Simulated Epidemics in 3D Protein Structures to Detect Functional Properties
Mattia Miotto, Lorenzo Di Rienzo, Pietro Corsi, Giancarlo Ruocco,, Domenico Raimondo, Edoardo Milanetti

TL;DR
This paper introduces a novel method using simulated epidemics on 3D protein structures' interaction networks to analyze protein stability and identify functional sites, bridging epidemiological models and structural biology.
Contribution
It applies epidemic spreading models to protein interaction networks, providing a new computational approach for assessing stability and detecting allosteric sites.
Findings
Effective in assessing thermal stability
Capable of identifying allosteric sites
Provides insights into protein functional properties
Abstract
The outcome of an epidemic is closely related to the network of interactions between the individuals. Likewise, protein functions depend on the 3D arrangement of their residues and on the underlying energetic interaction network. Borrowing ideas from the theoretical framework that has been developed to address the spreading of real diseases, we study the diffusion of a fictitious epidemic inside the protein non-bonded interaction network. Our approach allowed to probe the overall stability and the capability to propagate information in the complex 3D-structures and proved to be very efficient in addressing different problems, from the assessment of thermal stability to the identification of allosteric sites.
Click any figure to enlarge with its caption.
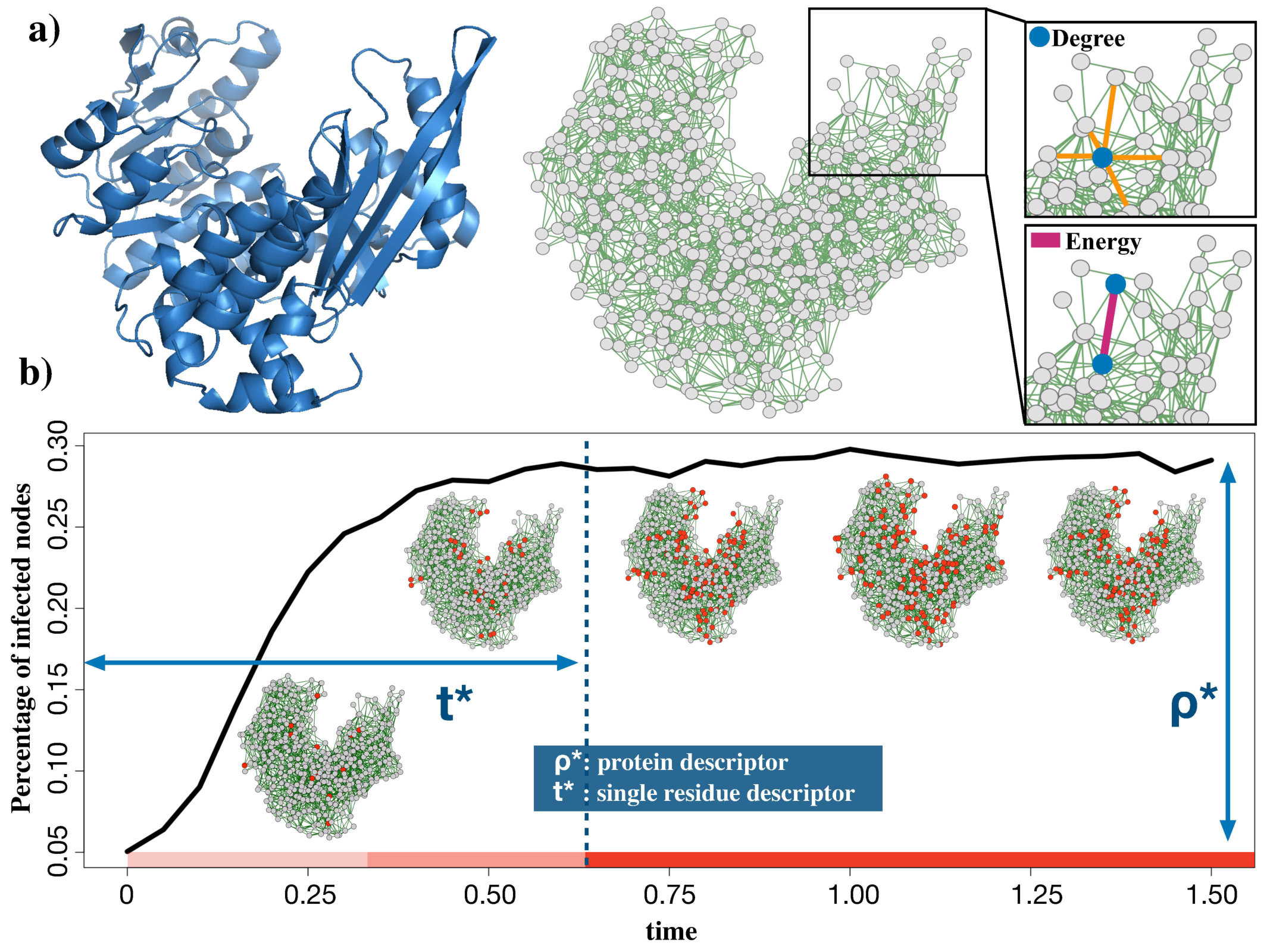 Figure 1
Figure 1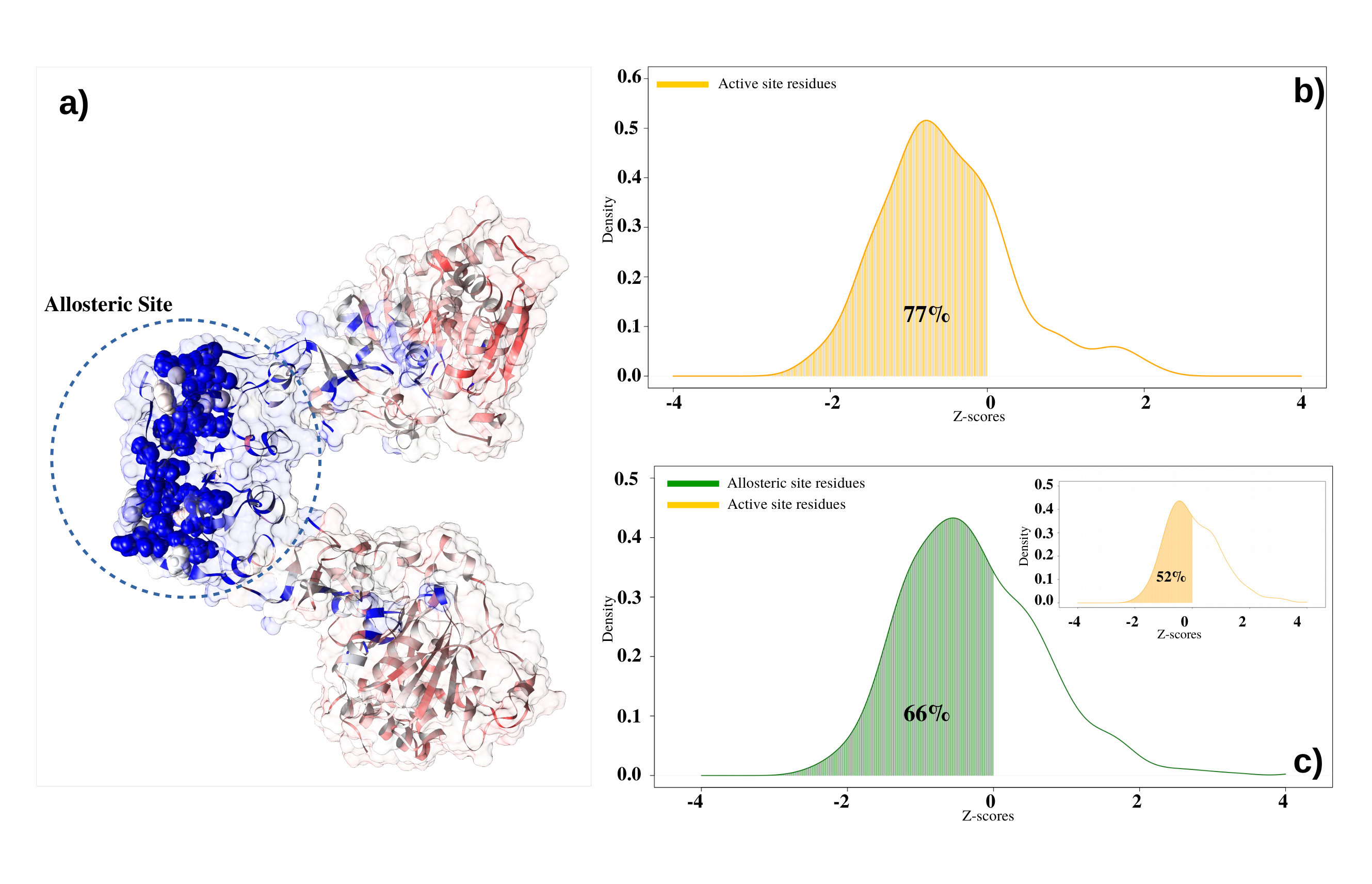 Figure 2
Figure 2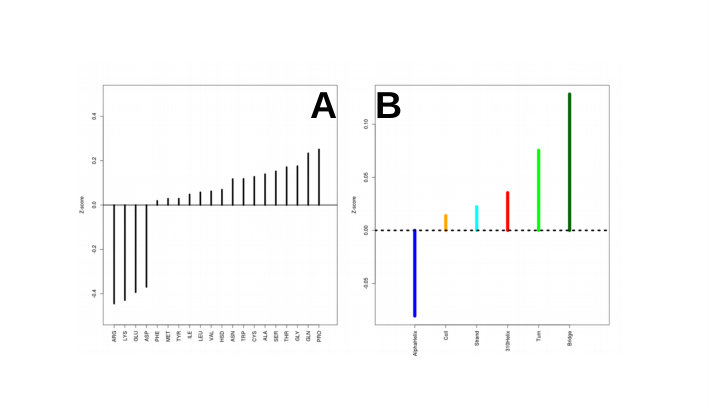 Figure 3
Figure 3 Figure 4
Figure 4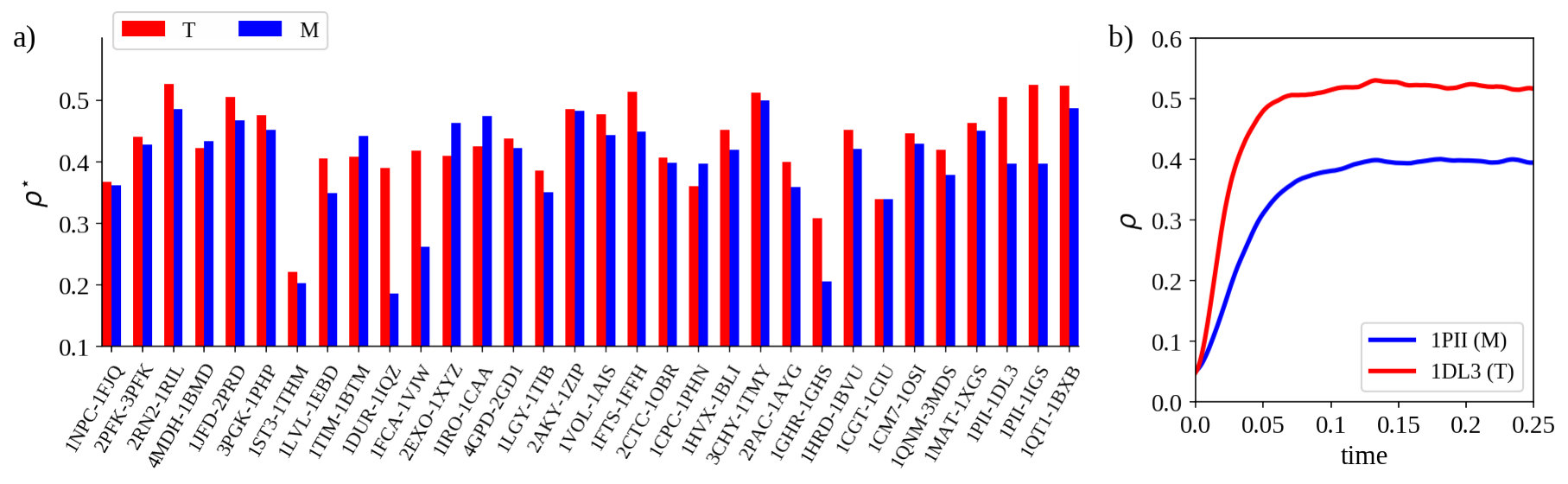 Figure 2
Figure 2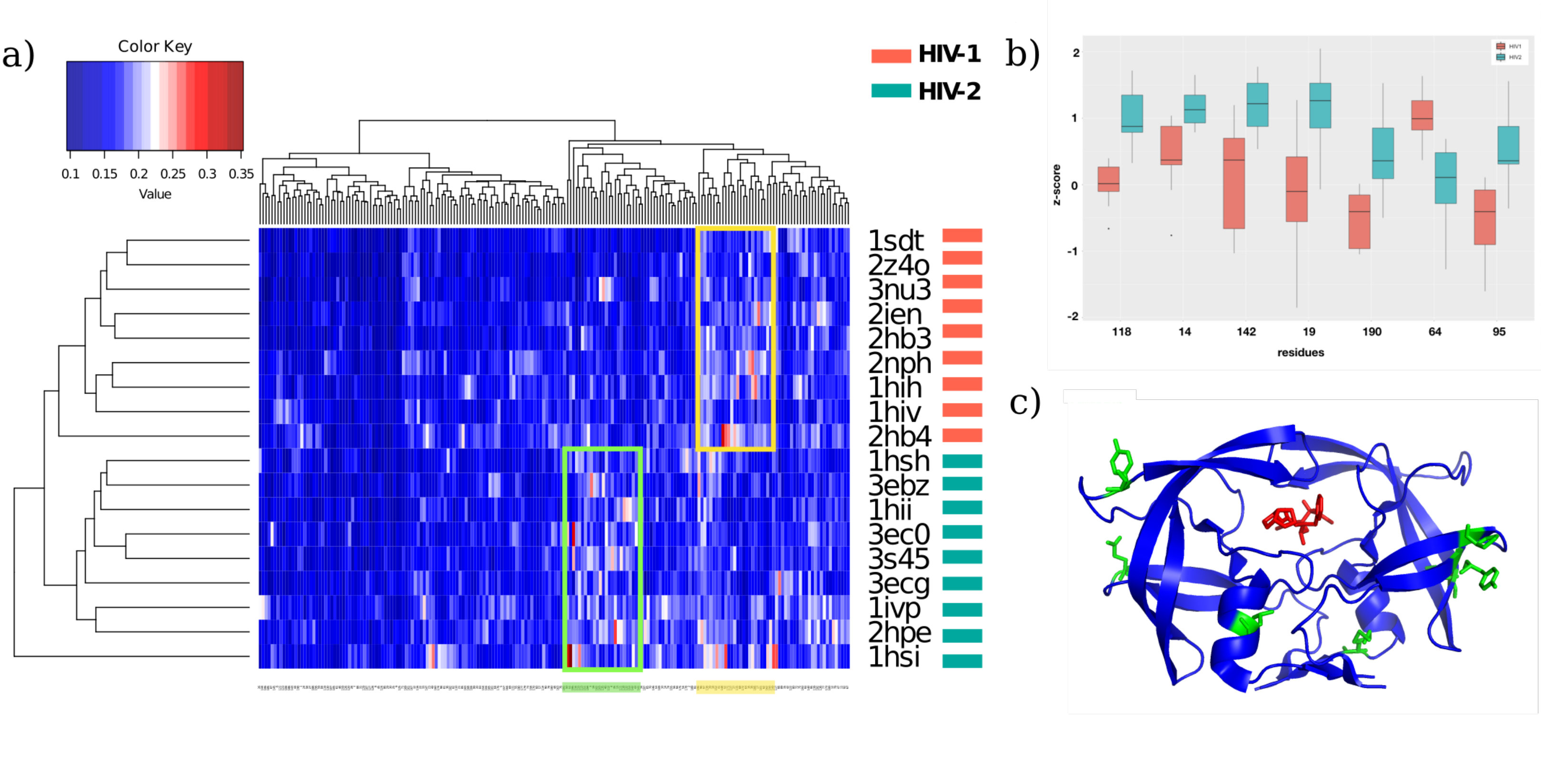 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Simulated Epidemics in 3D Protein Structures to Detect Functional Properties
Mattia Miotto111The authors contributed equally to the present work.
Department of Physics, Sapienza University of Rome, Rome, Italy
Center for Life Nanoscience, Istituto Italiano di Tecnologia, Rome, Italy
Lorenzo Di Rienzo11footnotemark: 1
Department of Physics, Sapienza University of Rome, Rome, Italy
Pietro Corsi
University “Roma Tre”, Department of Science, Rome, Italy
Giancarlo Ruocco
Department of Physics, Sapienza University of Rome, Rome, Italy
Center for Life Nanoscience, Istituto Italiano di Tecnologia, Rome, Italy
Domenico Raimondo
Department of Molecular Medicine, Sapienza University of Rome, Rome, Italy
Edoardo Milanetti
Department of Physics, Sapienza University of Rome, Rome, Italy
Center for Life Nanoscience, Istituto Italiano di Tecnologia, Rome, Italy
Abstract
The outcome of an epidemic is closely related to the network of interactions between individuals. Likewise, protein functions depend on the 3D arrangement of their residues and the underlying energetic interaction network. Borrowing ideas from the theoretical framework that has been developed to address the spreading of real diseases, we study the diffusion of a fictitious epidemic inside the protein non-bonded interaction network, aiming to study the features of the network connectivity and properties. Our approach allowed us to probe the overall stability and the capability to propagate information in the complex 3D-structures and proved to be very efficient in addressing different problems, from the assessment of thermal stability to the identification of functional sites.
diffusion, information, transmission, thermal stability, proteins, melting temperature, energy, structure
Introduction
Proteins are large bio-molecules responsible for the majority of live-sustaining tasks in cells Mannige (2014); Chothia et al. (1997). Their great versatility is due to the complex three-dimensional structure they can acquire, which arises as a result of physical and chemical interactions among all its constituent amino acids. In particular, the global structure is uniquely defined once the sequence of amino acids composing the molecule is specified Dill et al. (2008), with different sequences that can give, up to local rearrangements, the same overall 3D architecture Lesk and Chothia (1980); Ofran and Margalit (2006).
The peculiar structural conformation each protein assumes is the result of a long evolutionary optimization Debès et al. (2013). Proteins are adapted to carry on specific tasks, usually binding to other molecules while being embedded in a complex dynamical environment in the presence of both thermal and molecular noises. In this scenario what evolution does is to select sequences that allow proteins to exert their task more efficiently in the environment they live in while maintaining the same overall 3D architecture Domingues et al. (2000); Karshikoff et al. (2015).
Understanding which changes in the amino acid sequence can improve protein efficiency while preserving the biological function has both theoretical and practical implications. Many works investigated the role of different amino acids in the protein structure, folding, stability and dynamics Chakrabarty and Parekh (2016). In this respect, methods based on graph theory approaches have contributed considerably to the understanding of protein structural flexibility, their hierarchy of structures and in the identification of key residues Dokholyan et al. (2002); del Sol et al. (2006); Amitai et al. (2004); Vendruscolo et al. (2002); Aftabuddin and Kundu (2007). All those findings demonstrated that a network-based analysis can be pivotal to shed light on the complex aspects relative to the organization of protein structures Miotto et al. (2018). However, network approaches have often focused on a static description of the system while interesting properties, especially at the level of the single residue, are related to the dynamical behavior of the network Yang et al. (2013).
Theoretical epidemic modeling indeed is a typical approach to study the dynamical behavior of an interaction network, describing the evolution of a contagion process across a population Kermack and McKendrick (1927, 1932).
In the last decades, epidemic models have seen applications in several fields Vespignani (2011); Yang and Wang (2016) thanks to the growth of network sciences. From the spread of real diseases to the diffusion of news in social networks, epidemic models give a measure about the diffusion of information within either the whole network or from a particular node to any other.
Here, we combine a graph-based schematization of proteins with an epidemic diffusion approach to study the overall stability and the capability to propagate perturbations (or information) in their complex 3D-structures Castellano et al. (2009); Albert and Barabási (2002).
In particular, our novel approach proved to be very efficient in characterizing protein thermal stability and in identifying functional sites of proteins, where trivial static network descriptors exhibit a lower efficiency.
Methods
.1 Datasets
To investigate the capability of a diffusion protocol to grasp the essential feature of the protein structure and function, we defined four different datasets: ‘Thermal dataset’, ‘Enzyme dataset’, ‘Allosteric dataset’ and ‘HIV dataset’. Details regarding their collection are provided below.
- •
Thermal dataset. A set of 32 pairs of homologous proteins with different thermal properties was manually collected from literature Brinda and Vishveshwara (2005); Kannan and Vishveshwara (2000); Mozo-Villarias et al. (2003); hard Sterner and Liebl (2001). Experimentally determined structures were collected from the PDB Touw et al. (2015) and filtered according to method (x-ray diffraction), resolution (below 3 ), and percentage of missing residues (covering more than 95% of to the Uniprot Pundir et al. (2017) sequence). Proteins for which experimentally determined structures were only available in a bound state, i.e. in complex with either a ligand or an ion, were excluded. Further information is available in Table 1 of the Supplementary Material).
- •
Enzyme dataset. It was composed grouping all the enzymes present among the proteins of the Thermal dataset. For each enzyme, we retrieved information about the residues forming the active site (see Table 1 in the Supplementary Material), from the Enzyme Portal of EBI Alcántara et al. (2012).
- •
Allosteric dataset. We collected from Amor et al. (2016) proteins whose active and allosteric sites are both known.
- •
HIV dataset. It is composed of 2 apo (free) structures of the HIV-1 e HIV-2 proteases together with 16 holo (bound) PDB structures (8 of HIV-1 and 8 of HIV-2) being in complex with different ligands is taken from Triki et al. (2018).
All protein structures were minimized using the standard NAMD Phillips et al. (2005) algorithm and the CHARMM force field Vanommeslaeghe and MacKerell (2012) in vacuum. A 1 fs time step was used and structures were allowed to thermalize for 10000 time steps. This procedure aims at removing energetic clashes that may be present due to the crystallization procedure.
.2 Network representation
Protein structures are represented as Residue Interaction Networks Grewal and Roy (2015) (RINs in short), where each node represents a single residue . The nearest atomic distance between a given pair of residues and is defined as . Two RIN nodes are linked together if Phillips et al. (2005); Vanommeslaeghe and MacKerell (2012). Furthermore links are weighted by the sum of two energetic terms: Coulomb (C) and Lennard-Jones (LJ) potentials. The C contribution between two atoms, and , is calculated as:
[TABLE]
where and are the partial charges for atoms and , as obtained from the CHARMM force-field: is the distance between the two atoms, and is the vacuum permittivity. The Lennard-Jones potential is instead given by:
[TABLE]
where and are the depths of the potential wells of atom l and m respectively, and are the distances at which the potentials reach their minima. Therefore, the weight of the link connecting residues and is calculated by summing the contribution of the single atom pairs as:
[TABLE]
where and are the numbers of atoms of the i-th and j-th residue respectively.
.3 Diffusion model on the protein network
Epidemic modeling describes the dynamical evolution of the contagion process within a population. An individual (or node) is said susceptible (S) when it is healthy but could contract the disease, infected (I) when the contagion is transmitted by an adjacent node and recovered (R) when it manages to recover from the disease. In principle, recovered individuals are immunized and hence they are safe from other infections for a certain time. To study the evolution of the density of infected individuals we have to define the basic processes that rule the transition of individuals states, e.g.
[TABLE]
More in details, we must specify (i) the topology of the interaction network, i.e. which nodes directly interact with each other; (ii) the strength of the interaction which is linked to the transmission rate of the infection and (iii) the recovering rate (i.e. the probability, if present, of return healthy after having contracted the infection). Depending on the choices one makes for the set of transitions in Eq. 4, different models and processes can be simulated. A detailed description of the most studied models in classical epidemiology is given in [Pastor-Satorras et al. (2015).
In the present work, we simulated an epidemic diffusion over the protein RINs (see Figure 1). While we preserved the full topological information, we restrained to an SIS (susceptible-infected-susceptible) epidemic model, where each node (i.e. residue) once infected, can transmit the infection to near neighbor nodes in the network. A residue can recover from the infection, returning to the susceptible state (meaning that the transition is instantaneous). In this scenario, the probability of finding a node in infected state is given by:
[TABLE]
where is the rate with which node recovers from infection, while represents the infection rate of node given that node is infected at time Allen (1994).
Eq. 5 can not be solved analytically for complex topologies like the RIN ones but a numerical treatment is required. In the Supplementary Material, we provide a short treatment of the mean-field approximation, where instead it is possible to analytically solve Eq. 5 for different choices of the transitions in Eq. 4. In our case, for each RIN node we identify the recovering rate, , with the node degree. While the infection rate between node and , is given by the weight of the link ( as given by Eq. 3). Once defined the infection and recovering rates, we simulated the diffusion process into the 3D structure, starting from a specific set of residues or by picking an initially random set, and looking at the mean density of infected residues over time:
[TABLE]
where is the number of infected residue at time t, is the total number of protein residues and we indicated with the mean over the realizations of the diffusion process ( all results presented here, are obtained setting M=1000 in order to avoid large fluctuations that are unavoidable in a single realization). It has been found that, depending on the connectivity matrix architecture and the sets of and parameters, the system can exhibit different behaviors. As , the infection, starting from some nodes, propagates in the whole network and reaches a stationary regime where a certain density of nodes is constantly infected at each time, independently from the size and the identity of the initial set of infected nodes. Intuitively, if the number of nodes that recover from the infection overcomes those that become infected. On the other hand, when the infection is too aggressive. The nontrivial scenario () is achieved when the network architecture and the parameters allow having a balance between the number of nodes that become infected and the ones that recover.In the simulations, we defined the transient time as the time after which , with (see Figure 1b); in other words is the number of time steps needed by the epidemic to reach its stationary state.
Statistical analysis was performed by using R package stats R Core Team (2013). In particular, clustering analysis performed on the HIV dataset was made using the HeatMap function, applying the Euclidean distance matrix (given by the ‘dist’ function) and the ‘hclust’ method for the clustering algorithm.
Results
.4 Stationary epidemic behavior is a global measure of protein thermal stability
Different thermal behaviors in homologous proteins have long been studied and several features have been identified as responsible for those differences (such as salt bridges, charged amino acids disposition, etc. Amadei et al. (2017); Tartaglia et al. (2007); Vishveshwara et al. (2002); Vijayabaskar and Vishveshwara (2010); Lee et al. (2014); Folch et al. (2010, 2008)).
These features are very well defined in network representation, both in terms of network topology (structure) and link weights (energy). Here we exploit our epidemic-diffusion algorithm to assess the capability of the network to reflect the protein thermostability.
In particular, we compared the stationary state density of infected nodes between all the couples of the Thermal dataset. For each protein, the diffusion was simulated, starting each time from a randomly selected set of infected residues. In particular, of the nodes were infected at .
In 84% of cases (27 out of 32 comparisons), thermophilic proteins acquired a higher density of infected nodes with respect to their mesophilic counterparts, when epidemic diffusion reaches the equilibrium (Figure 2a). According to us this results reflects both the overall higher connectivity and the higher energy of the links in the thermophilic proteins compared to the mesophilic ones. Our diffusion-based approach is able to well capture this aspect, even better than the network analysis alone is able to do (see supplementary material). In Figure 2b, we reported an example of diffusion process results where the different steady states are very well visible (PDB id: 1PII-1DL3).
.5 Epidemic transient phase permits local characterization of protein structures
After demonstrating that we can properly apply the epidemic diffusion approach exploring global features of a three-dimensional structure of a protein, we investigated our diffusion approach at a single residue level. i.e. we tested if residues that functionally need to have strong communication with the rest of the protein are characterized by peculiar diffusive properties. In this framework, one of the most important challenges in computational biology is the characterization of the active and allosteric sites in proteins. Since the substrate-binding has to be detected also far from the binding region through a cascade of residue-residue interactions Amor et al. (2016), we hypothesize that the diffusive approach could be a perfect approach to capture this aspect.
So we investigated, in particular, the transient phase of the epidemic, i.e. the number of time steps necessary to reach the equilibrium (). The time varies according to some features of the infected initial nodes. In particular, if the epidemic starts from energetically interconnected residues, it is very likely that the stationary state will be obtained in a shorter time when compared with the other residues.
We simulated an epidemic originating from every single residue of all proteins in the Enzyme dataset. In particular, since epidemic originating from a single node usually are characterized by a fast extinction, for each residue, we selected also its two closest neighbors in sequence as a contagion starting point.
We normalized results over each protein size by using the -score, in order to make the comparison between proteins with sequences of different length possible.
The -score of the -th residue was defined as:
[TABLE]
where the over-bar represents the mean of over all the amino acids in the analyzed protein.
Charged residues exposed on the protein surface and core residues are obviously very fast in propagating the infection, because of the high energy interactions the charged residues are involved in and because of the high number of contacts the core residues have. We preliminary confirmed this (see supplementary material) as shown in Suppl. Figure 1. We also correlated protein secondary structure location (as calculated by STRIDE Frishman and Argos (1995)) of each residue with its because we can suppose that, on average, residues belonging to secondary structures are assembled in a dense part of the interaction network. As expected, Beta Strand and Alpha Helix components are characterized by lower (see supplementary material).
We then proceeded to apply the diffusion protocol in order to analyze values for 11 pairs of (thermostable-mesostable) enzymes in the Enzyme dataset for which we know residues forming the active site. The comparison between the values of the active site with that of the other residues clearly shows that the former is characterized by a statistically significant lower . The ‘functional information’ the protein receives after ligand binding into the active site needs to be fastly communicated to the whole protein and our diffusive method is able to well characterize this important biological aspect. We show in Figure 3b the distribution of -scores belonging to the active site residues. The of residues belonging to active sites presents a -score lower than 0, represented by the orange area under the curve. This means stronger connectivity of active site residues with the whole protein than the average value of all other residues.
Then we have considered the twenty allosteric proteins with known active and allosteric site residues (see Allosteric dataset in Methods). Even in this case, we applied our epidemic protocol in order to evaluate the number of time steps necessary to reach the equilibrium. As shown in Figure 3c, of allosteric residues shows a -score value lower than zero, demonstrating that allosteric residues are faster than average residues in propagating information inside the protein network due to their biological functional role in the 3D structure. Interestingly, the active sites of these proteins are not characterized by peculiar diffusive characteristics because just active sites reach -score values lower than 0. The reason for this behavior, different from what observed before, can be due to their different binding ‘state’: in the Thermal dataset the proteins were in the apo form, while in the Allosteric dataset the proteins are in the holo form, with the ligand occupying the active site and diffusive approach seems to be very sensitive to these two states.
.6 HIV-1 and HIV-2 Proteases can be discriminated by their epidemic diffusion profile
Finally, we used epidemic diffusion time analysis in order to discriminate HIV-1 and HIV-2 proteases. Despite the structural similarities, HIV-1 and HIV-2 proteases show dramatic disparities in susceptibility to HIV-1 protease inhibitors Triki et al. (2018). Each of the 18 HIV proteases (HIV dataset) has been represented by a diffusion time profile (e.g. the concatenation of single residue ) and all profiles have been easily compared since all protein sequences have the same length.
The heatmap reported in Figure 4a shows clustering analysis results performed on residues and proteins of the HIV dataset. All of the proteins are correctly identified as HIV-1 or HIV-2 protease demonstrating the possibility to use an epidemic diffusion approach in order to evaluate functional differences related to three-dimensional protein structures.
We then explored key residues responsible for discrimination of the two groups. For each residue of HIV-1 and HIV-2 proteases, we compared their scores distributions with a t-test. The 39 residues showing the most significant difference, when we set p-value threshold to 0.05 are: 14, 19, 22, 23, 40, 41, 43, 56, 61, 62, 64, 70, 72, 73, 84, 95, 103, 108, 114, 115, 116, 118, 120, 134, 136, 140, 142, 155, 160, 163, 165, 167, 168, 170, 179, 190, 191, 192, 193.
Notably, lowering the p-value threshold to 0.005, we identified a subgroup of seven residues, i.e. 14, 19, 64, 95,1 18, 142, 190, shown in Figure 4b. As we said before, although HIV-1 and HIV-2 proteases share a great deal of structural similarity the reasons for intrinsic protease inhibitor resistance in HIV-2 are not known. Very interestingly, the subgroup of seven residues we identified occur at sites distant from the active site (see Figure 4c) and two out of seven residues are also identical in the two proteases. This leads us to present a hypothesis that perhaps our epidemic approach could have been captured subtle structural changes, imparted by a limited number of residues, causing dramatic functional differences between homologous proteins (HIV-1 and HIV-2 proteases). That is the seven amino acids outside the HIV-2 protease active site may cause subtle changes in conformation and in long-range effects compared to HIV-1, which might impact protease inhibitors binding affinity.
I Discussion
Proteins are complex systems where evolution must be very proficient in tuning parameter (e.g. selecting mutations) to obtained more fitted proteins with respect to some features while maintaining the protein functional. For instance, optimizing enzymes to be more efficient at high temperatures (i.e. increasing their thermal stability) must not reduce enzyme flexibility and the ability to change configurations.
Graph theory-based methods represent a powerful approach to investigate protein topological and energetic properties. However, we could consider it a static view of the protein structures that does not allow us to describe their complexity in a complete way. To overcome this limitation several aspects of proteins were investigated through dynamical approaches, like molecular dynamics or perturbation-response approaches, which take into account the dynamical properties. A problem connected with these approaches is that they are typically characterized by a high computational cost.
In this work, we explored the possibility to adopt an epidemic diffusion-based method as an efficient way to study functional aspects (both local and global) of proteins strictly connected with their three-dimensional structural organization. The new idea we introduced with this approach was to investigate the dynamic properties of the interactions network of a protein structure by using epidemic diffusion-based algorithms, preserving in this way both the topology and the energy properties of the interactions. The most striking advantage of this method is that it is not very computationally expensive allowing for a fast exploration of complex problems related to protein function (diffusion on an average protein requires few minutes on a standard personal computer). Starting from the RIN formalism Sengupta and Kundu (2012); Böde et al. (2007), we studied the diffusion of a fictitious epidemic inside the protein structure represented as a network using energies and node degrees as proxies of infection and recovery rates.
A large number of mathematical models have been formulated to study the spread of infectious diseases, but most of these are just variants of Kermack and McKendrick epidemic model Kermack and McKendrick (1927, 1932). Reproducing different aspects of the spread of real diseases, all models ultimately provide a measure of the information diffusion throughout the entire network.
Simulations of diffusion processes were performed considering typical network parameters for calculating the probability of transmission of infection (proportional to the link energy) and the probability of each node of returning susceptible (proportional to node degree).
From diffusion simulations, two descriptors were defined, one () providing global information and the other () local one. In particular, a residue-specific descriptor is of fundamental importance because the identification of functional key residues in a protein structure is a useful aspect for protein design in many open biological questions.
Considering the stationary phase, the mean of the percentage of infected nodes is constant over the steps balancing the rate of infection and recovery. The The value of the stationary percentage of infected nodes is a very compact way to quantify the global properties of the entire protein related to residue-residue energetic interactions. A protein characterized by strong interconnectivity will have a very strong energetic coupling between its residues showing, at the equilibrium, a higher number of infected nodes.
Given an overall fold, the arrangement of side chains organizes the inter-molecular interaction to better resist the thermal noise. Therefore, we test the sensibility of this formalism applying it on a well-defined set of homologous protein pairs, one protein from a mesostable organism, the other one from a thermostable one.
We found that thermophilic proteins have a significantly higher percentage of infected residues than homologous mesophilic counterparts, meaning that thermophilic proteins organize their network of interactions in order to promote infection. We could, therefore, conclude that thermophilic proteins have, on average, a higher level of interconnectivity than mesophilic proteins.
Another important aspect we explored in this work was the local properties of proteins that often are generated by long-range effects. In this case, the problem was studied by taking into account the transient phase of the diffusion simulation, which is composed of steps between the initial infection and the stationary state.
The number of steps necessary in order to reach the stationary phase, , is depending on the choice of the starting infected nodes, expressing their centrality in the energy network. This local characterization can be utilized in order to identify which kind of residues (or domains) are more central in a protein, in terms of their connection with the rest of the protein. We clearly demonstrated that both residues belonging to enzyme allosteric and active sites typically reach the state of equilibrium with a number of steps smaller than any other residue.
We also investigated the local property of each residue of two HIV-1 and HIV-2 proteases. The method showed its perfect ability to separate the two classes of proteases, in terms of transient phase, elucidating non-trivial differences (HIV-1 and HIV-2 protease share of sequence identity) by analyzing the dynamic properties of residues represented as a network. The epidemic approach was also able to select seven residues responsible for the discrimination of the two groups, which might impact protease inhibitors binding affinity helping to understand the key differences between HIV-1 and HIV-2 infections. We believe that the study of the dynamical aspect of the protein structure network is, in general, a promising direction for the future. We intend to investigate how far our results can be generalized to other types of protein functional elements, other types of proteins like membrane proteins and hopefully to other kinds of macromolecules like nucleic acids.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mannige (2014) R. Mannige, Proteomes 2 , 128 (2014).
- 2Chothia et al. (1997) C. Chothia, T. Hubbard, S. Brenner, H. Barns, and A. Murzin, Annual Review of Biophysics and Biomolecular Structure 26 , 597 (1997).
- 3Dill et al. (2008) K. A. Dill, S. B. Ozkan, M. S. Shell, and T. R. Weikl, Annual Review of Biophysics 37 , 289 (2008).
- 4Lesk and Chothia (1980) A. M. Lesk and C. Chothia, Journal of Molecular Biology 136 , 225 (1980).
- 5Ofran and Margalit (2006) Y. Ofran and H. Margalit, Proteins: Structure, Function, and Bioinformatics 64 , 275 (2006).
- 6Debès et al. (2013) C. Debès, M. Wang, G. Caetano-Anollés, and F. Gräter, P Lo S Computational Biology 9 , e 1002861 (2013).
- 7Domingues et al. (2000) F. S. Domingues, W. A. Koppensteiner, and M. J. Sippl, FEBS Letters 476 , 98 (2000).
- 8Karshikoff et al. (2015) A. Karshikoff, L. Nilsson, and R. Ladenstein, FEBS Journal 282 , 3899 (2015).