van der Waals forces stabilize low-energy polymorphism in B2O3: Implications for the crystallization anomaly
Guillaume Ferlat, Maria Hellgren, Fran\c{c}ois-Xavier Coudert, Henri, Hay, Francesco Mauri, Michele Casula

TL;DR
This study uses advanced computational methods to show that van der Waals forces stabilize low-energy polymorphs of B$_2$O$_3$, explaining its tendency to form glassy structures instead of crystalline forms.
Contribution
It reveals the crucial role of van der Waals interactions in stabilizing B$_2$O$_3$ polymorphs and explains the crystallization anomaly through first-principles calculations.
Findings
Van der Waals forces stabilize the experimentally known B$_2$O$_3$ polymorph.
Many metastable structures are close in energy to the glass.
The best metastable polymorph shares motifs with the glass and a borosulfate compound.
Abstract
The cohesive energies and structural properties of recently predicted, and never synthesized, BO polymorphs are investigated from first principles using density functional theory and high-accuracy many-body methods, namely, the random phase approximation and quantum Monte Carlo. We demonstrate that the van der Waals forces play a key role in making the experimentally known polymorph (BO-I) the lowest in energy, with many competing metastable structures lying only a few kcal/mol above. Remarkably, all metastable crystals are comparable in energy and density to the glass, while having anisotropic and mechanically soft structures. Furthermore, the best metastable polymorph according to our stability criteria has a structural motif found in both the glass and a recently synthesized borosulfate compound. Our findings provide new perspectives for understanding the BO…
Click any figure to enlarge with its caption.
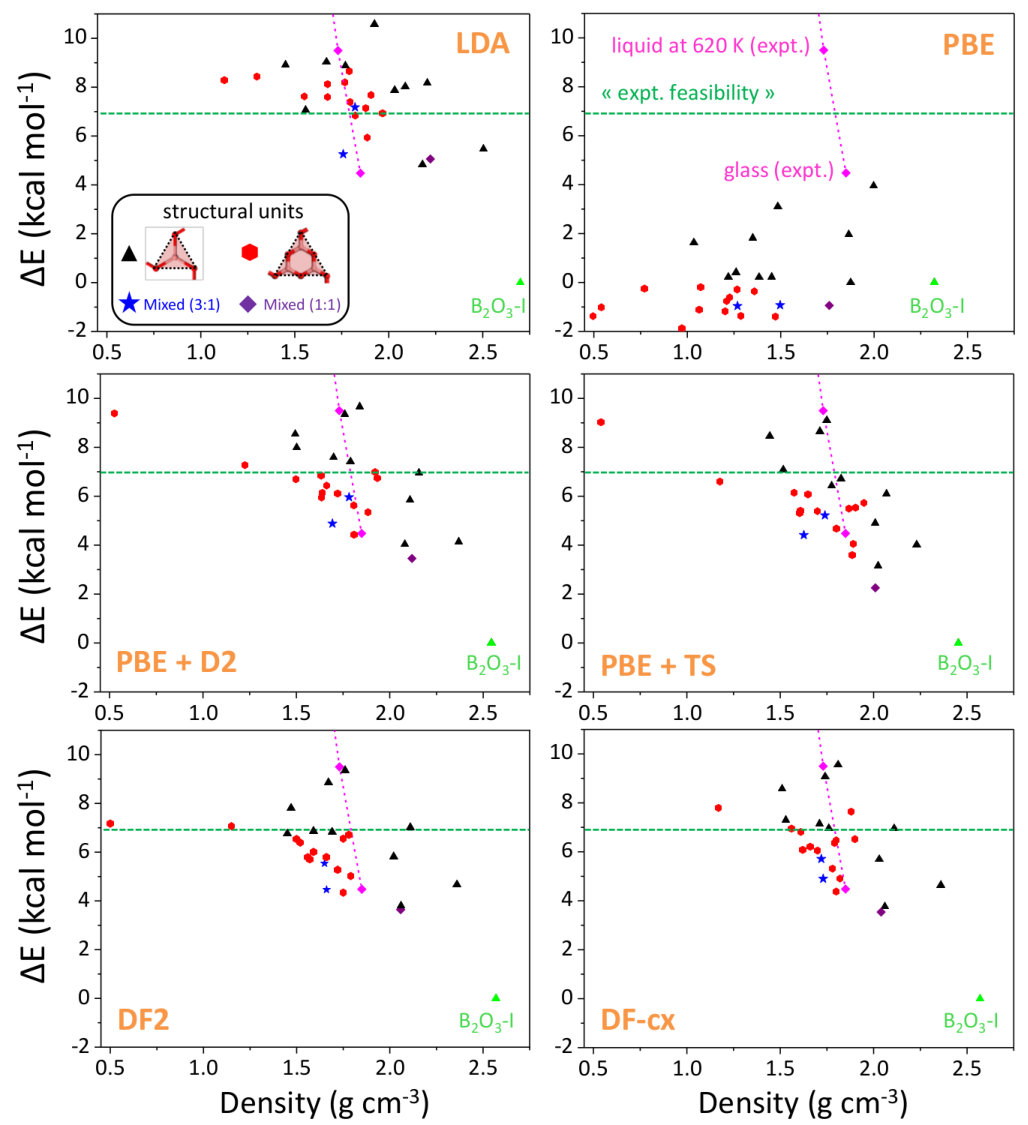 Figure 1
Figure 1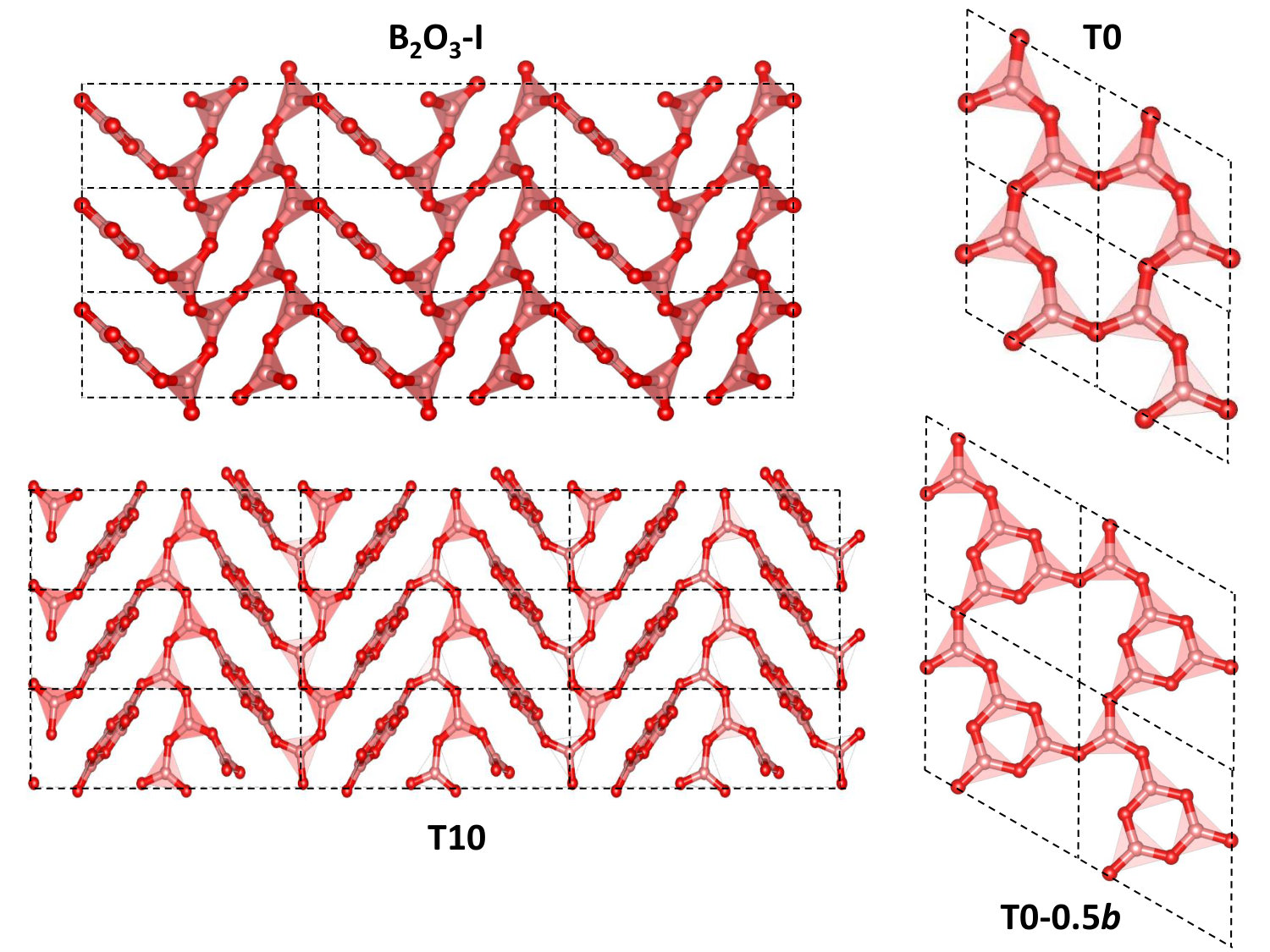 Figure 1
Figure 1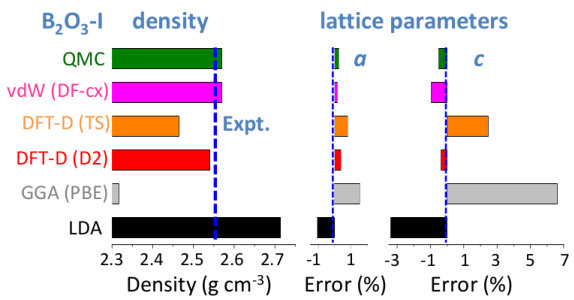 Figure 2
Figure 2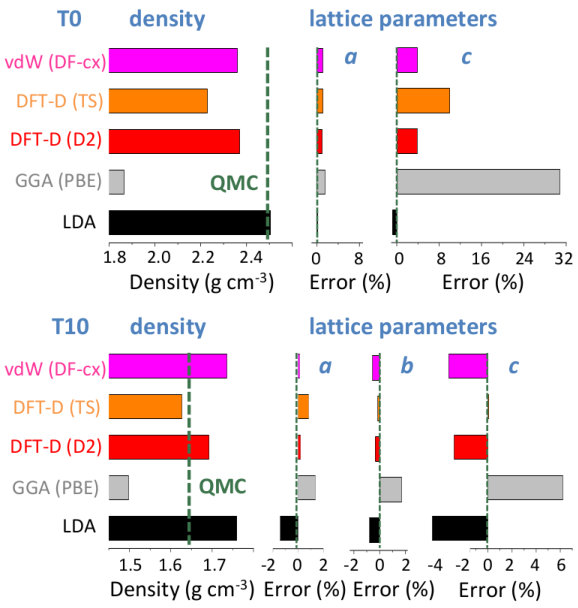 Figure 3
Figure 3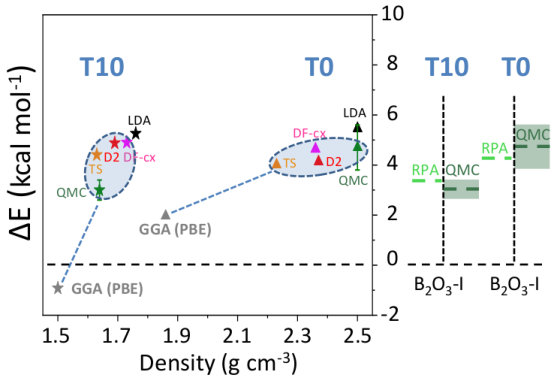 Figure 4
Figure 4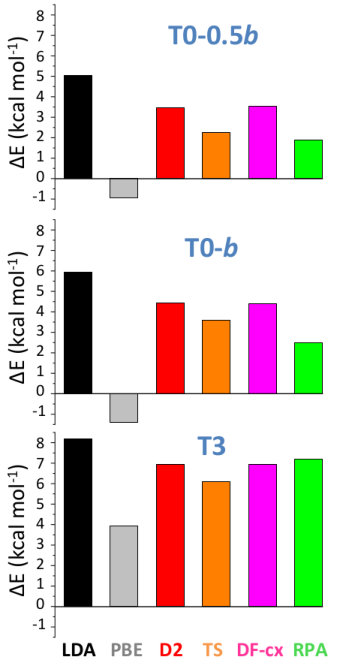 Figure 5
Figure 5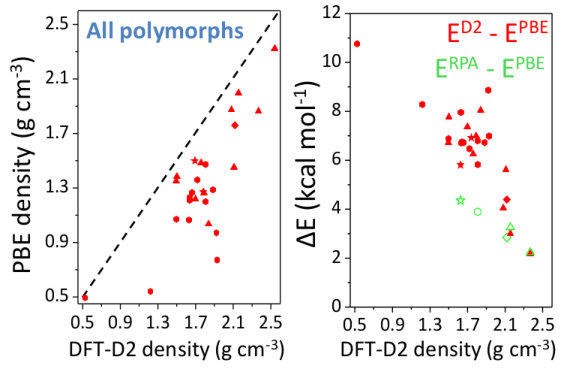 Figure 6
Figure 6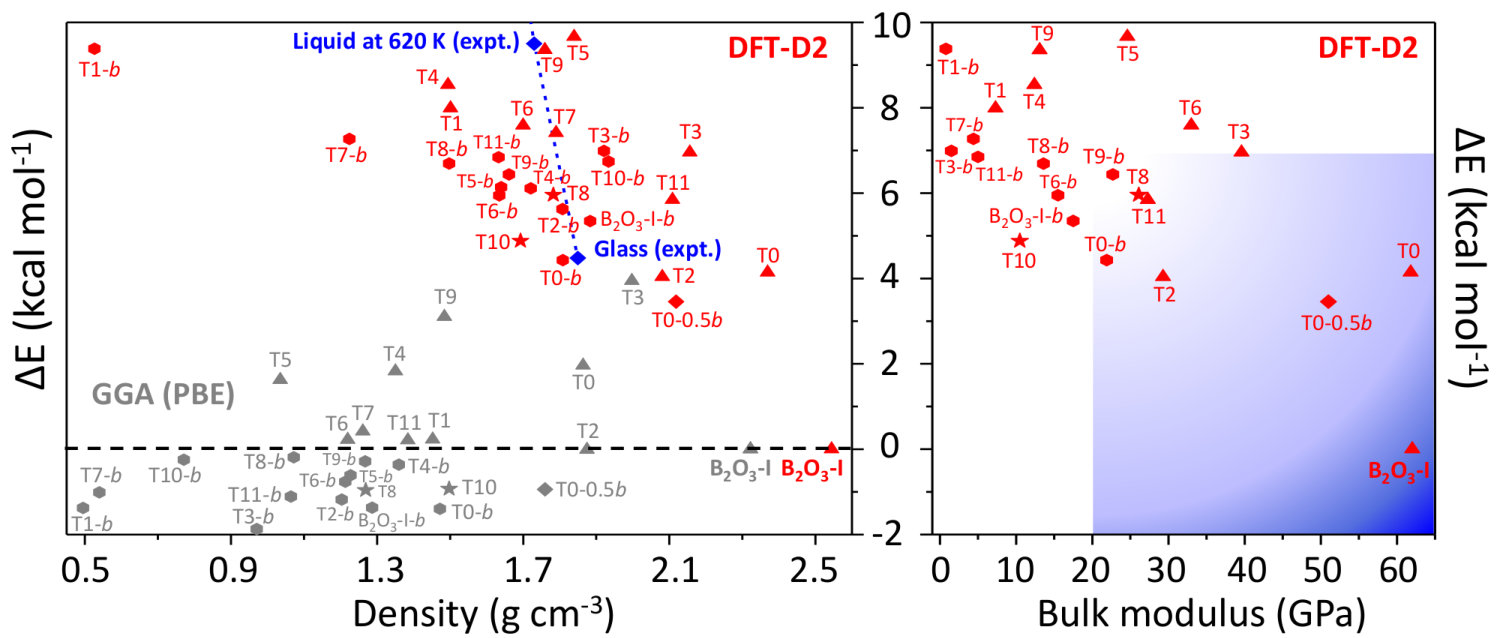 Figure 7
Figure 7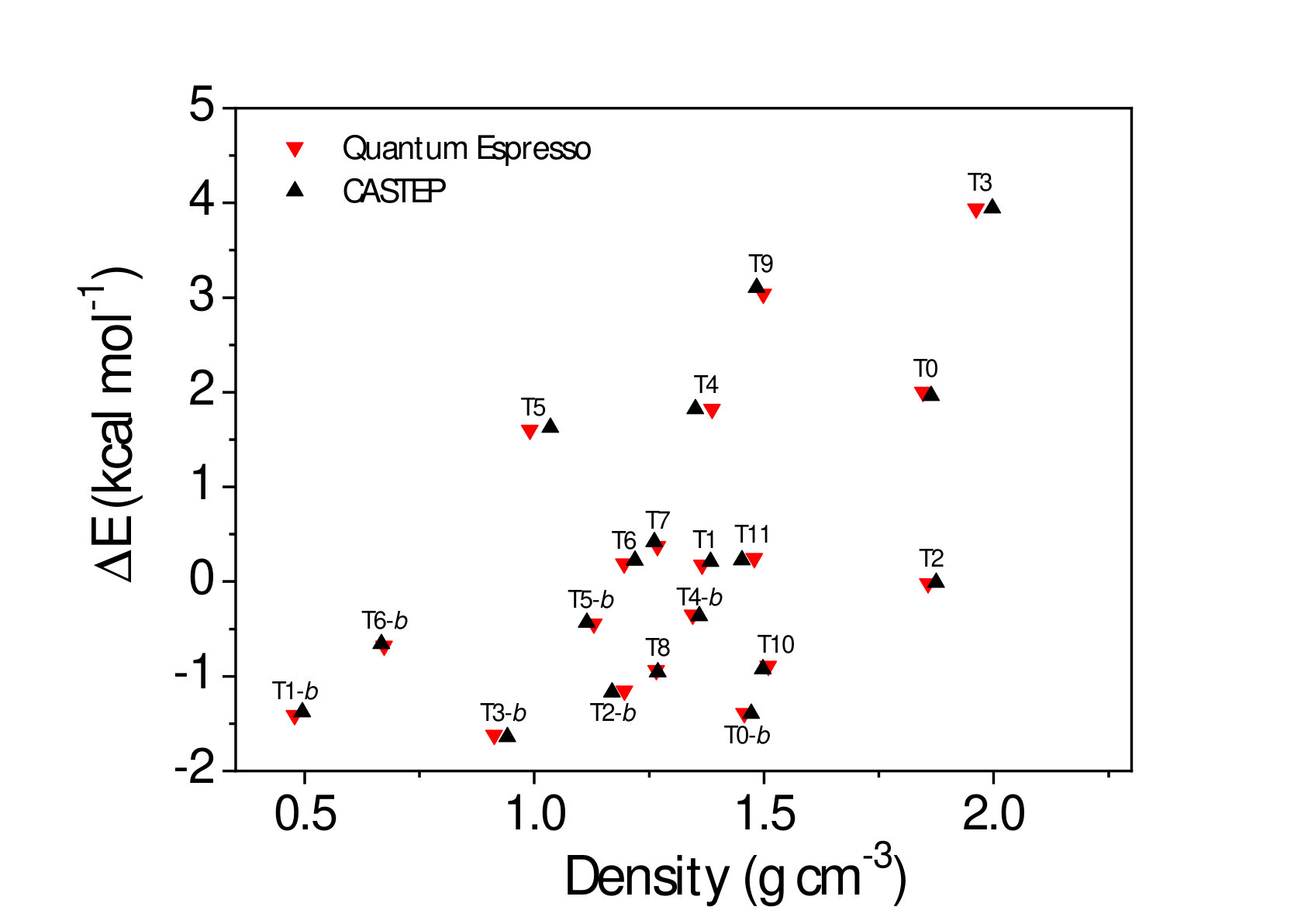 Figure 9
Figure 9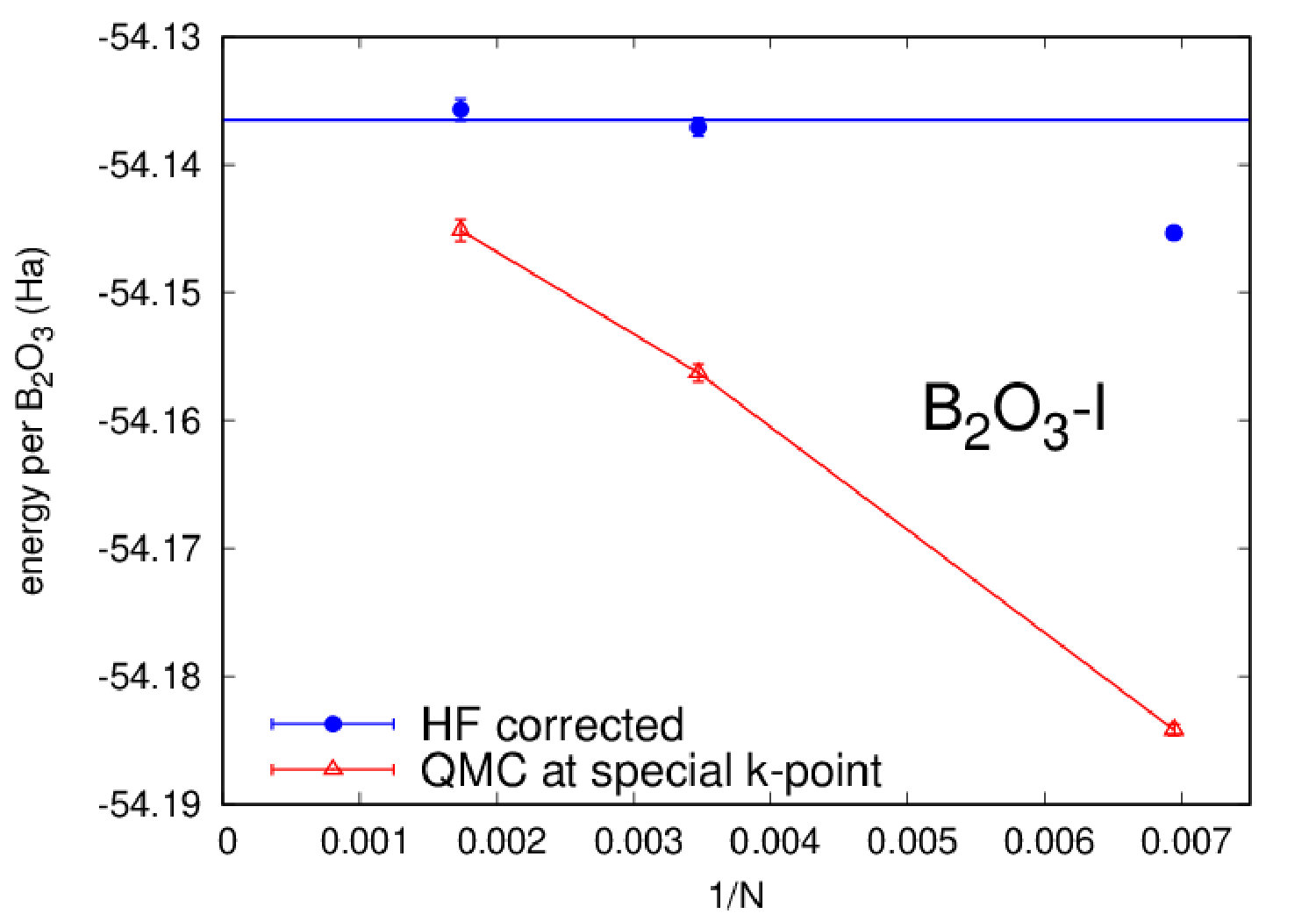 Figure 10
Figure 10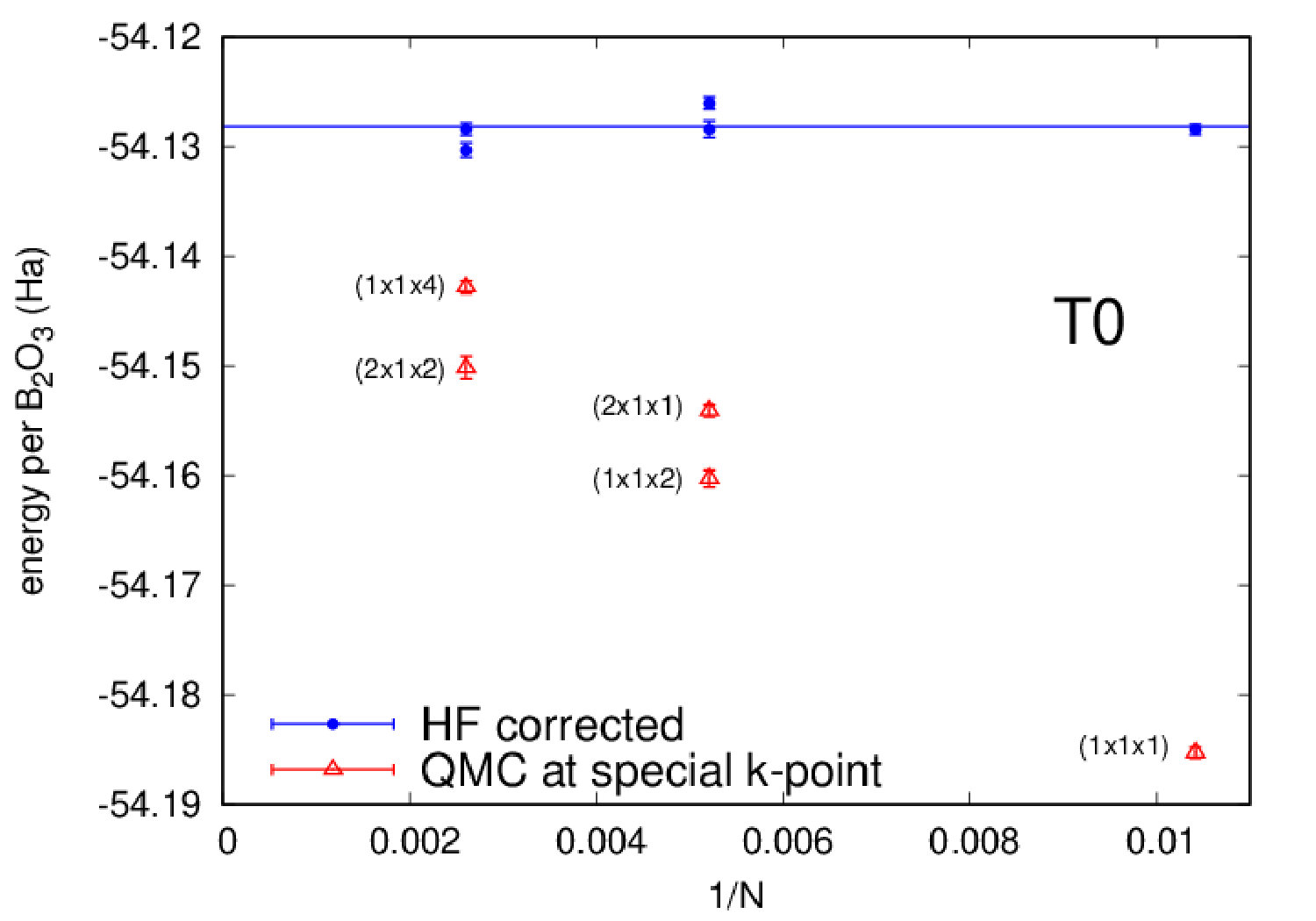 Figure 11
Figure 11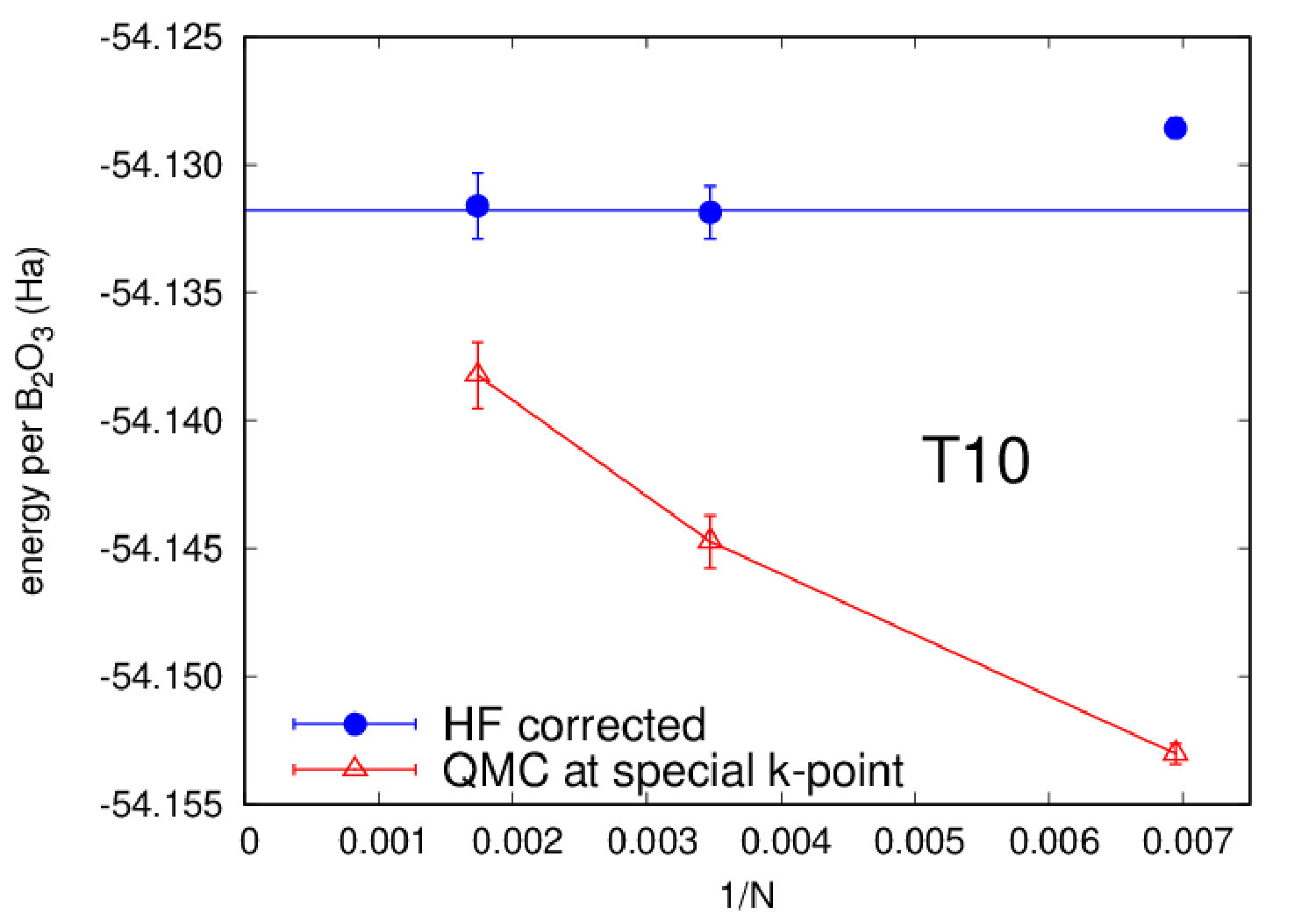 Figure 12
Figure 12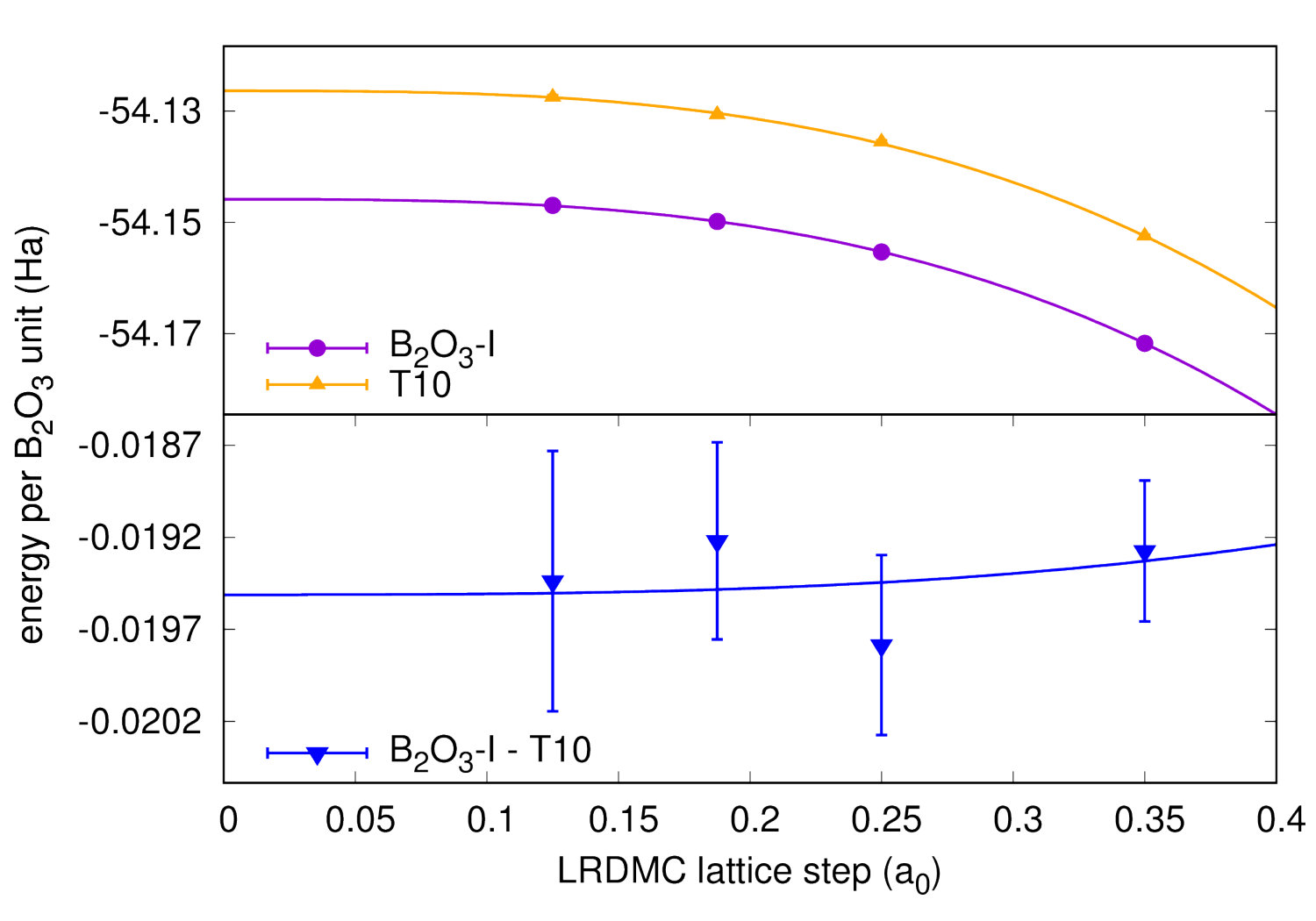 Figure 13
Figure 13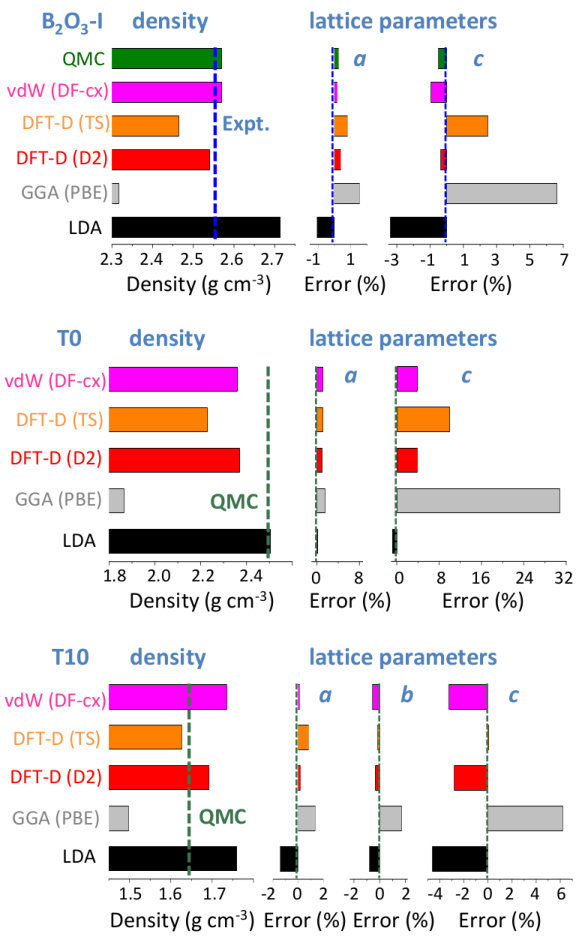 Figure 14
Figure 14Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
van der Waals forces stabilize low-energy polymorphism in B2O3:
Implications for the crystallization anomaly
Guillaume Ferlat
Sorbonne Université, MNHN, UMR CNRS 7590, IRD, IMPMC, F-75005 Paris, France
Maria Hellgren
Sorbonne Université, MNHN, UMR CNRS 7590, IRD, IMPMC, F-75005 Paris, France
François-Xavier Coudert
Chimie ParisTech, PSL University, CNRS, Institut de Recherche de Chimie Paris, 75005 Paris, France
Henri Hay
Sorbonne Université, MNHN, UMR CNRS 7590, IRD, IMPMC, F-75005 Paris, France
Francesco Mauri
Sorbonne Université, MNHN, UMR CNRS 7590, IRD, IMPMC, F-75005 Paris, France
Dipartimento di Fisica, Università di Roma La Sapienza, Piazzale Aldo Moro 5, I-00185 Roma, Italy
Michele Casula
Sorbonne Université, MNHN, UMR CNRS 7590, IRD, IMPMC, F-75005 Paris, France
(March 2, 2024)
Abstract
The cohesive energies and structural properties of recently predicted - and never synthesized - B2O3 polymorphs are investigated from first principles using density functional theory and high-accuracy many-body methods, namely, the random phase approximation and quantum Monte Carlo. We demonstrate that the van der Waals forces play a key role in making the experimentally known polymorph (B2O3-I) the lowest in energy, with many competing metastable structures lying only a few kcal/mol above. Remarkably, all metastable crystals are comparable in energy and density to the glass, while having anisotropic and mechanically soft structures. Furthermore, the best metastable polymorph according to our stability criteria has a structural motif found in both the glass and a recently synthesized borosulfate compound. Our findings provide new perspectives for understanding the B2O3 anomalous behavior, namely, its propensity to vitrify in a glassy structure drastically different from the known crystal.
I INTRODUCTION
Diboron trioxide (B2O3) not only is the second most used component of industrial glasses after silica (SiO2), but also a canonical network-forming system per se (see, e.g., Refs. Ferlat (2015); Wright (2018) for reviews). The originality of the low-pressure B2O3 networks, either crystalline or vitreous, stems from their building blocks, which are bidimensional (2D) trigonal (BO3) units. This is in contrast with most network formers, such as silica, based on three-dimensional (3D) tetrahedral units. Fully 3D networks are then formed by binding these rigid units through flexible cation-oxygen-cation bonds, which give B2O3 low-density structures, and a great potential for polymorphic transformations, under, e.g., high temperature or pressure. This is clearly reflected by various studies showing polyamorphic transformations in the glass Nicholas et al. (2004); Lee et al. (2005); Trachenko et al. (2008); Zeidler et al. (2014); Lee et al. (2018) and to a lesser extent in the liquid Brazhkin et al. (2010); Alderman et al. (2015).
However, our knowledge of the B2O3 crystalline forms remains very limited: up to now, only one low-pressure BO3-based crystal (B2O3-I) has been experimentally characterized Gurr et al. (1970) in addition to a high-pressure phase (B2O3-II), based upon BO4 tetrahedral units. The possible existence of another low-density polymorph, of unidentified structure, has been reported long ago Cole and Taylor (1935), but the status of this report remains unclear since subsequent attempts failed to reproduce it. This is in sharp contrast with the rich polymorphism found in other oxide systems: in silica Piccione et al. (2000), for instance, more than 20 low-pressure polymorphs (from coesite to zeolites) built upon the same basic units have been experimentally reported.
Another striking and very uncommon feature is the abnormal structural dissimilarity between the glassy (g-B2O3) and crystalline forms. In g-B2O3, about half of the BO3 elemental bricks are arranged into superstructural units, i.e. threefold rings referred to as boroxol rings Youngman et al. (1995); Umari and Pasquarello (2005), fully absent from B2O3-I (see Fig. 1). As a consequence, the glass density is considerably smaller ( -30%) than the B2O3-I one. Likely related Zanotto and Cassar (2017) but yet not understood, is the extremely high glass-forming ability of B2O3, arguably the best glass former. Indeed, the B2O3-I crystallization has never been observed from ambient pressure liquid, even if seeded with germs for months. The synthesis requires cooling the liquid under pressure, or alternatively using chemical routes, a behavior which has been coined as the crystallization anomaly Ulhmann et al. (1967).
Mostly inspired by the structural differences between g-B2O3 and B2O3-I, several theoretical works have predicted additional polymorphs Takada et al. (2003); Huang et al. (2007); Ferlat et al. (2012); Claeyssens et al. (2013). In particular, a set of 25 new crystals Ferlat et al. (2012), further complemented by two additional ones Claeyssens et al. (2013), has recently been obtained using density functional theory (DFT). It spans a very narrow energy range (a few kcal/mol), with values comparable to, or even lower than, B2O3-I, therefore challenging it as the ground state. Hence, high-level theories are needed to go beyond the standard approximations (LDA, GGA) that have been used in all previous DFT works. As a matter of fact, a drastically different physical picture emerges from our high-level calculations as will be shown later.
In the current work, we employ accurate many-body methods such as quantum Monte Carlo (QMC) and the random-phase approximation (RPA) to provide a definitive answer to the relative stability between B2O3-I and a subset of predicted polymorphs. In addition, we present results for a large set of polymorphs at different levels of DFT which demonstrate a huge effect of the van der Waals (vdW) forces on both the structures and energetics, an effect that has been neglected in all previous studies Takada et al. (2003); Huang et al. (2007); Ferlat et al. (2012); Claeyssens et al. (2013). We definitely assess B2O3-I as the ground state while we introduce a novel polymorph (T0-0.5), adapted from a recently synthesized borosulfate structure Daub and Hillebrecht (2015) which we reveal as the most stable among all putative structures. Its layers are decorated by triangle and boroxol units (Fig. 1) in equal proportions (1:1), making it a close, albeit crystalline, structural approximant of the glass.
II METHODS
Highly accurate QMC simulations have been carried out on three polymorphs (B2O3-I and two predictions T0 and T10). We used a Jastrow-Slater variational wave function, with Slater and Jastrow parts developed on a localized Gaussian basis set. The Jastrow factor contains correlation terms up to the four-body (electron-ion-electron-ion) form, able to capture van der Waals effects within variational Monte Carlo (VMC) Sorella et al. (2007). The wave function has been fully optimized (Slater orbitals together with Jastrow coefficients) by energy minimization Umrigar et al. (2007), starting from DFT-LDA generated one-body orbitals. Moreover, we have performed a complete structural relaxation for both cell parameters and internal coordinates at the VMC level Barborini et al. (2012). Then, using relaxed VMC geometries, we have carried out lattice-regularized diffusion Monte Carlo simulations Casula et al. (2005, 2010) and a very accurate finite-size extrapolation Dagrada et al. (2016) in order to provide energies with accuracy better than 1.0 kcal/mol. A careful convergence of all relevant criteria, i.e. basis set, geometry, finite-size effects and level of theory, is necessary, given the phase-space proximity of the B2O3 polymorphs. All QMC calculations have been performed using the TurboRVB code Sorella . Further details can be found in the Supplemental Material (SM).
Calculations with the RPA were performed on a larger set of six polymorphs (B2O3-I, T0, T10, T3, T0-0.5 and T0-). The RPA is a state-of-the-art density functional approach based on many-body perturbation theory and the adiabatic connection fluctuation-dissipation formula Langreth and Perdew (1975); Gunnarsson and Lundqvist (1976). By including polarization diagrams to infinite order in the Coulomb interaction the RPA captures vdW forces Marini et al. (2006); Lebègue et al. (2010). It also provides an accurate description of Hartree-Fock exchange, and an overall good description of correlation effects (at least as far as energy differences are concerned). RPA total energies were calculated with the VASP code Kresse and Furthmüller (1996); Kresse and Joubert (1999). Computational details can be found in the SM.
Dispersive interactions can also be added in a semiempirical form to standard DFT approximations such as in the DFT-D approaches: for instance, in D2 Grimme (2006) the vdW interactions coefficients () are fixed for a given atomic pair while in the Tkatchenko-Scheffler (TS) approach Tkatchenko and Scheffler (2009), they are calculated self-consistently according to the atomic neighborhood. At a more advanced level, vdW are accounted for in fully non-local functionals that include polarization effects from first-principles, such as in the vdW-DFT class of functionals Lee et al. (2010); Thonhauser et al. (2015). In this work, we used representative functionals from these different levels, namely, D2 Grimme (2006), TS Tkatchenko and Scheffler (2009) [added on top of the Perdew-Burke-Ernzerhof (PBE) functional Perdew et al. (1996), using the CASTEP code Clark et al. (2005)] and the recently derived DF-cx Thonhauser et al. (2015) vdW-DFT functional (using the Quantum Espresso package Giannozzi et al. (2009)). We checked explicitly that the use of different codes and pseudo-potentials does not affect the reported results (Fig. S1 of the SM).We also report results from LDA and GGA (PBE) functionals as references. In particular, the comparison between GGA and dispersion-corrected schemes (D2 and TS) built upon the same GGA, allows one to probe straightforwardly the relevance of vdW interactions. Thanks to the low computational cost of DFT, we studied a total of 27 polymorphs (those from Ref. Ferlat et al. (2012) supplemented with T0-0.5), which contain up to 135 atoms per unit cell Ferlat et al. (2012).
Finally, we studied the mechanical properties via DFT-D2 calculations of second-order elastic constant tensors following a methodology described elsewhere Coudert (2013); Hay (2016). See the SM for all the details of the DFT calculations.
III RESULTS AND DISCUSSION
We first assess the quality of the structural results obtained from the different schemes by relaxing the B2O3-I structure, and taking the experimentally known geometry as reference (Fig. 2). Not surprisingly, the lattice parameters obtained with LDA are underestimated (and the density overestimated by +6%) while the opposite is true for PBE (density error of -10%). This reflects the well-documented tendencies Haas et al. (2009); Hay et al. (2015) of LDA to overbind and of PBE to underbind. Note, however, that the size of these errors is large for a non-layered inorganic system, placing B2O3-I in the topmost range (95th percentile) of volume errors for inorganic materials of the Materials Project database Sun et al. (2016). Interestingly, these deficiencies are largely cured not only by QMC (+0.5% error on density) but also by all the vdW-corrected schemes used here, which show a systematic improvement.
Although B2O3-I is a fully connected 3D network made of strong interatomic bonds, the importance of the vdW corrections stems from the structural porosity, arising from locally planar regions - on the scale of a few building units - arranged in a zig-zag pattern thanks to the B-O-B angular flexibility (Fig. 1). This leads to a softer direction, perpendicular to the locally planar regions, and nearly parallel to the c direction, as reflected by larger errors in this lattice parameter (Fig. 2). The existence of such a softer direction is found in all but two polymorphs (see also Fig. 3).
We now focus on the densities and energies of two previously predicted Ferlat et al. (2012) polymorphs, namely T0 and T10 using QMC results as reference in the absence of experimental data. We report these results in Figs. 3 and 4. In the following, all crystals’ energies are expressed with respect to the B2O3-I one. Similarly to the experimentally known case, PBE severely underestimates the densities. This impacts the energies, which are also underestimated. Note that T10, which in the PBE original predictions Ferlat et al. (2012) had a slightly lower energy ( kcal/mol) than B2O3-I, turns out to be metastable ( kcal/mol). On the contrary, the vdW-corrected DFT schemes perform reasonably well in both densities and energies. Moreover, the RPA energies agree well with QMC (Fig. 4, right panel).
We also calculated RPA energies for the T0-0.5, T0-, and T3 structures, shown in Fig. 5 together with those from the different DFT schemes. We note that all three vdW-corrected functionals yield essentially the same results, with a typical variability of 2 kcal/mol on the energies, and compare well with the more advanced RPA. This gives strong confidence in the overall picture. A complete account for the full set of 27 B2O3 polymorphs, using the various DFT frameworks, is reported (Fig. S2) in the SM.In the following we shall take D2 as representative of the vdW-corrected functionals BM_ , in order to make a thorough comparison with PBE, and show the impact of the dispersive interactions on the B2O3 polymorphism.
Figure 6 highlights the density and energy corrections brought back by the vdW contributions. Being attractive, the vdW forces systematically provide denser structures (Fig. 6, left panel), on average by 40% and by up to 150% for some polymorphs, such as T8-. Such high figures reflect the fact that for many polymorphs the vdW scheme allows one to find a qualitatively different geometry from the PBE one (e.g. internal voids and large-scale rings tend to be less symmetric and more puckered). This densification effect, which acts effectively as an internal negative pressure, results in an increasing enthalpic penalty with decreasing density (Fig. 6, right panel). Overall, the more porous the polymorph, the larger the vdW contribution to the energy. This trend is supported by the RPA results presented in the same figure, and is also in line with studies of e.g. silica zeolites Hay et al. (2015). In other words, the energies of the predicted polymorphs are more impacted by vdW than the B2O3-I one, because of their lower density.
The energy diagram (Fig. 7, left panel) resulting from the vdW inclusion provides several major outcomes. First, all predicted polymorphs energies fall above B2O3-I, while in the PBE picture Ferlat et al. (2012); Claeyssens et al. (2013) many polymorphs are possible candidates for the ground state. Note that the overall correlation between metastability and density is now in line with the one observed for silica polymorphs Coudert (2013). Although the results are presented at 0 K, we checked that the picture obtained at 300 K is unchanged within 1 kcal/mol (by computing finite-temperature contributions to the vibrational free energy within the DFT-D2 scheme; see the SM).A second important point is that, despite being metastable, most of the predicted polymorphs are still within a thermodynamically accessible energy range, estimated at 7 kcal/mol upp . In particular, polymorphs such as T0- and T0- could be amenable to synthesis. Remarkably, the layers that constitute these polymorphs are found experimentally in several chemically complex borates Wang et al. (2007); Liu et al. (2009); Daub and Hillebrecht (2015).
However, assessing the synthesizability of a given polymorph is a very challenging task, since the energy is not the sole ingredient at play. Following an earlier work on silica zeolites which associated experimentally observed structures with good mechanical properties Coudert (2013), we undertook a comprehensive characterization of the mechanical properties of the B2O3 polymorphs, including bulk (), Young (), and shear () moduli as well as linear compressibility and Poisson’s ratio Hay (2016). In all the calculated moduli, and as illustrated in Fig. 7 (right panel) for the bulk modulus, there is a rather clear distinction between B2O3-I ( 60 GPa) and most of the other polymorphs which show much smaller values ( 0-30 GPa). For illustrative purposes, the shaded-blue rectangle in the energy-versus- parameter space delimits the ranges of values in experimentally realized silica polymorphs. Noticeably, this area excludes most of the predicted B2O3 polymorphs, with a few noticeable exceptions including T0- and T0-0.5.
Further, for most polymorphs, and , which are directional quantities, show high anisotropies, implying that there is one direction much weaker than the others with respect to an applied stress. This is in line with the aforementioned soft direction in these structures. In silica zeolites Coudert (2013), proposals of feasibility criteria were derived from the lowest values of these mechanical quantities: strictly transferring these criteria mod to B2O3 leaves only B2O3-I as a realizable structure with, however, a handful of additional candidates (T3, T11, B2O3-I-, T0, and T0-0.5) close to the criteria thresholds. Thus, many of the predicted structures suffer from mechanical stability issues and this may be one of the reasons why they have not been experimentally realized yet.
Our findings have strong implications for understanding the B2O3 behavior at the glass transition. Indeed, the vast majority of the predicted polymorphs are clustered close to the liquid and glass states in the energy-density diagram (dashed-blue line in Fig. 7, left panel). Since the glass structure is generally expected to resemble that of the underlying crystals, Fig. 7 provides a framework to understand the glass state and its apparent anomalous properties, such as its low density, its high fraction of rings, and its strong structural dissimilarity with B2O3-I. In addition, the existence of many competing polymorphs of similar energies, as observed here, has been shown to correlate with the glass forming ability; a situation reported for a large range of systems Perim et al. (2016); Goodman (1975) and models Naumis (2012); Ronceray and Harrowell (2017); Russo et al. (2018).
These aspects, which combine the energetics with the mechanical properties, allow one to propose the following explanation for the B2O3 crystallization anomaly. As the low-density liquid is cooled and gets closer to the region of high polymorphic degeneracy, it hovers over a rugged energy landscape, i.e., dominated by many local minima of similar energies. These minima are associated to low-density and mechanically weak structures prone to collapse into an amorphous framework. Although there exists a ground state (B2O3-I) of higher density, the driving force (energy separation between the metastable states and B2O3-I) is small enough and the energy barriers (associated to the topological reconfiguration required to reach the B2O3-I density) are sufficiently high so that the system is kinetically trapped, in accordance with the very sluggish kinetics found in this system Zanotto and Cassar (2017). Applying pressure to the melt favors higher density and stiffer structures, and thus results in the B2O3-I crystallization.
IV CONCLUSIONS
We have revealed that B2O3 is a system remarkably sensitive to vdW interactions in both energies and structures. The importance of vdW in B2O3 is striking and unexpected, particularly when compared to SiO2, a system commonly considered similar for which, however, only mild effects from vdW have been reported Hay et al. (2015). Thus, the set of polymorphs studied in this work constitutes a valuable test bed to develop new methods for the accurate treatment of electronic correlation. At the same time, the account of vdW allows one to retrieve a polymorphic picture which not only supports the generally admitted experimental knowledge (B2O3-I is the ground-state) but also brings the metastable polymorphs closer to the glass in the energy-density phase-space. From the simultaneous characterization of energies and mechanical properties, we provide here a semi-quantitative map of the likeliness of synthesizability. Not only is it in agreement with the experimental observations - the structural motif from the most robust predicted polymorph (T0-0.5) has been found in synthetized borates - but also it provides a framework to explain the B2O3 intriguing anomalies. The glass has a low density and a high amount of boroxol rings because the supercooled liquid acquires those structural characteristics from the closest crystalline polymorphs. It, however, fails to crystallize in any of these because of both their energies degeneracy (which induces frustration) and their weak mechanical properties.
We thank Harald Hillebrecht for communicating the results of Ref. Daub and Hillebrecht (2015) prior to their publication. This work was performed using HPC resources from GENCI-TGCC/CINES/IDRIS (Grants No. A0030906493, No. A0030801875, No. A0010906493, and No. A0010907625) and from PRACE (project 2016143322). Financial supports from the French National Research Agency program PIPOG ANR-17-CE30-000 and from the program Emergence-Ville de Paris are acknowledged.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ferlat (2015) G. Ferlat, Rings in Network Glasses: The B 2 O 3 Case (Springer, Switzerland, 2015), chap. 14, p. 367.
- 2Wright (2018) A. C. Wright, Phys. Chem. Glasses: Eur. J. Glass Sci. Technol. B 59 , 65 (2018).
- 3Nicholas et al. (2004) J. Nicholas, S. Sinogeikin, J. Kieffer, and J. Bass, Phys. Rev. Lett. 92 , 215701 (2004).
- 4Lee et al. (2005) S. K. Lee, K. Mibe, Y. Fei, G. D. Cody, and B. O. Mysen, Phys. Rev. Lett. 94 , 165507 (2005).
- 5Trachenko et al. (2008) K. Trachenko, V. V. Brazhkin, G. Ferlat, M. T. Dove, and E. Artacho, Phys. Rev. B 78 , 172102 (2008).
- 6Zeidler et al. (2014) A. Zeidler, K. Wezka, D. A. J. Whittaker, S. P. Salmon, A. Baroni, S. Klotz, H. E. Fischer, M. C. Wilding, C. L. Bull, M. G. Tucker, et al., Phys. Rev. B 90 , 024206 (2014).
- 7Lee et al. (2018) S. K. Lee, Y.-H. Kim, P. Chow, Y. Xiao, J. Cheng, and G. Shen, Proc. Natl. Acad. Sci. USA 115 , 5855 (2018).
- 8Brazhkin et al. (2010) V. V. Brazhkin, I. Farnan, K. I. Funakoshi, M. Kanzaki, Y. Katayama, A. G. Lyapin, and H. Saitoh, Phys. Rev. Lett. 105 , 115701 (2010).