 7Kheifets (2013) A. S. Kheifets, Time delay in valence-shell photoionization of noble-gas atoms, Physical Review A 87 , 063404 (2013) . · doi ↗
- 8Guénot et al. (2014) D. Guénot, D. Kroon, E. Balogh, E. W. Larsen, M. Kotur, M. Miranda, T. Fordell, P. Johnsson, J. Mauritsson, M Gisselbrecht, K. Varjù, C. L. Arnold, T. Carette, A. S. Kheifets, E. Lindroth, A. L’Huillier, and J. M. Dahlström, Measurements of relative photoemission time delays in noble gas atoms, Journal of Physics B: Atomic, Molecular and Optical Physics 47 , 245602 (2014) . · doi ↗