Unraveling the Mott-Peierls intrigue in Vanadium dioxide
Francesco Grandi, Adriano Amaricci, Michele Fabrizio

TL;DR
This paper investigates the complex interplay of electronic correlations and lattice effects in VO$_2$, proposing a comprehensive model solved with dynamical mean-field theory to clarify the metal-insulator transition and the existence of a monoclinic metallic phase.
Contribution
It introduces a minimal model for VO$_2$ that includes key physical ingredients and solves it using dynamical mean-field theory, revealing new insights into the phase transition.
Findings
Identification of a distorted metal phase between insulator and metal
Rich thermodynamics with multiple minima in the Born-Oppenheimer potential
Clarification of the monoclinic metallic phase evidence
Abstract
Vanadium dioxide is one of the most studied strongly correlated materials. Nonetheless, the intertwining between electronic correlation and lattice effects has precluded a comprehensive description of the rutile metal to monoclinic insulator transition, in turn triggering a longstanding "the chicken or the egg" debate about which comes first, the Mott localisation or the Peierls distortion. Here, we suggest that this problem is in fact ill-posed: the electronic correlations and the lattice vibrations conspire to stabilise the monoclinic insulator, and so they must be both considered not to miss relevant pieces of the VO physics. Specifically, we design a minimal model for VO that includes all the important physical ingredients: the electronic correlations, the multi-orbital character, and the two components antiferrodistortive mode that condenses in the monoclinic insulator. We…
Click any figure to enlarge with its caption.
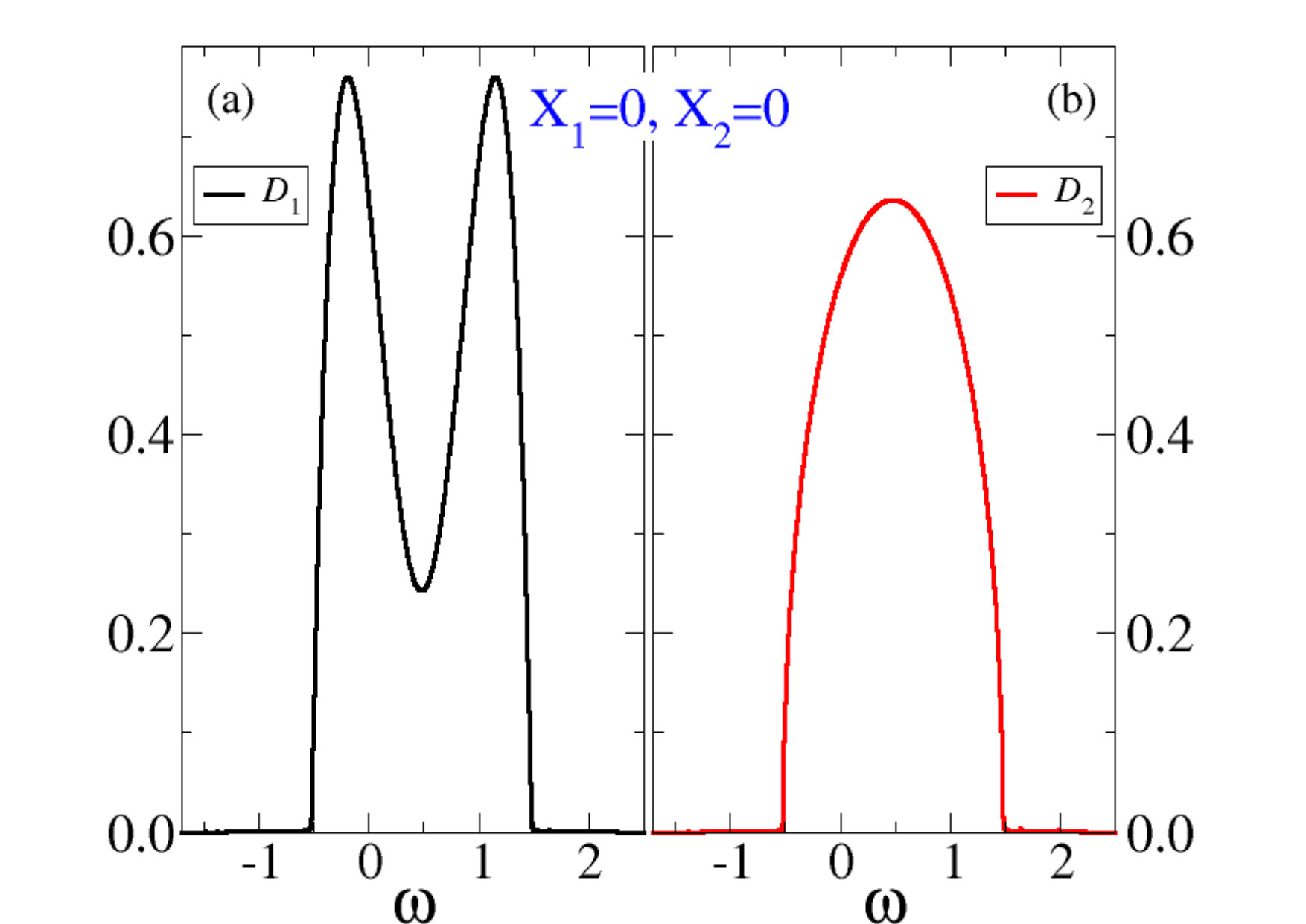 Figure 1
Figure 1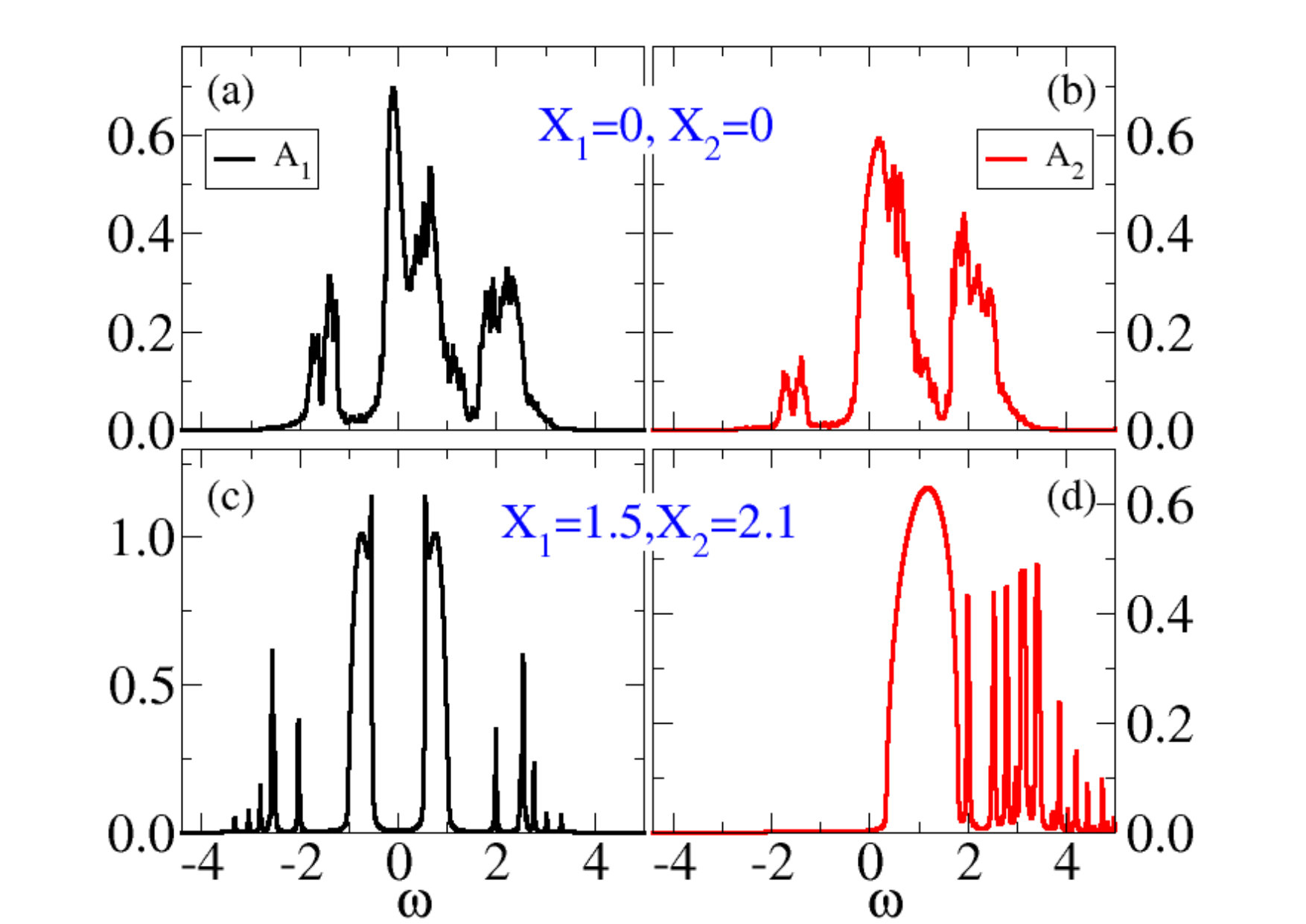 Figure 2
Figure 2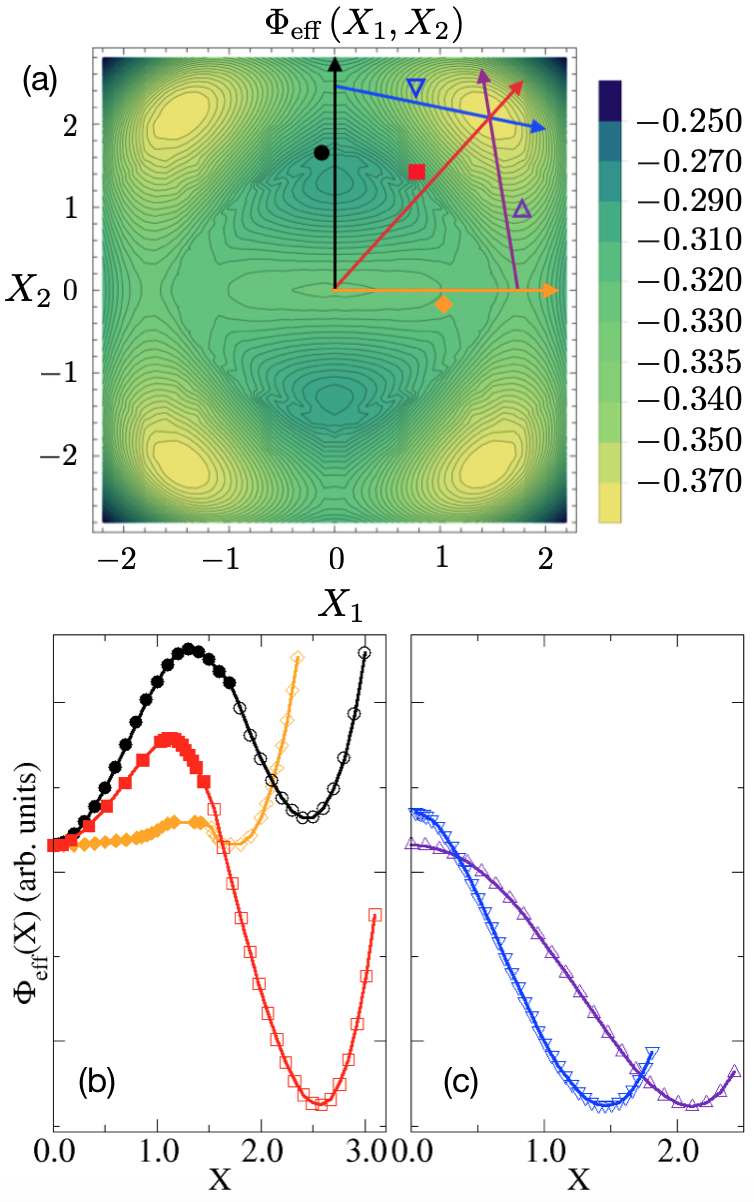 Figure 3
Figure 3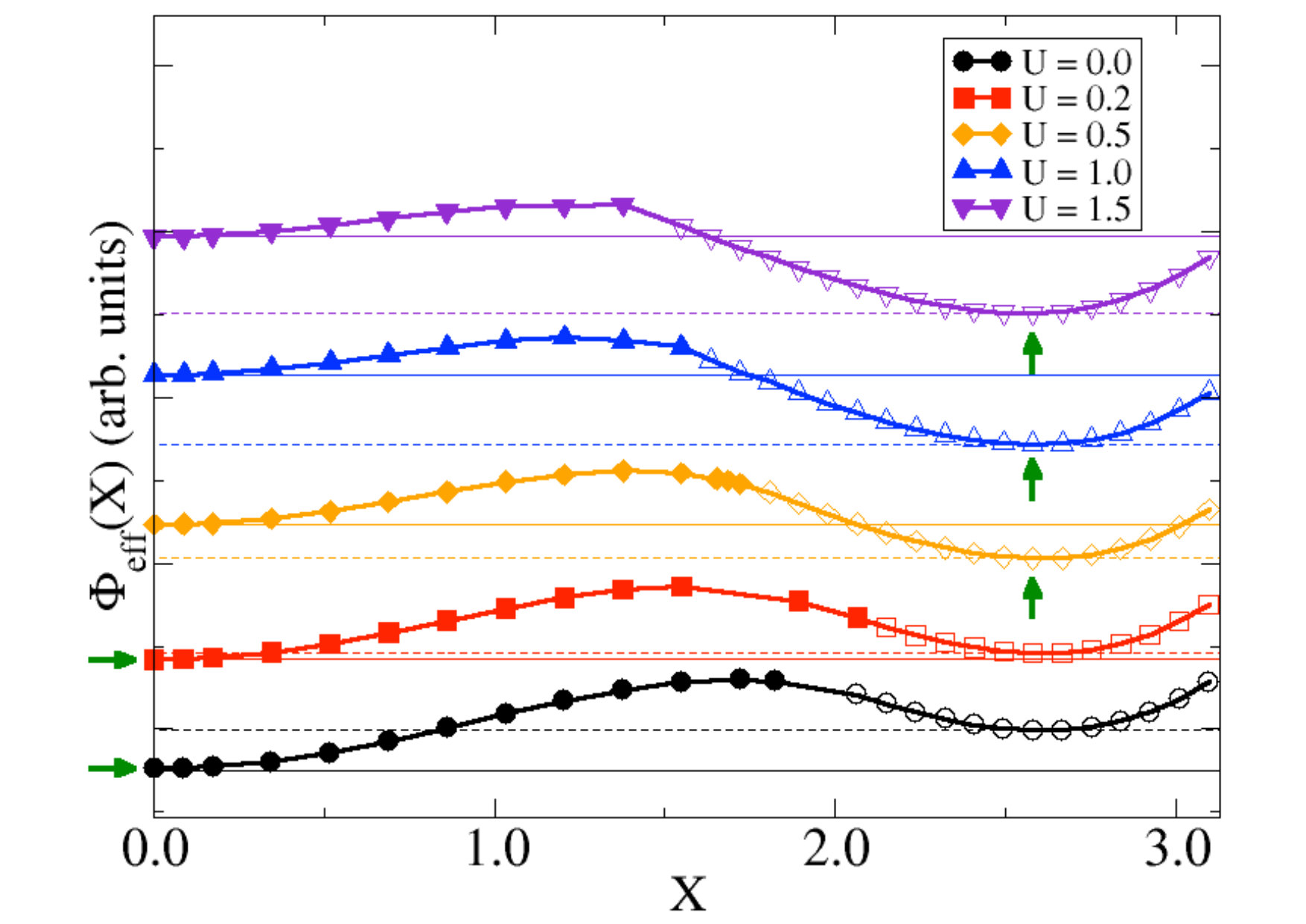 Figure 4
Figure 4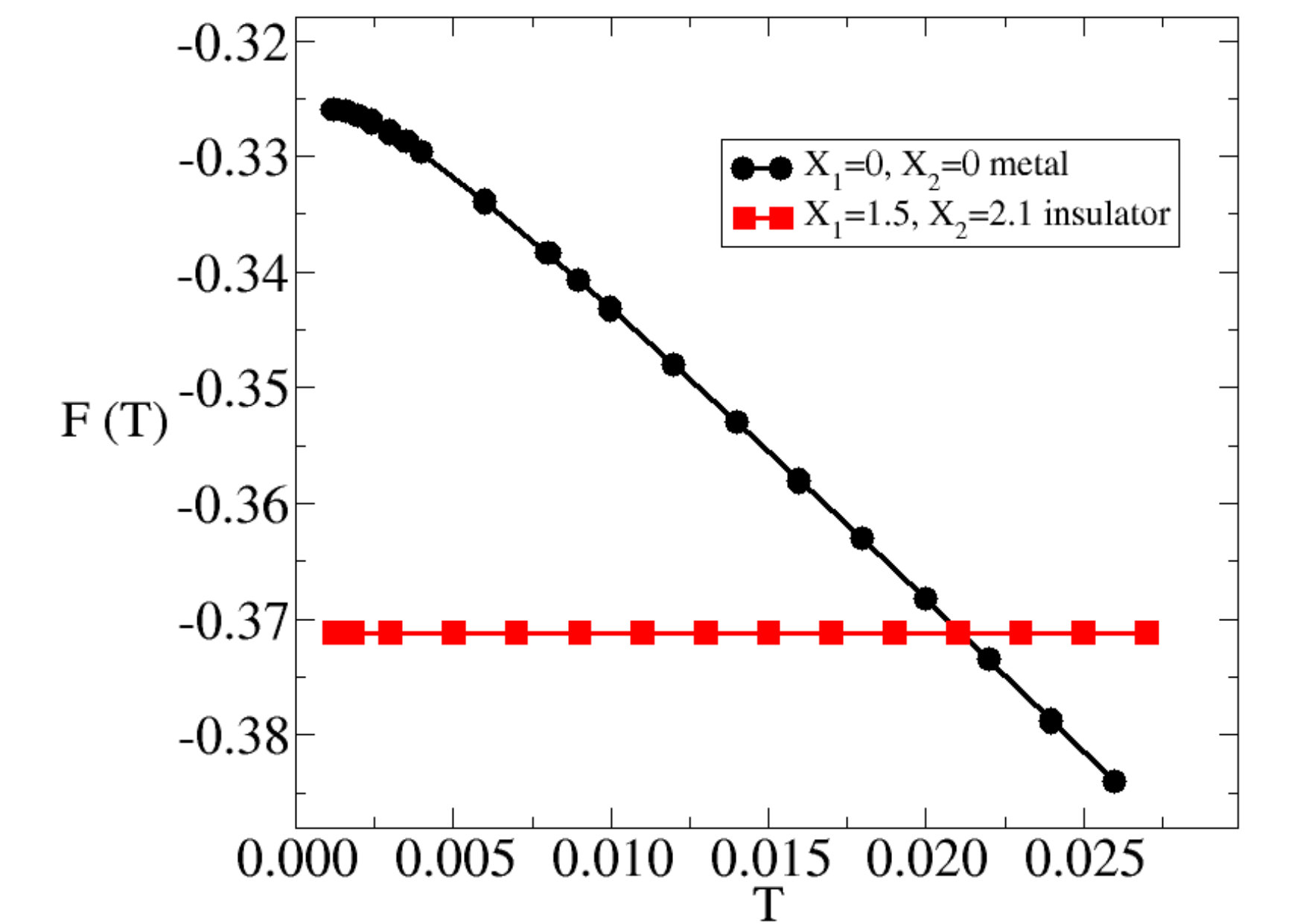 Figure 5
Figure 5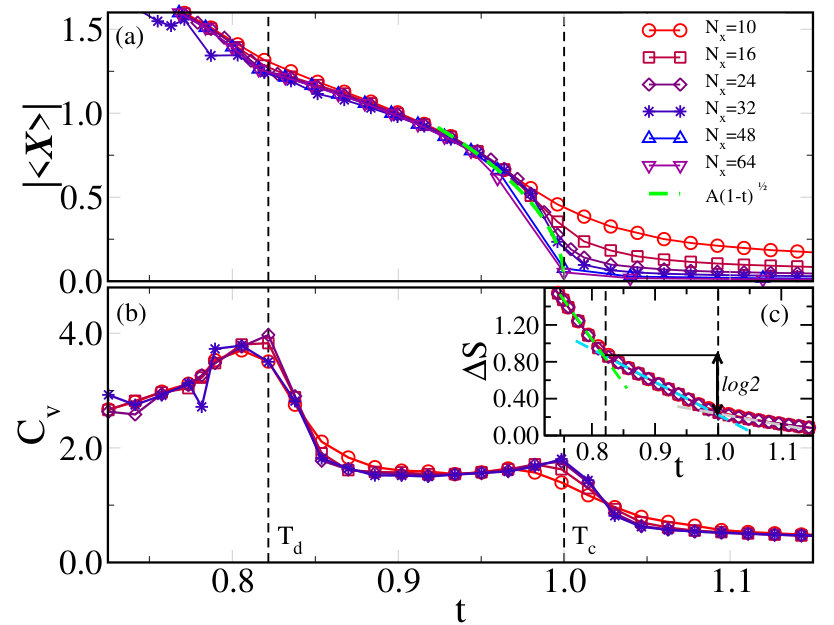 Figure 6
Figure 6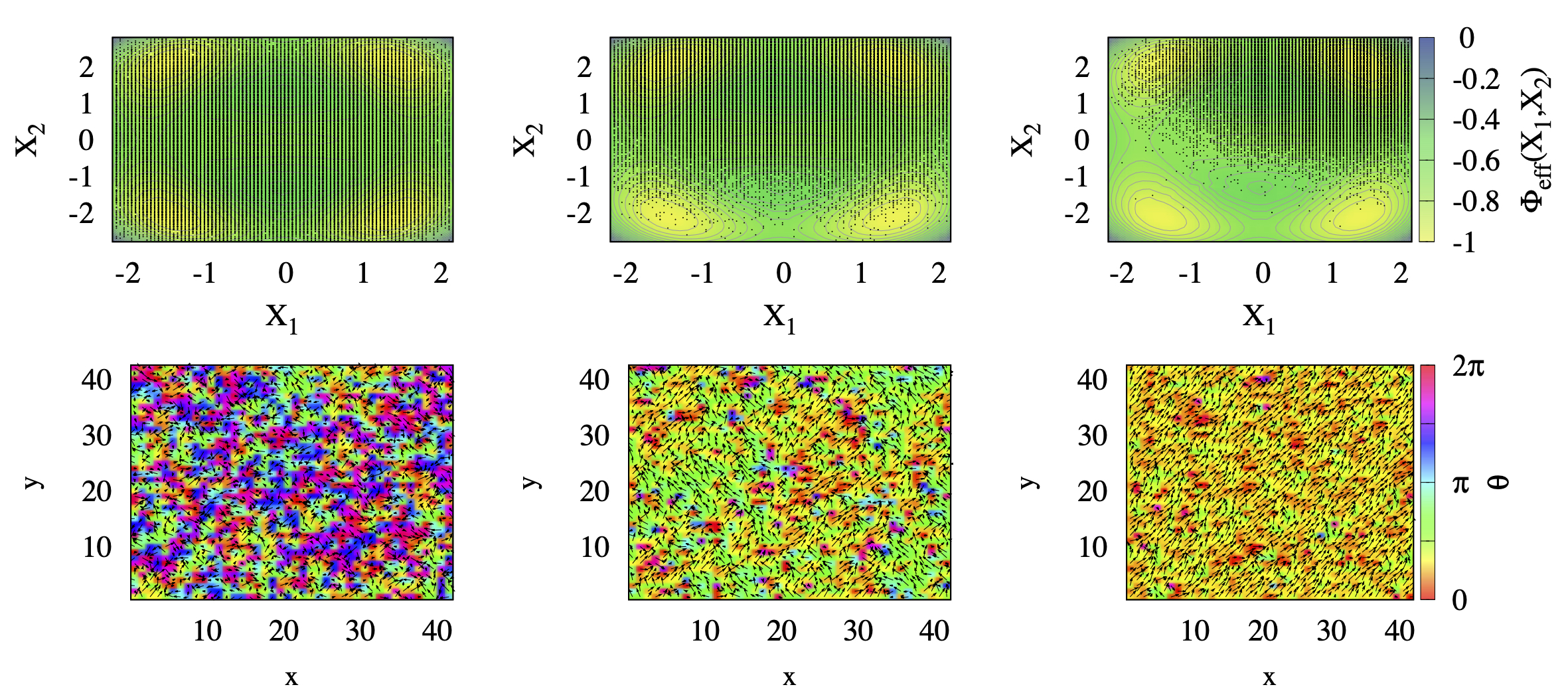 Figure 7
Figure 7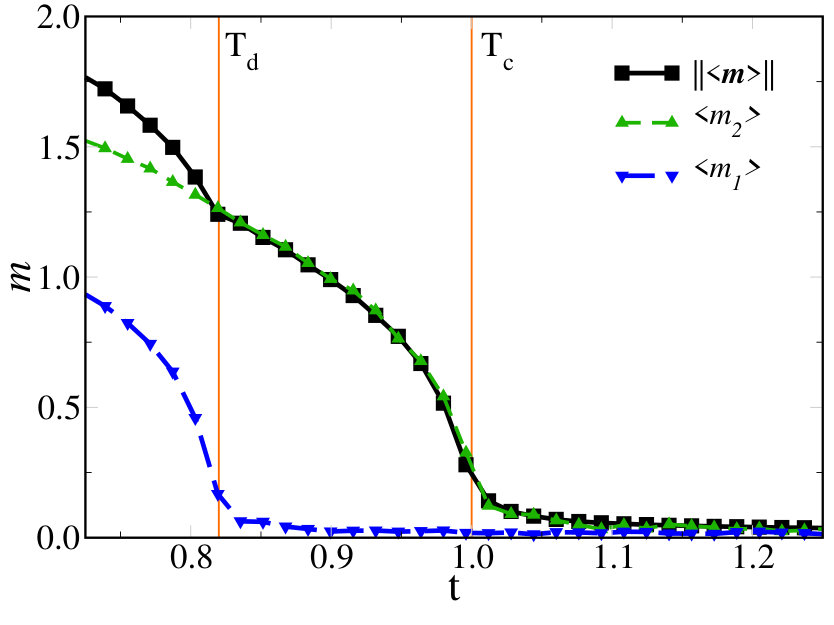 Figure 8
Figure 8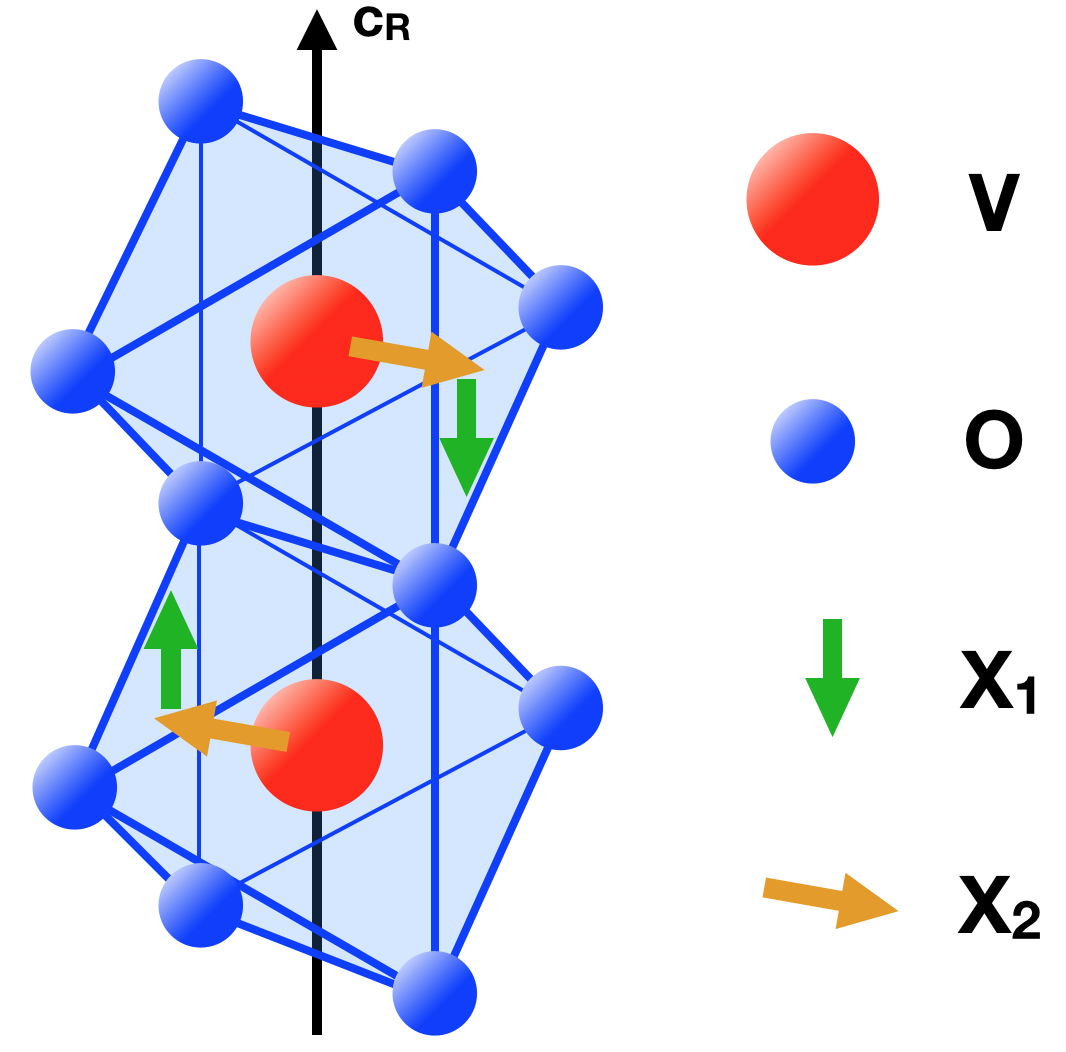 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Unraveling the Mott-Peierls intrigue in Vanadium dioxide
F. Grandi
Scuola Internazionale Superiore di Studi Avanzati (SISSA), Via Bonomea 265, I-34136 Trieste, Italy
Department of Physics, University of Erlangen-Nürnberg, 91058 Erlangen, Germany
A. Amaricci
Scuola Internazionale Superiore di Studi Avanzati (SISSA), Via Bonomea 265, I-34136 Trieste, Italy
CNR-IOM DEMOCRITOS, Istituto Officina dei Materiali, Consiglio Nazionale delle Ricerche, Via Bonomea 265, I-34136 Trieste, Italy
M. Fabrizio
Scuola Internazionale Superiore di Studi Avanzati (SISSA), Via Bonomea 265, I-34136 Trieste, Italy
(March 12, 2024)
Abstract
Vanadium dioxide is one of the most studied strongly correlated materials. Nonetheless, the intertwining between electronic correlation and lattice effects has precluded a comprehensive description of the rutile metal to monoclinic insulator transition, in turn triggering a longstanding “the chicken or the egg” debate about which comes first, the Mott localisation or the Peierls distortion. Here, we suggest that this problem is in fact ill-posed: the electronic correlations and the lattice vibrations conspire to stabilise the monoclinic insulator, and so they must be both considered not to miss relevant pieces of the VO2 physics. Specifically, we design a minimal model for VO2 that includes all the important physical ingredients: the electronic correlations, the multi-orbital character, and the two components antiferrodistortive mode that condenses in the monoclinic insulator. We solve this model by dynamical mean-field theory within the adiabatic Born-Oppenheimer approximation. Consistently with the first-order character of the metal-insulator transition, the Born-Oppenheimer potential has a rich landscape, with minima corresponding to the undistorted phase and to the four equivalent distorted ones, and which translates into an equally rich thermodynamics that we uncover by the Monte Carlo method. Remarkably, we find that a distorted metal phase intrudes between the low-temperature distorted insulator and high-temperature undistorted metal, which sheds new light on the debated experimental evidence of a monoclinic metallic phase.
I Introduction
Vanadium dioxide (VO2) is a transition metal compound with tremendous potential for technological applications, essentially in reason of its nearly room temperature metal-to-insulator transition Yang et al. (2011); Driscoll et al. (2009); Zhou et al. (2013); Huang et al. (2010); Ke et al. (2018); Liu et al. (2012); Zheludev and Kivshar (2012); Kats et al. (2013); del Valle et al. (2019); Zhang et al. (2019). Over the years, VO2 has been subject to an intense investigation, which dates back to the first decades of the last century Hoschek and Klemm (1939); Cook (1947); Andersson (1954); Archer et al. (1954); Magnéli and Andersson (1955); Andersson (1956); Rüdorff et al. (1958); Morin (1959); Westman (1961); Klemm and Grimm (1939), but that is yet alive Gray et al. (2018); Wall et al. (2018); Chen et al. (2019) and, to some extent, debated Biermann et al. (2005); Eyert (2011); Brito et al. (2016); Nájera et al. (2017); Plašienka et al. (2017); Nájera et al. (2018); Kim et al. (2013). At the critical temperature and ambient pressure, VO2 undergoes a first-order transition from a metal () to an insulator () Park et al. (2013); Chen et al. (2017), both phases being paramagnetic Alarich (1964); Villeneuve and Hagenmuller (1985); Berglund and Guggenheim (1969). In concomitance with the metal-insulator transition, a structural distortion occurs from a high-temperature rutile (R) structure to a low temperature monoclinic (M1) one.
The crystal structure of rutile VO2 is formed by equally spaced apart Vanadium atoms sitting at the centre of edge-sharing oxygen octahedra that form linear chains along the R -axis, which we shall denote as cR, see Fig. 1. The tetragonal crystal field splits the -manifold into two higher eg and three lower t2g levels. In the oxidation state V4+, the single valence electron of Vanadium can, therefore, occupy any of the three t2g orbitals, which are in turn distinguished into a singlet (or ) and a doublet (or ), having, respectively, bonding and non-bonding character along the cR-axis. The M1 phase is instead characterised by an anti-ferroelectric displacement of each Vanadium away from the centre of the octahedra, see Fig. 1, so that the above-mentioned chains, from being straight in the R phase, turn zigzag and dimerise Jo et al. (2016); Baum et al. (2007).
A simple portrait of the transition in VO2 was proposed in 1971 by Goodenough Goodenough (1971a). According to his proposal, the basal-plane component of the anti-ferroelectric distortion raises the energy of with respect to the Marcelli et al. (2017). In addition, the cR component of the distortion, which drives the chain dimerisation, opens a hybridisation gap between bonding and anti-bonding combinations of the . For large enough crystal field splitting and hybridisation gap, the bonding combination of the fills completely, while the anti-bonding as well as the get empty, hence the insulating behaviour. The Goodenough’s mechanism for the metal-insulator transition in VO2 thus relies on a single-particle description: the Peierls instability of the quasi-one-dimensional band that becomes half-filled after the growth of the crystal field drained the orbital.
However, Pouget et al. Pouget et al. (1974) and later Zylbersztejn and Mott Zylbersztejn and Mott (1975) soon after argued that the role of electronic correlation cannot be neglected as in the Goodenough’s scenario. Indeed, a tiny substitution of V with Cr changes the low-temperature insulator from the M1 crystal structure to a new monoclinic phase, named M2, where dimerised and zigzag chains alternate Pouget and Launois (1976); Villeneuve and Hagenmuller (1985). The M2 phase can be also stabilised under hydrostatic pressure or uniaxial stress Berglund and Jayaraman (1969); Pouget et al. (1974, 1975); Park et al. (2013); Chen et al. (2017); Quackenbush et al. (2016). In addition, a triclinic (T) phase with intermediate structural properties Pouget and Launois (1976) was shown to intrude between M1 and M2. The zigzag undimerised chains in M2 are still insulating and display magnetic properties akin those of a spin- antiferromagnetic Heisenberg chain Pouget et al. (1974); Marezio et al. (1972); Pouget and Launois (1976). This likeness can be rationalised only invoking sizeable electronic correlations. Given the low concentration of substitutional Chromium or the small value of uniaxial stress required to stabilise M2, it is reasonable to conclude that M1 must be as correlated as M2 Rice et al. (1994); Mott and Friedman (1974); Pergament (2003); Zinamon and Mott (1970).
We believe that, even though electronic correlations are likely necessary, they are nonetheless not sufficient to explain the phase diagram of VO2. It is known that a strong enough repulsion may drive a Mott transition in a three-band Hubbard model at the density of one electron per site Werner et al. (2008). Therefore, it is well possible that the insulating phase of VO2 is driven by correlations alone, and that the structural distortion below is just the best way the Mott insulator can freeze the residual spin and orbital degrees of freedom to get rid of their entropy. However, should that be the case, VO2 would most likely remain insulating even above , which is not the case, all the more so because is more than one order of magnitude smaller than the optical gap in the M1 phase Qazilbash et al. (2008). For the same reason, we must exclude a transition merely driven by the larger electronic entropy of the metal.
We are thus inclined to believe that the structural distortion is also necessary to stabilise the insulating phase in VO2, but, once again, not sufficient in view of the behaviour of the M2 phase, and of the bad metal character of the R phase Lee et al. (2017); Qazilbash et al. (2007, 2006). It is therefore quite likely that Goodenough’s scenario is after all correct, though it requires an active contribution from electronic correlations.
Indeed, different DFT-based calculations, which should properly account for the effects of the lattice distortion on the electronic structure, though within an independent-particle scheme, do not agree one with another, and none explains at once all experiments. For instance, straight LDA or GGA methods do not find any gap opening in M1 and M2 phases Eyert (2002a); Liebsch et al. (2005). Such gap is instead recovered by GW Continenza et al. (1999); Gatti et al. (2007); Sakuma et al. (2009) or LDA+U Korotin et al. (2002); Kozhevnikov et al. (2007); Yuan et al. (2012), in all its variants. However, GW does not give easy access to the total energy, and therefore it does not explain why low temperatures should favour the M1 distorted phase against the rutile undistorted one. In turns, LDA+U or GGA+U calculations, known to overemphasise the exchange splitting, predict the existence of local moments even in the rutile phase Korotin et al. (2002); Kozhevnikov et al. (2007); Yuan et al. (2012), not observed in experiments Zhang et al. (2018). Relatively recent calculations based on HSE hybrid functionals bring even worst results: both rutile and M1 phases are predicted to be magnetically ordered insulators, with the former lower in energy Grau-Crespo et al. (2012); Coulter et al. (2013), even though earlier calculations were claimed to be more in accordance with experiments Eyert (2011). In turn, mBJ exchange potentials seem to predict the proper conducting behaviour of the R and M1 phases, as well as their lack of magnetism Zhu and Schwingenschlögl (2012), which is erroneously predicted to occur also in the M2 phase Yuan et al. (2012). This suggests that suppression of magnetic moments is somehow the rule of mBJ functionals applied to VO2, which only by chance is the correct result for R and M1 phases. Finally, calculations based on PBE0 hybrid functionals properly account for the magnetic and electronic properties of M1 and M2 phases, but predict ferromagnetism in the rutile structure, at odds with experiments Alarich (1964), as well as the existence of a never observed ferromagnetic and insulating monoclinic phase, dubbed M0 Xu et al. (2017), also predicted by PBEsol functionals Moatti et al. (2019).
One might expect that combining ab-initio techniques with many-body tools, e.g., DFT with dynamical mean-field theory (DMFT) Kotliar et al. (2006), should work better and finally provide uncontroversial results in accordance with experiments. Unfortunately, different calculations by state-of-the-art DFT+DMFT methods do not even agree about an unanimous view of the M1 monoclinic phase. Specifically, M1 has been regarded from time to time as a correlation-assisted Peierls insulator Biermann et al. (2005); Belozerov et al. (2012), or, vice versa, as a Peierls-assisted Mott insulator Weber et al. (2012a), or, finally, as a genuine Mott insulator Laad et al. (2006); Brito et al. (2016, 2017).
In view of the above controversial results, we think it is worth desisting from describing VO2 straight from first principles, and rather focusing on a minimal model, which can include all the ingredients that are, by now, widely accepted to be essential. As we mentioned, electron-electron correlations must play an important role and thus need to be included and handled in a truly many-body scheme. At the meantime, the coupling of the electrons to the lattice is equally important and must be included as well. We earlier mentioned that the monoclinic distortion in the M1 phase actually entails two different antiferrodistortive components: the basal-plane displacement of V from the octahedron centre, resulting into a zigzag shape of the formerly straight chains, and the out-of-plane displacement that produces the chain dimerisation. The two phenomena may actually occur separately, as indeed proposed by Goodenough Goodenough (1971a), who argued that, generically, the basal-plane distortion should appear at higher temperatures than dimerisation. Indeed, time-resolved spectroscopy measurements during a photoinduced monoclinic-to-rutile transition have shown that dimerisation melts on earlier time-scales than the basal-plane displacement Baum et al. (2007); Kumar et al. (2017); Yuan et al. (2013), which therefore must be distinct and actually more robust than the former. We must mention, however, that this conclusion does not agree with other experiments Morrison et al. (2014); Otto et al. (2019); Tao et al. (2012); Corr et al. (2010); Otto et al. (2019). More convincing evidence is offered by the monoclinic metal that intrudes, under equilibrium conditions, between rutile metal and monoclinic insulator at ambient pressure Umeda et al. (1966); Laverock et al. (2014); Yao et al. (2010); Lee et al. (2018), and nor just above a critical pressure as originally believed Arcangeletti et al. (2007). This phase might correspond to a crystal structure where dimerisation is almost melted unlike the zigzag distortion Yao et al. (2010); Moatti et al. (2019), so that are still above the , though the dimerisation is too weak to stabilise at that temperature an hybridisation gap within the band Nájera et al. (2017). Even the disappearance prior to the metal-insulator transition Gray et al. (2016) of the so-called singlet peak, which is associated to dimerisation and observed in optics, can be regarded as a consequence of the melting of dimerisation preceding the complete monoclinic-to-rutile transformation. All the above experimental facts point to the need to treat separately the basal-plane displacement and the out-of-plane one. Finally, the importance of the basal plane antiferrodistortive mode suggests the last ingredient to be considered: the multi-orbital physics. This aspect was originally emphasised by Goodenough Goodenough (1971a) and successively confirmed by many optical measurements Qazilbash et al. (2008); Aetukuri et al. (2013); Huffman et al. (2017).
To summarise, we shall consider a microscopic model which includes the following relevant features:
the electron-electron correlations and the coupling to the lattice distortion Marezio et al. (1972); Budai et al. (2014); Lee et al. (2017); Paul (1970); Lederer et al. (1972); Gatti et al. (2015); Hwang et al. (2017); McWhan et al. (1973, 1974); Srivastava and Chase (1971); Sommers et al. (1975); Hyland (1968); Gupta et al. (1977); Stefanovich et al. (2000); Maurer et al. (1999); Pintchovski et al. (1978); Wall et al. (2012); Kübler et al. (2007); Wegkamp and Stähler (2015); Schilbe and Maurer (2004); Srivastava and Chase (1971); Brews (1970); Cavalleri et al. (2001); He and Millis (2016); Matsuda et al. (2020); 2. 2.
the existence of two different antiferrodistortive components, each playing its own distinctive role Goodenough (1971a); Baum et al. (2007); Kumar et al. (2017); 3. 3.
the multi-orbital physics Goodenough (1971a); Qazilbash et al. (2008); Aetukuri et al. (2013); Huffman et al. (2017).
with the minimal requirement of capturing, at least at a qualitative level, the following aspects of the VO2 physics:
- A.
the existence of an undistorted paramagnetic metal and a monoclinic distorted insulator Choe et al. (2017); Goodenough (1960); Berglund and Jayaraman (1969); Barker et al. (1966); Adler (1968);
- B.
the first-order character of the transition between them Sasaki and Watanabe (1964); Ladd and Paul (1969); Everhart and MacChesney (1968); Bongers (1965); Morin (1959); Liu et al. (2011); Nakano et al. (2012); Okuyama et al. (2014); J. Hyland and W. B. Taylor (1966); Kawakubo and Nakagawa (1964); Chandrashekhar et al. (1973); Mitsuishi (1967);
- C.
the possible existence of an intermediate monoclinic metal Umeda et al. (1966); Kim et al. (2008); Nag et al. (2012); Yao et al. (2010); Laverock et al. (2014); Zhang et al. (2009); Kim et al. (2006); Cocker et al. (2012); Kumar et al. (2014); Ilinskiy et al. (2012a, b); Chen et al. (2008);
Many models have been already put forth to describe VO2. However, most of them focus either on the role of the electron-electron correlations, or on that of the electron-lattice coupling Mattis and Langer (1970); Hearn (1972a); Tomczak et al. (2008); Nájera et al. (2017, 2018); Kawakubo (1965); Hearn (1972b); Hyland (1970); Adler et al. (1967); Mitra et al. (1971); Chattejee et al. (1972); Caruthers et al. (1973); Caruthers and Kleinman (1973); Woodley (2008); Netsianda et al. (2008); Tselev et al. (2010), and thus do not allow accessing in a single framework the whole VO2 phase diagram, e.g., the points A., B. and C. above. Despite that, we must mention that the purely electronic Dimer Hubbard Model presented in Nájera et al. (2017), which by construction is not able to capture the monoclinic to rutile phase transition, is nevertheless able to describe some of the observed features of the monoclinic metal, like the MIR peak in the optical conductivity observed in Qazilbash et al. (2007). There are actually some exceptions where electron-electron and electron-lattice interactions have been considered on equal footing de Graaf and Luzzi (1968); Paquet and Leroux-Hugon (1980); Shi et al. (1991). In particular, the model studied in Paquet and Leroux-Hugon (1980) includes explicitly all ingredients listed above. However, therein it is assumed a small bandwidth of the -derived band as compared to the one, which contradicts LDA calculations Eyert (2002a). Moreover, Paquet and Leroux-Hugon (1980) includes the two distinct effects of the monoclinic distortion, but parametrized by a single displacement variable. In this way they preclude the possibility to describe the emergence of the monoclinic metal that seems to be observed experimentally. Furthermore, the mean-field treatment of the electron-electron interaction, despite its strength being comparable to the conduction bandwidth, yields not surprisingly to the formation of local moments in the rutile metal, not in accordance with magnetic measurements Zhang et al. (2018). This negative result, highlighted by the same authors of Ref. [Paquet and Leroux-Hugon, 1980], solicits for a more rigorous treatment of the interaction.
This is actually the scope of the present work, which is organised as follows. In Sec. II we introduce a simple model that includes the three ingredients previously outlined, which we believe should capture the main physics of Vanadium dioxide. In Sec. III we discuss the dynamical mean-field theory (DMFT) approach to the model Hamiltonian, and presents in Sec. III.1 its ground state phase diagram. In Sec. IV we discuss the insulator-metal transition that occurs in our model upon raising the temperature. In Sec. IV.1 we discuss the case in which such transition is driven solely by the electronic entropy, hence neglecting the lattice contribution to entropy, whereas in Sec. IV.2 the opposite case. We will show that the latter situation is rather suggestive, since it foresees different transition temperatures of the two antiferrodistortive components, as predicted by Goodenough Goodenough (1971a). In turn, this result might explain the evidence supporting the existence of a monoclinic metal phase. Finally, Sec. V is devoted to concluding remarks.
II The model
As we mentioned, the orbitals that are relevant to describe the physics of VO2 are the Vanadium ones, comprising the singlet and doublet, which host a single conduction electron. We believe that in this circumstance the doublet nature of the is not truly essential; what really matters is the distinction between and based on their bonding character with the ligands and response to atomic displacement. Therefore, in order to simplify our modelling without spoiling the important physics, we shall associate the doublet with just a single orbital Sandri et al. (2013); Hearn (1972a), which, together with the other orbital mimicking the singlet, give rise to two bands, and , which accommodate one electron per site, i.e., they are quarter filled.
The other ingredient that is necessary to properly describe VO2 is the electron-electron Coulomb interaction. However, since the main role that Coulomb repulsion is believed to play is to suppress charge fluctuations on , we shall ignore the long range tail and replace Coulomb repulsion with a short-range interaction.
Finally, we need to include the coupling to the lattice. For simplicity, we shall focus our attention only on the rutile and monoclinic M1 phases, as such ignoring the M2 phase, which is actually regarded by some as just a metastable modification of the M1 structure Pouget et al. (1974, 1975); Woodley (2008). Under this assumption, we can model the lattice antiferrodistortion through a two-component zone boundary mode at momentum , with displacement and classical potential energy \Phi\big{(}X_{1},X_{2}\big{)}. The and components model, respectively, the dimerising out-of-plane displacement and the band-splitting basal-plane one, see Fig. 1 Sboychakov et al. (2010); Mattis and Langer (1970).
The model Hamiltonian is thus written as the sum of three terms:
[TABLE]
is the purely electronic component reading:
[TABLE]
where is the occupation number at momentum of the band , the electron number operator at site , the chemical potential used to enforce the quarter filling condition and, finally, is the on-site Hubbard repulsion.
With the aim to reduce the number of independent Hamiltonian parameters, we assume that the density-of-states (DOS) and , of the band 1 and 2, respectively, have same bandwidth and centre of gravity, which we shall take as the zero of energy. In addition, we consider both DOS symmetric with respect to their centre, and such that , where is the wave-vector of the antiferrodistortive mode . This assumption actually overestimates the dimerisation strength, since it entails that any is able to open a hybridisation gap in the middle of band 1, which, we remark, does not coincide with the chemical potential unless band 2 is pushed above it. This implies that a finite hybridisation gap within band 1 does not stabilise an insulator so long as band 2 still crosses the Fermi energy. Therefore our simplified modelling does not spoil the important feature that a distorted insulating phase may occur only above a critical threshold of the Hamiltonian parameters, although it affects the value of that threshold, whose precise determination is however behind the scope of the present model-study.
In order to emphasise the bonding character of the , band 1, along the axis, as opposed to the more isotropic , band 2, we choose the following forms of the two corresponding DOS’s:
[TABLE]
with and a normalisation factor. We take so that has a double-peak structure evocative of a one-dimensional DOS Paquet and Leroux-Hugon (1980); Haverkort et al. (2005); Belozerov et al. (2012). Hereafter, we take the half bandwidth as our energy unit, and fix and . The resulting DOS’s are shown in Fig. 2 (a) and (b). There we note the two-peak structure of the band 1 DOS, mimicking the Van Hove singularities of a quasi one-dimensional band structure, in contrast to the structureless band 2 DOS.
We highlight that the electron-electron interaction in Eq. (2) only includes the monopole Slater integral , and not higher order multipoles responsible of Hund’s rules. This approximation, that makes the analysis more transparent, requires some justification. The Coulomb interaction of a single Vanadium projected onto the t2g manifold, which effectively behaves as an atomic shell, can be written in terms of two Slater integrals as:
[TABLE]
where , and are the total occupation, spin, and angular momentum, respectively. Common values of the parameters are and Biermann et al. (2005). Denoting as the lowest energy with electrons in the t2g shell, the effective Hubbard for V4+ can be defined through:
[TABLE]
to be compared with the VO2 bandwidth of about Eyert (2002a). In units of the half-bandwidth, , the value we shall use hereafter Shih et al. (2012); Fujimori et al. (1992). We observe that the Coulomb exchange has no effect on the configurations with , while it splits those with in three multiplets, with , which are spread out over an energy , about a quarter of the full bandwidth. Such small value is not expected to qualitatively alter the physical behaviour, see, e.g., de’ Medici et al. (2011), which justifies our neglect of the exchange splitting in the model Hamiltonian (2).
We model the potential energy \Phi\big{(}X_{1},X_{2}\big{)} using a Landau functional for improper ferroelectrics Levanyuk and Sannikov (1974); Kumar et al. (2017); Eyert (2002a) expanded up to the sixth order in the lattice displacements:
[TABLE]
where is the number of sites and the couplings to are all positive. The terms proportional to , i.e. the harmonic part of the potential, and that proportional to have full rotational symmetry in the – plane. On the contrary, favours a lattice distortion only along one of the two components, whereas a distortion with . In the specific case of VO2, , and thus it is preferable to equally displace both modes Kumar et al. (2017) rather than just one of them.
Finally, we write the electron-lattice coupling as:
[TABLE]
where creates an electron at momentum in orbital with spin , and we recall that, by construction, . The dimerisation induced by the out-of-plane displacement is controlled by the coupling constant , while parametrises the strength of the crystal field splitting generated by the basal-plane displacement . By symmetry, the coupling between the field and the electron dimerization is at leading order linear Mattis and Langer (1970); Shi et al. (1991). The quadratic coupling in is intentional and has a physical explanation. Indeed, corresponds to the Vanadium displacement parallel to the diagonal of the rutile basal plane away from the centre of the Oxygen octahedron. As a result, the hybridisation between the and the Oxygen ligands closer to the new Vanadium position increases, whereas the hybridisation with the further Oxygens diminishes Paquet and Leroux-Hugon (1980). At linear order in the V-displacement , the two opposite variations of the hybridisation cancel each other, but, at second order, they add up to a net rise in energy of the level, hence the expression in Eq. (7). The Hamiltonian Eq. (1) is invariant under the transformations , reflecting a ZZ2 (also known as or “Vierergruppe”) symmetry.
Despite the great simplification effort, the model Hamiltonian Eq. (1) has still several parameters to be fixed. We emphasise that our main goal is to reproduce qualitatively the physics of VO2, without any ambition of getting also a quantitative agreement. Nonetheless, just to be sure not to explore a Hamiltonian parameter space completely detached from the real VO2 compound, we choose parameters in line with the existing literature. We already mentioned our choice of , in units of the half-bandwidth, which is in line with the value used in realistic calculations Biermann et al. (2005); Hyland (1969); Lazarovits et al. (2010); Paquet and Leroux-Hugon (1980); Mott and Friedman (1974); Sommers and Doniach (1978). The other parameters involve the phonon variables. We shall choose , , , , and . Those values permit to reproduce the inter-band character of the band gap experimentally observed for the monoclinic insulator Aetukuri et al. (2013) and to obtain a size of it close enough to the experimental findings, see Sec. III.1.1 for further details on the spectral properties of the system. Moreover, we checked a posteriori that we can reasonably reproduce the size of the electron-phonon interaction Okazaki et al. (2004); Hearn (1972a); van Veenendaal (2013) and the lattice energy change across the rutile-to-monoclinic transition van Veenendaal (2013) as they were obtained in previous experiments or theoretical analysis. As a concluding remark, we point out that the direct experimental fits of the coupling constants is satisfactorily in agreement with previous estimations of the same Kumar et al. (2017); Lysenko et al. (2017), further corroborating our choice of the parameter set.
III DMFT solution
We solve the model Hamiltonian Eq. (1) by means of DMFT Georges et al. (1996) within the adiabatic Born-Oppenheimer approximation. This approach will allow us to treat correlation effects non-perturbatively beyond an independent-particle description. Exploiting the Born-Oppenheimer approximation, we solve the electronic problem at a fixed displacement . To any choice of the displacement it corresponds to a different electronic problem through the electron-phonon coupling discussed above. Within DMFT such resulting interacting lattice electrons problem is mapped onto a quantum impurity model constrained by a self-consistency condition, which aim to determine the bath so to describe the local physics of the lattice model. The effective bath is described by a frequency dependent Weiss field , with the orbital index. The self-consistency condition relates the Weiss fields to the local self-energy function , obtained from the solution of the effective quantum impurity model, and the local interacting Green’s function
[TABLE]
where . The self-consistency conditions read:
[TABLE]
Once a Weiss field is given, the solution to the DMFT equations is obtained iteratively as follows. We solve the effective quantum impurity problem associated to the given Weiss field using Exact Diagonalization technique Caffarel and Krauth (1994); Weber et al. (2012b). To this end, we discretize the effective bath into a number of electronic levels Caffarel and Krauth (1994); Capone et al. (2007); Weber et al. (2012b). The resulting Hamiltonian is diagonalized using Lanczos method and the ground-state (at zero temperature) or low lying states in the spectrum (at finite temperature) are determined Capone et al. (2007). The impurity Green’s functions are then obtained using the dynamical Lanczos technique Georges et al. (1996); Capone et al. (2007). The self-energy is obtained by solving the Dyson equation for the impurity problem . The self-energy is used to evaluate the local interacting Green’s function and, finally, to update the Weiss fields by means of the self-consistency condition. The procedure is iterated until the overall error on the determination of the Weiss field falls below a threshold, which in our calculations was set to . In this work, we use as the total number of bath sites, corresponding to a finite system of electronic levels or spins. Yet, we tested our results with respect to larger values of without finding significant differences.
Using the DMFT method we computed the electronic properties for several values of the displacement . The part of Eq. (7) related to the tilting enters in the single-particle term of the impurity hamiltonian as the usual crystal field splitting, while the dimerization acts directly on the density of states of band , see the expression of appearing in Eq. (3), and it opens a gap of size in correspondence to its center of gravity.
We calculate the total electronic energy, or the free-energy at finite temperature, which renormalizes the Born-Oppenheimer adiabatic potential of the displacement through:
[TABLE]
We shall restrict our analysis to the paramagnetic sector forcing spin symmetry. However, we did check that magnetic solutions are higher in energy. We first present results at zero-temperature , and then move to those at .
III.1 Ground state phase diagram
In Fig. 3a we show the adiabatic potential in (8) calculated by DMFT at . The energy landscape shows five minima. A local minima is located at the origin , and corresponds to an undistorted metal that we identify with the R phase of Vanadium dioxide. Four degenerate global minima are instead located at and , which are related to each other by the ZZ2 symmetry and represent the four equivalent lattice distortions. We find that these global minima describe an insulating phase, and thus realize a two-band version of the Goodenough scenario Goodenough (1971a) for the M1 phase, in qualitative agreement with ab-initio calculations of VO2 Woodley (2008); Netsianda et al. (2008). A detailed discussion of the electronic properties of all minima is postponed to the next Sec. III.1.1.
In figures 3b and 3c we instead show the evolution of the adiabatic potential along some specific lines, as indicated in Fig. 3a. We note that along the horizontal and vertical cuts, marked by a diamond and a circle in Fig. 3a, respectively, the energy landscape shows a saddle point, i.e., a minimum along the cut direction, but maximum in the perpendicular one. Within our model description, the effect of a uniaxial tensile strain would be taken into account by adding to the Hamiltonian Eq. (1) terms like: or (), depending on the direction of the applied stress Mogunov et al. (2019); Cao et al. (2009); Strukov and Levanyuk (1998). In presence of such terms, the saddle points observed in Fig. 3a along the lines or may turn into additional minima of the energy landscape Tselev et al. (2010), which can possibly describe the occurrence of the M2 phase in the framework of the same model Hamiltonian.
In order to understand what is the role of the Hubbard interaction in stabilising the insulating solution, we studied the evolution of for several values of , along the line in the – plane connecting the rutile local minimum with one of the monoclinic global minima (the diagonal cut in Fig. 3a marked by a diamond symbol). Our results are reported in Fig. 4. We note that already at the energy has two minima. One is at the origin and corresponds to the undistorted metal. The other is located at finite , and thus represents a distorted phase that must evidently be also insulating in order to be a local energy minimum. Therefore at small , the stable phase is the undistorted metal at in Fig. 4, while the local minimum at (monoclinic insulator) is metastable. However, for larger , the situation is reversed: the distorted insulator becomes the global minimum, while the undistorted metal a local one, entailing the typical scenario of a first-order metal-insulator transition driven by interaction. The above results show that electron-electron interaction is crucial to stabilise the distorted insulator, though the active contribution of the lattice is equally essential. Indeed, the interaction strength, the half-bandwidth, is too small to drive on its own the metal-insulator transition de’ Medici et al. (2011). In other words, the picture that emerges from Fig. 4, with the interaction and the coupling to the lattice both necessary to stabilise the insulator, fully confirm our expectation in Sec. I.
III.1.1 Spectral functions
Further insights into the properties of the metal-insulator transition can be gained by looking at the spectral functions:
[TABLE]
where and is the local interacting Green’s function obtained within the DMFT solution of the model. In Fig. 5 we show at the different minima in 3a, with measured with respect to the chemical potential. We note that already in the absence of interaction, , the different shapes of the DOS’s, see Fig. 2, lead to a larger occupation of band than band . Such population unbalance is increased by , which effectively enhances the crystal field, leading to an even larger occupation of band at expenses of Sandri et al. (2013); Sandri and Fabrizio (2015); Grandi et al. (2018). This is evident in the spectral function of the undistorted metal at , reported in Fig. 5(a) and Fig. 5(b), where the occupied part of overwhelms that of more than in the case of Fig. 2. We also note in the figures 5(a) and 5(b) side peaks that correspond to the precursors of the Hubbard bands.
The scenario is radically different in the insulating solution, see Fig. 5(c) and Fig. 5(d). Here we observe the formation of a hybridisation gap opening at the chemical potential inside the band 1. Two coherent-like features flank the gap. The band 2 is instead pushed above the Fermi energy, and therefore is empty. We still observe precursors of the Hubbard sidebands in , as well as signatures of the precursor of the upper Hubbard band in , though rather spiky because of the bath discretisation.
We note that in the insulating solution the lowest gap corresponds to transferring one electron from band 1 to band 2, i.e., from to in the VO2 language, and has a magnitude of about , for a realistic value of the half-bandwidth of 1.3 eV Eyert (2002a). This value of the gap is not too far from the experimental one, Berglund and Guggenheim (1969); Qazilbash et al. (2008); Liu et al. (2011). Therefore, our simplified modelling yields results that are not only qualitatively correct but, rather unexpectedly, also quantitatively not far off the actual ones. The band 1 band 1 transition, i.e., , though being slightly higher in energy, has a much steeper absorption edge since it involves the two coherent peaks in Fig. 5(c), already observed in previous works Biermann et al. (2005); Brito et al. (2016); Lazarovits et al. (2010); Nájera et al. (2017). This result is in loose agreement with XAS linear dichroism experiments Koethe et al. (2006); Gray et al. (2016) that are able to distinguish the two absorption processes.
In order to assess the degree of electronic correlations, we calculate the quasiparticle residue of each band in the undistorted metal phase, defined by:
[TABLE]
with . We find that the two bands at show almost the same value , not inconsistent with more realistic calculations Lazarovits et al. (2010); Biermann et al. (2005); Belozerov et al. (2011); Weber et al. (2012a). Such agreement, a priori not guaranteed, gives further support to our simple modelling.
IV Phase transition at finite temperature
Our main scope here is however to describe the first-order phase transition upon heating from the low-temperature M1 monoclinic insulator to the high-temperature rutile metal. In general, we can envisage a phase transition primarily driven either by the electron entropy or by the lattice one.
Indeed, we note that the electron free energy of the metal solution, which is metastable at , must drop faster upon raising temperature than the insulator free energy since the metal carries more electron entropy than the insulator. This effect alone, that is ignoring lattice entropy, would be able to drive a first-order transition when insulator and metal free energies cross. On the other hand, since the distorted ground state breaks the symmetry of the adiabatic lattice potential in Fig. 3, we might expect such symmetry to be recovered by raising temperature only because of lattice entropy effects, i.e., ignoring the electronic contribution to entropy.
In reality, both effects should combine to drive the transition. However, dealing together with lattice and electron entropies within our computational scheme would imply to calculate the adiabatic potential at any temperature, which is a rather heavy task. For this reason, in what follows we shall analyse separately electron and lattice entropy effects, and at the end argue what would happen should they act together.
IV.1 Electron-driven transition
Let us first neglect the lattice entropy and study the temperature evolution of the free energies of the two inequivalent minima that we found at zero temperature. For that, we need to evaluate the electronic entropy, which can be obtained through:
[TABLE]
The last equality corresponds to a change of integration variable from the temperature to the adiabatic potential , which is also the internal energy.
From the entropy we can estimate the free energy:
[TABLE]
which, we emphasise once more, does not include the lattice contribution to entropy. We shall compare the free energy of the undistorted metal solution at , with that of the distorted insulator at . In principle, the equilibrium displacement in the insulator should change with temperature. In practice, since the entropy of the insulator is negligible for all temperatures under consideration, we shall fix at the value. The temperature evolution of the metal and insulator free energies so obtained are shown in Fig. 6. As expected, the larger entropy of the metal pushes its free energy below the insulator one at relatively low temperature, , substantially smaller than the insulating gap, and thus justifying our assumption of frozen . identifies the insulator-metal transition, which is evidently first order since the two free energies cross with different slopes. Incidentally, in half-bandwidth units, corresponds to for a realistic bandwidth of , which has the right order of magnitude when compared with the true critical temperature of . However, we should mention that a different choice of the parameters appearing in Eq. (1), keeping them still in a regime representative of the physics of Vanadium dioxide, would have produced a different value of .
IV.2 Lattice driven transition
We now move to study the properties of the lattice-driven transition. For that, we first need to model the lattice dynamics. However, since the tetragonal R to monoclinic M1 transition is a complex structural transformation, with martensitic features, especially in films de Almeida et al. (2000); Lopez et al. (2002); Pan et al. (2004); Claassen et al. (2010); Viswanath and Ramanathan (2013), our modelling ought to be oversimplified, and aimed just to get qualitatively reasonable results, with no pretension of quantitative accuracy.
As a first step, we must relax our previous assumption of a global antiferrodistortive mode, and instead introduce a displacement field, i.e., a site dependent displacement \mathbf{X}_{i}=\big{(}X_{1i},X_{2i}\big{)}. We assume that feels the local adiabatic potential \Phi_{\text{eff}}\big{(}{\bf X}_{i}\big{)} of Fig. 3a, temperature independent since we are neglecting the electron entropy. In addition, we suppose that the displacements of nearest-neighbour sites are coupled to each other by an invariant term that tends to minimise the strain. With those assumptions the classical Hamiltonian reads:
[TABLE]
where denotes a configuration of all the displacement vectors. The model (13) is equivalent to a generalized -model, where plays the role of two-component spin of variable length, while is the conventional spin stiffness. \Phi_{\text{eff}}\big{(}{\bf X}_{i}\big{)} is the effective anisotropic potential obtained from the solution of the electron problem. Both the length and the direction of the local distortion are controlled by the effective potential \Phi_{\text{eff}}\big{(}{\bf X}_{i}\big{)}, which is not invariant under but under separate and transformations, i.e., . The phase diagram of an model in presence of an anisotropy term that lowers down to is already known Schneider and Stoll (1976); José et al. (1977); Lou et al. (2007). In particular, the anisotropy for is a dangerously irrelevant perturbation that does not change the universality class of the transition José et al. (1977); Lou et al. (2007). Our specific case study, where , has not been considered yet, at least to our knowledge, but it should most likely change the universality class, which is what we are going to investigate in the following.
We study the classical model Eq. (13) at different temperatures using standard Monte Carlo (MC) method Binder and Heermann (2019). We consider the model on a three-dimensional cubic lattice of side . The average value of a given observable, i.e. , where , is therefore estimated statistically using MC algorithm to explore the configuration space. New configurations are generated and accepted/rejected using Metropolis algorithm with local updates. Each local update corresponds to a shift of one of the two component , chosen with equal probability. Within our calculations we use , yet we checked that smaller values do not change the accuracy of the calculations. In addition, we include the possibility of global moves of the type \mathbf{X}_{i}\rightarrow\big{(}-X_{1i},X_{2i}\big{)}, \big{(}X_{1i},-X_{2i}\big{)} or \big{(}-X_{1i},-X_{2i}\big{)} with a total probability equally distributed among the three cases, i.e. with probability each. The local updates require the evaluation of the effective potential \Phi_{\text{eff}}\big{(}{\bf X}_{i}\big{)} at the new value of . To speed-up execution, we pre-evaluate all the interpolated values of the effective potential at any possible point compatible with the size of the shift using a bi-cubic spline method. A new configuration of the system is obtained after a full sweep of the lattice sites. The statistical error is thus controlled by the number of sweeps , to which it corresponds a number of MC steps. In order to avoid self-correlation problems, we measure the average of any observables every sweeps and in any case after a warm-up period of sweeps. In all our calculations, the number of sweeps is of the order of . We further minimize the statistical error by executing the numerical computation in parallel with cpu. The resulting statistical error is within the symbols in all our plots.
Before discussing the results, we have to mention that some details might depend on the precise form of the coupling between different sites. In the model Hamiltonian Eq. (13) we have choosen the simplest possible one, i.e. a nearest neighbor coupling, thus disregarding longer range interactions.
In Fig. 7(a) we plot the modulus of the average displacement, \big{|}\langle\,\mathbf{X}\,\rangle\big{|}, as function of the temperature. For small system size (e.g. ) \big{|}\langle\,\mathbf{X}\,\rangle\big{|} shows a smooth crossover in temperature. However, increasing unveils the existence of a continuous phase-transition at a critical value of the temperature. The actual value of depends by the size of the coupling constant , that somehow acts as a unit of measure for the energy. More involved calculations are necessary for the evaluation of in Vanadium dioxide from first-principle calculations and we postpone them for a future work. For that reason, we have preferred to use as the unit of temperature in Fig. 7 and in those that follow. In order to better reveal the second order character of the transition, we also show in Fig. 7(a) the fit with a mean-field square-root behaviour. The fit is rather good, although we known that close to the transition the actual critical behaviour must deviate from mean-field.
A closer look to the temperature dependence of the order parameter uncovers a non-trivial two-step evolution, which is more evident in Fig. 7(b), where we show the specific heat vs. . Indeed, clearly displays two peaks that are suggestive of two distinct transitions. The first transition at , below which \big{|}\langle\,\mathbf{X}\,\rangle\big{|} acquires a finite value, is followed by a second one at lower .
From the knowledge of the specific heat at constant volume , we can compute the change of the vibrational entropy in the region where the two transitions occur as . This quantity is displayed in Fig. 7(c) and it shows an almost linear behavior in the whole explored temperature range. However, as shown by the dashed lines there, the slopes of the line is different in the three regions , and .
In order to understand the nature of both transitions, in Fig. 8 we show at , left panels, , middle panels, and , right panels, the endpoint distribution after MC sweeps of the lattice of the displacement vectors superimposed to the potential landscape in the space (top panels), and a real space snapshot within a single layer of the cubic lattice (bottom panels). At high temperature, , the ’s cover homogeneously the whole potential landscape, see top-left panel, without any appreciable spatial correlation, see the bottom-left panel. Lowering slightly below , we observe a significant change in the displacement distribution, see middle panels. Specifically, the system seems to break ergodicity first along , in the simulation corresponding to the figure it localises in the half-plane, while it is still uniform along . Consequently, clusters of parallel displacement vectors form in real space. The alignment direction has for all clusters, while the component changes from cluster to cluster, see bottom-middle panel. Only below , full ergodicity breakdown occurs, with the system trapped around just one of the four equivalent minima, in the figure that with and . In other words, the symmetry of the model Eq. (13) gets broken in two steps upon cooling: first, the symmetry spontaneously breaks, and next, the residual symmetry, leading to two consecutive Ising-like transitions. This is summarised in Fig. 9, where we see that at becomes finite, and thus also \big{|}\langle\mathbf{X}\rangle\big{|}, while is still zero. Only below also acquires a finite average value. Accordingly the vibrational entropy loss , shown in Fig. 7(c), changes of a quantity between the two temperatures and . This is consistent with an increase of the available phase space for the system of a factor of two by moving from one temperature to the other.
Translated in the language of VO2, these results suggest the existence of an intermediate monoclinic phase for where the V atoms are displaced only within the basal plane, i.e., the chains are tilted but not yet dimerised. In our model Hamiltonian (1), such phase with describes a monoclinic metal, which, as discussed in Sec. I, has been reported in several experiments Umeda et al. (1966); Kim et al. (2008); Nag et al. (2012); Yao et al. (2010); Laverock et al. (2014); Zhang et al. (2009); Kim et al. (2006); Cocker et al. (2012); Kumar et al. (2014); Ilinskiy et al. (2012a, b); Chen et al. (2008). Only below , both components of the antiferrodistortive displacement are finite, leading to the M1 insulating phase.
In conclusion, without including the electron entropy we find two transitions that look continuous and in the Ising universality class: one at between a monoclinic insulator and a monoclinic metal, and another at from the monoclinic metal to a rutile one. On the contrary, neglecting the lattice entropy and just including the electronic one, we found in Sec. IV.1 a single first-order transition at , directly from the monoclinic insulator to the rutile metal. We can try now to argue what we could have obtained keeping both entropy contributions still within the Born-Oppenheimer adiabatic approximation.
In that case, we expect that the depth of the rutile minimum in the Born-Oppenheimer potential of Fig. 3 becomes a growing functions of , unlike the depth of the insulating minima, since in the insulator the electronic entropy is negligible with respect to that in the metal. For the same reason, we expect that the height of the two equivalent saddle points at but , see black and blue lines in panel (b) and panel (c), respectively, of Fig. 3, lowers with increasing , since these points with large crystal field splitting but without dimerisation just describe the monoclinic metal, eventually turning these saddle points into local minima. This effect might well turn the monoclinic-insulator to monoclinic-metal transition at into a first order one, all the more if we better modelled the martensitic features of the structural distortion. However, even in that case we still expect a further transition at into the rutile metal, unless the latter has such a large entropy compared to the monoclinic metal to drive a first order transition from the insulator directly into the rutile metal, as it would occur if , i.e., if the electronic entropy gain far exceeds the lattice one.
The experimental evidences supporting the existence of a monoclinic metal phase intruding between the M1 insulator and R metal Umeda et al. (1966); Kim et al. (2008); Nag et al. (2012); Yao et al. (2010); Laverock et al. (2014); Zhang et al. (2009); Kim et al. (2006); Cocker et al. (2012); Kumar et al. (2014); Ilinskiy et al. (2012a, b); Chen et al. (2008) suggest that, should our modelling be indeed representative of VO2, then the Hamiltonian parameters should be such that . This also entails a substantial release of lattice entropy across the transition, in accordance with experiments Budai et al. (2014); Chen et al. (2018) and theoretical Mellan et al. (2019) proposals. We emphasise that does not mean that correlations play a minor role, but rather the opposite, since it would imply the insulator, whose internal energy is substantially contributed by electronic correlations, would survive up to much higher temperature if it were not for the lattice.
V Conclusions
We have constructed a minimal model that we believe contains all essential ingredients to correctly capture the physics of the metal-insulator transition in vanadium dioxide.
The model comprises two orbitals per site, one mimicking the and the other the , thus neglecting the twofold nature of the latter, which broaden into two bands. The band has a double peak structure reflecting its bonding character along the rutile -axis, while the one is structureless. Both have the same bandwidth and centre of gravity. The density corresponds to one electron per site, i.e., the two bands are at quarter filling. The electrons feel an on-site Hubbard repulsion, and are coupled to two zone-boundary lattice modes, corresponding, respectively, to the basal plane component, i.e., the tilting of the Vanadium chains, and out-of-plane component, responsible of the chain dimerisation, of the antiferrodistortive displacement that acquires a finite expectation value below the transition from the high temperature rutile structure to the low temperature monoclinic one (M1). Using realistic Hamiltonian parameters and assuming the Born-Oppenheimer adiabatic approximation, we find at low temperatures phase coexistence between a stable distorted insulator, the monoclinic M1 insulator, and a metastable undistorted metal, the rutile metal. Upon rising temperature, our model description suggests a two-step transition. First, the dimerisation component of the antiferrodistortive displacement melts, leading to a transition from the monoclinic insulator to a monoclinic metal. At higher temperature also the tilting component disappears, and the monoclinic metal turns into the rutile one. Such a two-transition scenario, not in disagreement with experiments, is mostly driven by the lattice entropy, also in accordance with experiments.
One of the messages of our model calculation is that the electron-electron interaction has the role to effectively enhance the coupling to the lattice, stabilising a distorted phase otherwise metastable in the absence of interaction. This also implies that we could have obtained similar results with weaker electronic correlations but stronger electron-lattice coupling. This conclusion is actually supported by the phenomenology of Niobium dioxide NbO2, which, mutatis mutandis, is akin to that of VO2. NbO2 also undergoes a metal-insulator transition, though at substantially higher temperature of Janninck and Whitmore (1966); Beebe et al. (2017); Seta and Naito (1982); Rao et al. (1973). There is some experimental evidence of separate structural and electronic phase transitions occurring in this compound Seta and Naito (1982); Rao et al. (1973); Goodenough (1971b); Tolédano and Tolédano (1977), with a transition temperature for the structural change Sakata et al. (1967), from a high-temperature rutile structure to a low-temperature body centred tetragonal (BCT) one that locally resembles the M1 phase of VO2 Goodenough (1971b); Dhamdhere et al. (2016); Bolzan et al. (1994); Hiroi (2015). It has been proposed that the mismatch between and can be justified by invoking a melting of the dimerization component of the structural distortion in the BCT insulator at smaller temperature as compared to the melting of the tilting component Goodenough (1971b); Rao et al. (1973); Tolédano and Tolédano (1977); Sakata (1969). The metallic solution that appears in between the two transition temperatures should be mostly metallic along the cR axis, since the almost one dimensional band gives the most relevant contribution to the spectral weight at the Fermi level in this intermediate phase. This expectation is not in disagreement with some experimental findings in which they measure, above , a metallic conductivity along cR while a semiconducting one in the orthogonal direction Bélanger et al. (1974). However, we should point out that not all the experiments confirm this scenario Wahila et al. (2019). We believe that a similar anisotropy in the conduction properties should be displayed also by the monoclinic metallic phase of Vanadium dioxide. The single -electron in Nb4+ is expected to be less correlated than the -electron in V4+. This loss of correlations, testified by the VO2 M2 phase having no counterpart in NbO2 Haines et al. (1999), and by the efficacy of ab initio methods to describe NbO2 Eyert (2002b); Brito et al. (2017); O’Hara et al. (2014); O’Hara and Demkov (2015), is actually overcompensated by the increase in covalency due to the broader spatial distribution of the orbitals Wong et al. (2014), which, in turn, yields a stronger coupling with the zone-boundary lattice modes, and thus a higher transition temperature.
Acknowledgements
F.G. likes to thank Sergiy Lysenko for the useful discussions about the thermodynamic potential for the two phononic modes, as well as for the choice of the parameters appearing there. F.G. thanks also Maja Berović and Daniele Guerci for the discussions concerning the manuscript. A.A. thanks Massimo Capone and Sandro Sorella for useful discussions. We thank Martin Eckstein for fruitful debatings. We acknowledge support from the H2020 Framework Programme, under ERC Advanced Grant No. 692670 “FIRSTORM”. A.A. also acknowledges financial support from MIUR PRIN 2015 (Prot. 2015C5SEJJ001) and SISSA/CNR project ”Superconductivity, Ferroelectricity and Magnetism in bad metals” (Prot. 232/2015).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yang et al. (2011) Zheng Yang, Changhyun Ko, and Shriram Ramanathan, “Oxide electronics utilizing ultrafast metal-insulator transitions,” Annual Review of Materials Research 41 , 337–367 (2011) . · doi ↗
- 2Driscoll et al. (2009) T. Driscoll, Hyun-Tak Kim, Byung-Gyu Chae, Bong-Jun Kim, Yong-Wook Lee, N. Marie Jokerst, S. Palit, D. R. Smith, M. Di Ventra, and D. N. Basov, “Memory metamaterials,” Science 325 , 1518–1521 (2009) . · doi ↗
- 3Zhou et al. (2013) J. Zhou, Y. Gao, Z. Zhang, H. Luo, C. Cao, Z. Chen, L. Dai, and X. Liu, “Vo 2 thermochromic smart window for energy savings and generation,” Scientific Reports 3 , 3029 (2013) . · doi ↗
- 4Huang et al. (2010) Wan-xia Huang, Xiao-gang Yin, Cheng-ping Huang, Qian-jin Wang, Teng-fei Miao, and Yong-yuan Zhu, “Optical switching of a metamaterial by temperature controlling,” Applied Physics Letters 96 , 261908 (2010) . · doi ↗
- 5Ke et al. (2018) Yujie Ke, Shancheng Wang, Guowei Liu, Ming Li, Timothy J. White, and Yi Long, “Vanadium dioxide: The multistimuli responsive material and its applications,” Small 14 , 1802025 (2018) . · doi ↗
- 6Liu et al. (2012) M. Liu, H. Y. Hwang, H. Tao, A. C. Strikwerda, K. Fan, G. R. Keiser, A. J. Sternbach, K. G. West, S. Kittiwatanakul, J. Lu, S. A. Wolf, F. G. Omenetto, X. Zhang, K. A. Nelson, and R. D. Averitt, “Terahertz-field-induced insulator-to-metal transition in vanadium dioxide metamaterial,” Nature 487 , 345 (2012) . · doi ↗
- 7Zheludev and Kivshar (2012) N. I. Zheludev and Y. S. Kivshar, “From metamaterials to metadevices,” Nature Materials 11 , 917 (2012) . · doi ↗
- 8Kats et al. (2013) Mikhail A. Kats, Romain Blanchard, Shuyan Zhang, Patrice Genevet, Changhyun Ko, Shriram Ramanathan, and Federico Capasso, “Vanadium dioxide as a natural disordered metamaterial: Perfect thermal emission and large broadband negative differential thermal emittance,” Phys. Rev. X 3 , 041004 (2013) . · doi ↗