Finding reaction pathways with optimal atomic index mappings
Deb Sankar De, Marco Krummenacher, Bastian Schaefer, Stefan, Goedecker

TL;DR
This paper introduces a novel method using a permutation-invariant penalty function and Minima Hopping to efficiently find reaction pathways and intermediate states in complex molecular reactions, reducing computational effort.
Contribution
It proposes a new approach combining a permutation-invariant bias and global optimization to identify reaction pathways with fewer intermediate states.
Findings
Successfully applied to LJ38 benchmark system, identifying relevant intermediate states.
Reduced the number of states needed to find the lowest energy pathway compared to previous methods.
Applied to C60 and C20H20, revealing valuable synthesis pathways.
Abstract
Finding complex reaction and transformation pathways, involving many intermediate states, is in general not possible on the DFT level with existing simulation methods due to the very large number of required energy and force evaluations. This is due to a large extent to the fact that for complex reactions, it is not possible to determine which atom in the educt is mapped onto which atom in the product. Trying out all possible atomic index mappings is not feasible because of the factorial increase in the number of possible mappings. By using a penalty function that is invariant under index permutations, we can bias the potential energy surface in such a way that it obtains the characteristics of a structure seeker whose global minimum is the product. By performing a Minima Hopping based global optimization on this biased potential energy surface we can rapidly find intermediate states…
Click any figure to enlarge with its caption.
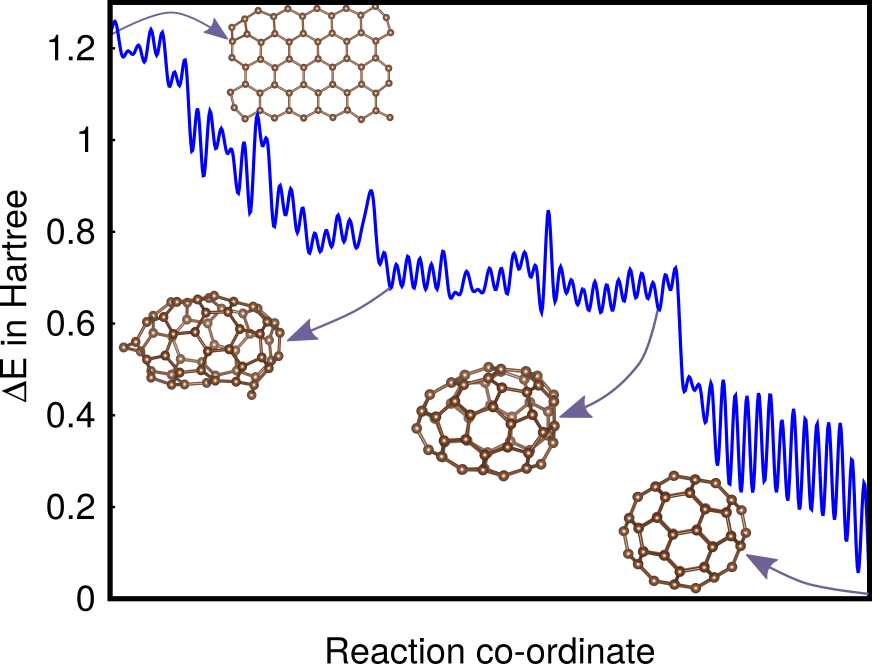 Figure 1
Figure 1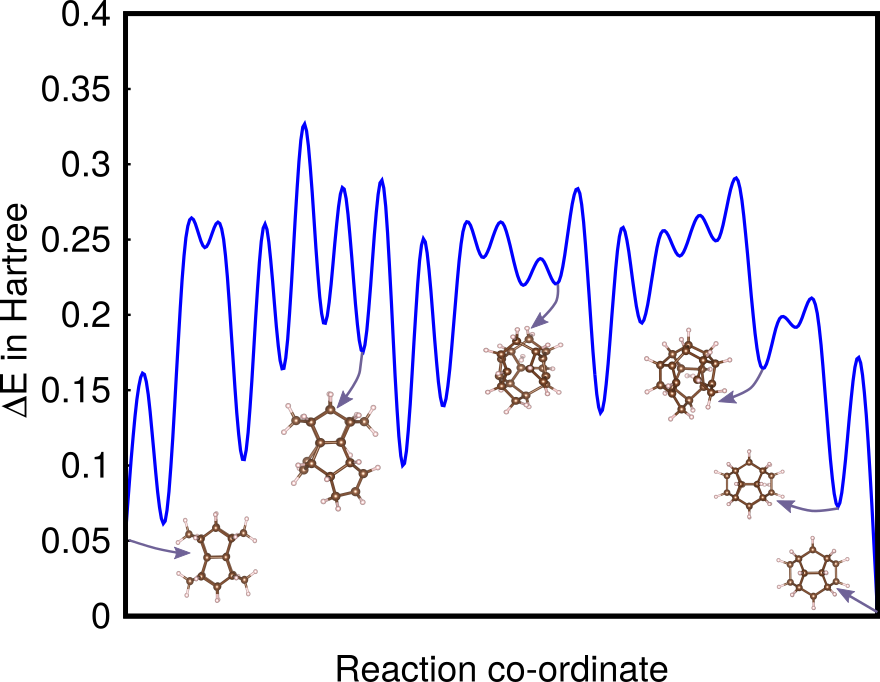 Figure 2
Figure 2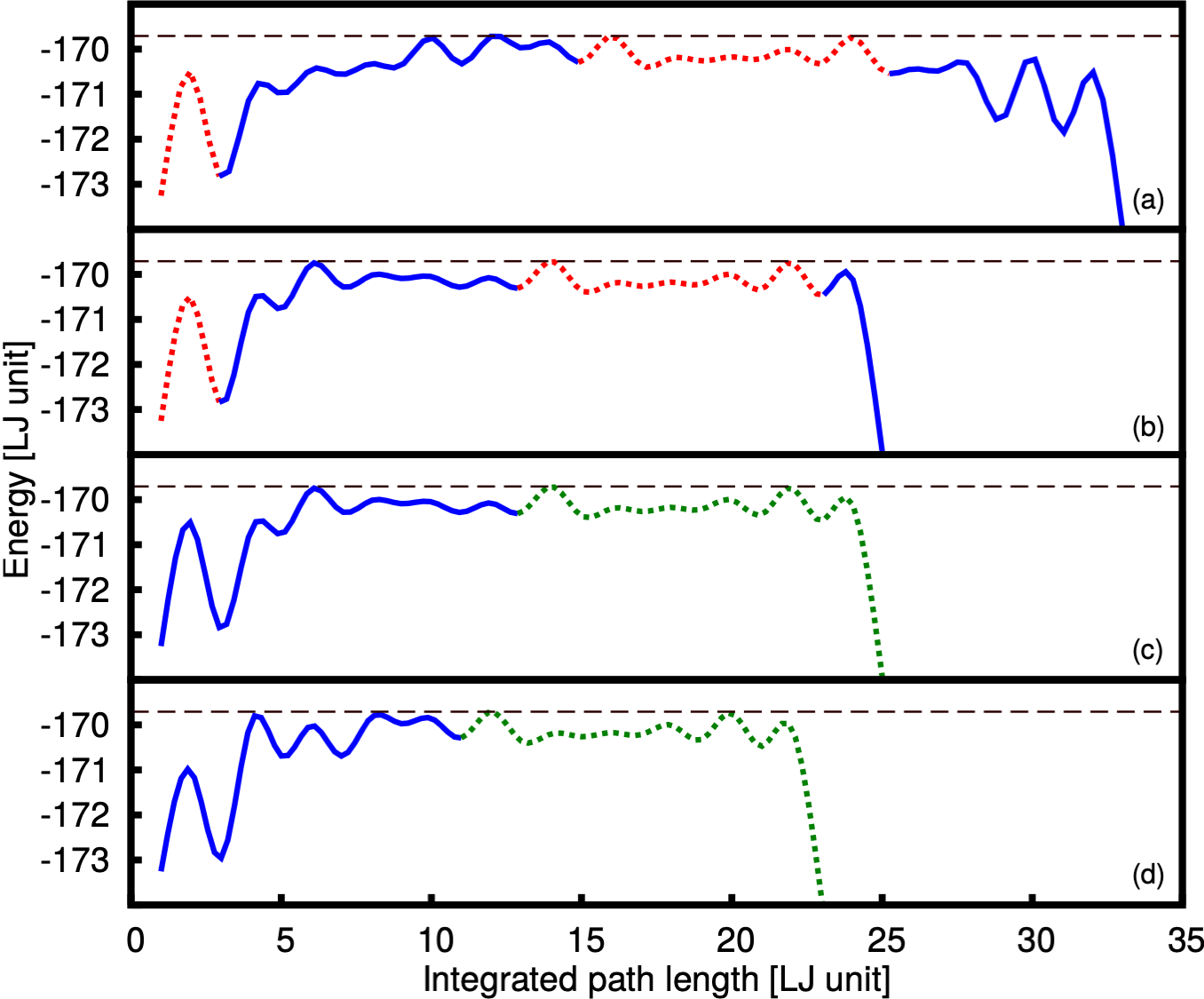 Figure 3
Figure 3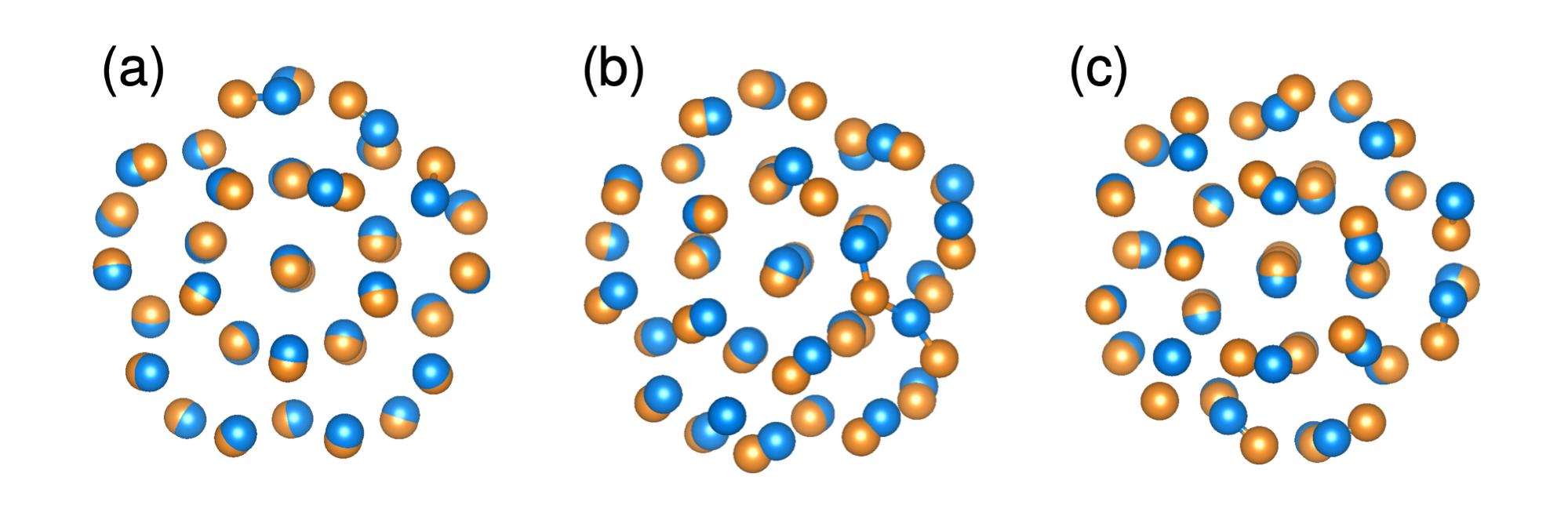 Figure 4
Figure 4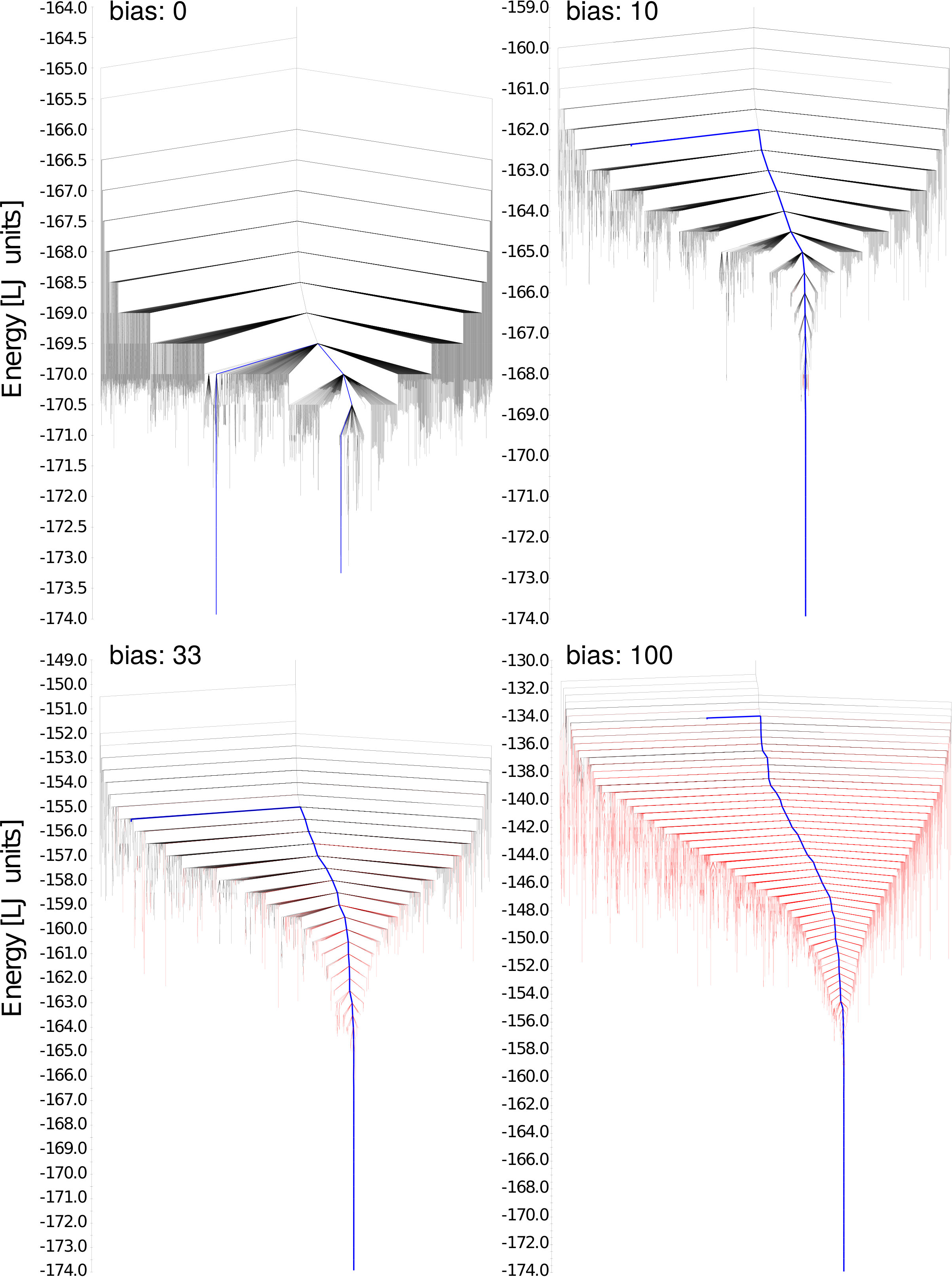 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Finding reaction pathways with optimal atomic index mappings
Deb Sankar De
Department of Physics, Universität Basel, Klingelbergstr. 82, 4056 Basel, Switzerland
Marco Krummenacher
Department of Physics, Universität Basel, Klingelbergstr. 82, 4056 Basel, Switzerland
Bastian Schaefer
Department of Physics, Universität Basel, Klingelbergstr. 82, 4056 Basel, Switzerland
Stefan Goedecker
Department of Physics, Universität Basel, Klingelbergstr. 82, 4056 Basel, Switzerland
Abstract
Finding complex reaction and transformation pathways, involving many intermediate states, is in general not possible on the DFT level with existing simulation methods due to the very large number of required energy and force evaluations. This is due to a large extent to the fact that for complex reactions, it is not possible to determine which atom in the educt is mapped onto which atom in the product. Trying out all possible atomic index mappings is not feasible because of the factorial increase in the number of possible mappings. By using a penalty function that is invariant under index permutations, we can bias the potential energy surface in such a way that it obtains the characteristics of a structure seeker whose global minimum is the product. By performing a Minima Hopping based global optimization on this biased potential energy surface we can rapidly find intermediate states that lead into the global minimum. Based on this information we can then extract the full reaction pathway. We first demonstrate for a benchmark system, namely that our method allows to preferentially find intermediate states that are relevant for the lowest energy reaction pathway and that we therefore need a much smaller number of intermediate states than previous methods to find the lowest energy reaction pathway. Finally we apply the method to two real systems, \ceC60 and \ceC20H20 and show that the found reaction pathway contains valuable information on how the system can be synthesized.
pacs:
61.90.+d, 31.15.A
The indistinguishability of atoms manifests itself in various ways. In a chemical reaction the indistinguishability means that in principle any atom of a certain type in the educt can be mapped onto any atom of the same type in the product. If the system undergoing the chemical reaction consist of atoms of type 1, atoms of type 2, etc, then there are consequently different possible mappings. For simple reactions it is often obvious which atom in the educt is mapped onto which atom of the product. In more complicated chemical reactions or similar processes such as phase or shape transformations in nano-particles the best mapping can however not be foreseen. Actually in such a context many atomic index mappings may exist which lead to different low barrier reaction pathways.
Determining the transition states of a reaction/transformation is of great importance in chemistry, physics and materials sciences. A large number of methods have therefore been proposed to solve this problem as reviewed for instance by Reiher Simm et al. (2018). In the so-called two-sided methods Henkelman et al. (2000); Granot and Baer (2008); Ghasemi and Goedecker (2011) one has not only to provide the educt and the product but also the correct atomic index mapping, i.e. it has to be known beforehand which atom in the educt is mapped on which atom in the product. In principle one could of course perform such a transition state search for all possible atomic index mappings. In practice this is of course prohibitive for large systems because the factorial increase in the number of possibilities.
In the so-called one sided methods Henkelman and Jónsson (1999); Abashkin and Russo (1994) one finds a transition state that is in a certain sense closest to the initial guess. Once arrived at the transition state one can find the adjacent minima by performing two simple local geometry optimizations starting from a point to the left and a point to the right of the saddle point along the line of negative curvature one can thus find the two minima that are connected by this saddle point. In this way one obtains automatically the correct index mapping, but it is not guaranteed that the two local minima found in this way are the desired educt and product.
There is a method that would allow in principle to study any chemical reaction or transformation, namely Molecular Dynamics (MD). In this approach there is no index mapping problem since any atom of the educt is mapped onto its corresponding atom in the product via its trajectory. However, since most chemical reactions are rare events, the number of MD time steps is prohibitive. Meta dynamics Laio and Parrinello (2002) methods speed up processes that are slow on the time scale of a typical MD time step. They are driven by collective variables that “push” the system in a desired direction. The standard collective variables can target certain phases or general structural motifs, but they can not target a single configuration. A typical collective variable that is used to drive chemical reactions in a desired direction is for instance a certain bond length, whose value in the product is known and different from the value in the educt. But it is very likely that there are other stable configurations that have the same bond length and so the collective variable can drive the system into a configuration that is different from the product. In the field of protein folding, certain averages of the dihedral angles that give rise to Ramachandran plots are frequently used as collective variable. Again, it is obvious that many configurations of the protein may give rise to the same average angles. Also very recently proposed collective variables, such as approximate entropies Piaggi and Parrinello (2018) do not allow to target a single configuration.
Temperature accelerated dynamics Sørensen and Voter (2000) is another way to speed up rare events that would be impossible to observe with standard MD. This class of methods has the advantage that it does not need collective variables, but on the other hand it is again not possible to steer the reaction pathways in a certain desired direction. High temperature MD trajectories are used in a similar spirit in the TSSCDS method Vázquez et al. (2018) to detect saddle points.
Minima hopping guided path search Schaefer et al. (2014) (MHGPS) is another unbiased method to explore the entire Potential Energy Surface (PES) and to find a large number of local minima and transition states. Essentially the same information can be obtained by combining basin hopping Wales and Doye (1997) with an eigenvector following approach Doye and Wales (1997) to find saddle points. So all these methods provide in principle the information required to construct any reaction pathway with a correct index mapping, but do not allow for steering. For small systems exploring the entire PES is feasible and of interest to find a more or less complete set of minima together with the saddle points connecting them. For large systems such an approach is however numerically too expensive and it is advantageous to concentrate on a subset of minima and saddle points that are of interest in a certain context. Transition path sampling Bolhuis et al. (2002) is yet another method that allows for the calculation of multiple reaction pathways between given initial and final configurations. However again the initial atomic index matching has to be known beforehand.
In this paper we will use configurational distances derived from fingerprints that are invariant under atomic index permutations Sadeghi et al. (2013) as a driving force towards the desired final configuration. In contrast to the standard collective variables, this fingerprint can uniquely identify a single configuration. The fingerprint distance is zero if and only if the configuration is exactly identical to the desired final configuration up to translations, rotations and index permutations. Along the reaction pathway the distance varies continuously until it vanishes at the final configuration. Adding the distance as a penalty to the true PES gives a ‘biased’ PES + w . The parameter is in this context a suitable chosen weight and are the cartesian coordinates of the atoms in the system. The penalty is based on a fingerprint obtained from the eigenvalues of an overlap matrix describing the configuration Sadeghi et al. (2013). Each pair of atoms (i , j) in the configuration gives rise to a block of matrix elements given by . The indices and specify the type of the orbitals. We used Gaussian type orbital of , , and character. Hence is a 4 by 4 matrix block. Denoting by the vector containing the sorted eigenvalues of the overlap matrix characterizing configuration p and by the analogous vector of configuration q, the distance between the two configurations was defined as
[TABLE]
in the original publication Sadeghi et al. (2013). This definition of the distance can lead to discontinuties in the first derivative with respect to the atomic positions when eigenvalues cross. Such discontinuities make a numerical optimization of the penalty term in the biased potential energy more difficult. We therefore removed these discontinuities by introducing smeared out eigenvalues given by
[TABLE]
The smeared eigenvalues do also cross, but at the crossing they have identical derivatives. Our regularized distance serves then as the penalty function:
[TABLE]
All derivatives of this function are continuous. Adding the gradient with respect to the atomic positions of this penalty term to the physical forces, gives biased forces that drive the system from the present configuration towards the desired final configuration . These forces are invariant with respect to index permutations.
In this work we concentrate on complex reaction pathways, where the system has to cross a substantial number of saddle points. By adding the penalty we transform the original PES into a biased one, denoted by . With the right choice of the parameter , this biased PES has the appearance of a PES of a structure seeker whose global minimum is the desired final state as can be seen from the disconnectivy graphs in Fig. 2) Becker and Karplus (1997); Miller et al. . This means that the downhill barriers (the barriers that one has to overcome when one hops from one minimum into another one with lower energy) are reduced but have not disappeared. Therefore a local optimization of the biased PES is not sufficient to find the final configuration. A global optimization is required. However, we exploit the fact that for a structure seeker it is easy to find the global minimum with the minima hopping method. We start from an initial configuration (educt) and then visit with the minima hopping methods consecutive intermediate states until we find the ground state. Because of the stochastic nature of minima hopping different intermediate states can be visited in different runs. With a reasonable choice for the weight of the penalty all these intermediate configurations will have low energies and are therefore physically possible intermediate states that are accepted during a Minima Hopping run. This means that one can find with this approach not only one reaction pathway but all physically relevant low energy reaction pathways.
Since the hops from one local minimum to the next in Minima Hopping are based on MD Roy et al. (2008); Sicher et al. (2011) followed by local geometry optimizations Schaefer et al. (2015), the identity of individual atoms of the same type can be traced back for any hop and one obtains in this way the correct mapping of the atomic indices for the entire complex reaction pathway. The MD trajectories cross barriers and so one could take some configurations along the MD trajectory as a starting point for a one sided saddle point search. The MD trajectory can however cross over several barriers within one hop of a MH run and in such a case it is not clear which point along the MD trajectory should be chosen as the starting point for the saddle point search. We therefore used a recursive variant of the freezing string method Behn et al. (2011) as described in reference Schaefer et al. (2014) to connect the sequence of accepted local minima by saddle points. The method described so far will be referred to as Biased Minima Hopping Guided Pathway Search (BMHGPS) in the following.
We will first apply our new method to a benchmark system, the Lennard Jones cluster with 38 atoms (), for which the lowest energy reaction pathway from the lowest energy icosahedral structure to the fcc ground state is most likely known, since it was studied previously Doye et al. (1999a); Neirotti et al. (2000); Mandelshtam and Frantsuzov (2006); Sehgal et al. (2014). Finding this transformation pathway is quite difficult since it requires a complete overall rearrangement of all the atoms in the system Doye et al. (1999a).
To find the biased PES that has the strongest structure seeker character, we used four different weights of the penalty function (). Choosing gives back the unbiased MHGPS method, which was included for comparison. For each weight 100 different MH runs were performed. As reference structure the fcc global minimum was taken in order to bias the minimization towards it.
As can be observed in the disconnectivity graphs in Fig. 2 the typical double funnel structure of the PES disappears by adding the penalty function to the energy. For all non-zero weights, the disconnectivity graph has the shape of a structure seeker for which it is quite easy to find the global minimum. The more the PES is biased the more fcc like minima can be observed. On the non-biased PES, no fcc structure except the global minimum can be seen (shown in this case in blue and not red since it represents the transformation pathway). For large weights fcc like structures start to dominate. Choosing even larger weights would result in a glassy landscape of fcc like structures where it would again be more difficult to find a pathway into the global minimum. In addition too large weights will destroy a faithful mapping between the local minima on the biased PES and the true physical PES. Fig. 1 illustrates this mapping for the icosahedron starting structure. It can be seen that the displacement of the surface atoms due to the bias is larger than for the core atoms. Following the entire transformation obtained by our method indeed shows that it starts by some kind of surface premelting which then propagates towards the centre.
All the MH runs were stopped once the global minimum was found. The number of distinct minima that were visited before the global minimum was found was greatly reduced by the bias. Averaging over 100 runs we visited on average 363 minima without any bias, 19 for , 10 for and 16 for . In order to obtain reaction pathways the accepted local minima from the MH run on the biased PES are transformed back to the physical PES by a local geometry optimization. Reaction pathways were then generated as described in reference Schaefer et al. (2014). All our pathways shown in Fig. 3 have a highest energy barrier of -169.709 in LJ energy units which is identical to the lowest highest barrier height found in previous studies Doye et al. (1999a, b). The reaction pathway found in these two studies was however based on a data set of 140 000 saddle points whereas we could obtain this result thanks to the bias with a much smaller data set. We used all the saddle points obtained in the 100 runs and collect in this way 3238 saddle points for , 2068 for and 4183 for . Without a bias we used a data set of similar size, as the previous studies, namely 83 000 saddle points. This reduction in the number of required saddle points is due to the fact that our biasing allows us to preferentially find the intermediate states and saddle points in the region of configurational space that is in between the educt and the product. Those states are the relevant ones for our wanted reaction pathway from the educt to the product.
Because of the significant efficiency gains obtained by BMHGPS, it can also be applied on the Density Functions Theory (DFT) level and we will use it to find highly complex reaction pathways for \ceC60 and \ceC20H20. The DFT calculations were performed with the BigDFT code Genovese et al. (2008) using dual space Gaussian pseudopotentials Willand et al. (2013). An initial pathway was obtained with the self-consistent charge density functional tight-binding method (SCC-DFTB) as implemented in DFTB+ Aradi et al. (2007) with s and p atomic orbitals for carbon. These pathways were then further refined with BigDFT calculations.
Several MD studies of \ceC60 formation failed to find a route from high-energy structures to the Buckyball ground state, presumably because the necessary time scales are not accessible to MD simulations Ballone and Milani (1990); Chelikowsky (1991, 1992); Jing and Chelikowsky (1992); Yi and Bernholc (1993); Xu and Scuseria (1994); Marcos et al. (1997). Only a semiclosed \ceC60 pseudocage was found after a simulation time of 250 ns using a classical Brenner potential and a adpative temperature MD method Maruyama and Yamaguchi (1998).
Using our new method, we were able to find a reaction pathway from an intial structure that was a planar graphene flake to the Buckyball ground state. Good weights are in the range of 0.2 to 0.3 which means that the energy difference between the ground state and the first Stone-Wales defect is increased 0.06 Ha to 0.08 Ha and the energy difference between the initial flake and the ground state is increased from 1.21 Ha to 1.97 Ha. The low energy reaction pathway shown in Fig. 5 is based on a data set of 9000 saddle points that were obtained from 4 MH runs that were stopped once the global minimum was found. Even though the barriers along the pathway are quite high, there is a clear tendency to lower the energy as one approaches the ground state. This is also true for other representative pathways that we have found. This explains why \ceC60 can be synthesized in a one-step procedure by graphite vaporization at high temperatures Krätschmer et al. (1990).
Using the same approach as for \ceC60 we also obtained the reaction pathway of \ceC20H20 from its pagodane configuration to its dodecahedron ground state. The reaction pathway, obtained from a data base of 900 saddle points, is shown in Fig 4. In contrast to \ceC60 there is no systematic lowering of the energy of the intermediate structures as one approaches the ground state. This reflects the fact that there is indeed no one step synthesis recipe to obtain the dodecahedron from the pagodane structure Prinzbach et al. (2006). The experimental synthesis procedure consist of 11 single steps, each of which involves other ingredients that modify the PES and are therefore expected to give the necessary driving force towards the ground state Fessner et al. (1987a); Prakash et al. (1988); Fessner et al. (1987b).
In summary, we have introduced a bias that is invariant under atomic index permutations and that can target a single well defined configuration as the final configuration of a chemical reaction or physical transformation. In this way we can overcome the index mapping problem that would require an exponentially large number of applications of standard two sided saddle point search methods, since one has to try out in principle all possible index permutations. The forces arising from the bias, by construction, do not depend on the indexing of the atoms. We have thus reduced the combinatorial atomic indexing problem, that has an exponential scaling, to a global minimization problem on a biased PES involving an indexing invariant penalty function. For suitably chosen weights the biased PES has the form of a structure seeker and the optimization problem can be solved quite rapidly in practice by global optimization methods such as Minima Hopping. In contrast to standard order parameters that can drive the system only into certain regions of a low dimensional order parameter space, this method can pull the system towards a single configuration in the full dimensional configuration space. Collective variables are in general system specific and finding them can be non-trivial. The penalty function we proposed is universal and can be applied to any reaction or transformation in molecules or clusters. We expect that this method will give atomistic insight into complex reaction pathways found for instance in catalysis as well as complex phase and shape transformations in nano-particles. Such processes play for instance an important role in non-classical nucleation Lee et al. (2016) where the particles undergo transformations similar to the ones studied here for the Lennard Jones cluster. Since the cost of our method is comparable to the cost of a Minima Hopping structure prediction run, the method can be applied to systems of the same size. This means that on the density functional level systems with up to about 100 atoms can be studied. With cheaper methods such as machine learning based force fields much larger systems will become accessible. Permutationally invariant distances could also be useful in other reaction path search methods where the atomic index mapping is a limiting factor.
Acknowledgements.
This work was done within the NCCR MARVEL. Computer resources were provided at CSCS under project s707 and at the Scicore computing center of the university of Basel. We also thank Dr. Luigi Genovese for support in adapting the BigDFT code.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Simm et al. (2018) G. N. Simm, A. C. Vaucher, and M. Reiher, J. Phys. Chem. A 123 , 385 (2018).
- 2Henkelman et al. (2000) G. Henkelman, B. P. Uberuaga, and H. Jónsson, J. Chem. Phys. 113 , 9901 (2000) . · doi ↗
- 3Granot and Baer (2008) R. Granot and R. Baer, J. Chem. Phys. 128 , 184111 (2008) . · doi ↗
- 4Ghasemi and Goedecker (2011) S. A. Ghasemi and S. Goedecker, J. Chem. Phys. 135 , 014108 (2011) . · doi ↗
- 5Henkelman and Jónsson (1999) G. Henkelman and H. Jónsson, J. Chem. Phys. 111 , 7010 (1999) . · doi ↗
- 6Abashkin and Russo (1994) Y. Abashkin and N. Russo, J. Chem. Phys. 100 , 4477 (1994).
- 7Laio and Parrinello (2002) A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. 99 , 12562 (2002) . · doi ↗
- 8Piaggi and Parrinello (2018) P. M. Piaggi and M. Parrinello, Proc. Natl. Acad. Sci. 115 , 10251 (2018) . · doi ↗