Indirect but Efficient: Laser-Excited Electrons Can Drive Ultrafast Polarization Switching in Ferroelectric Materials
Chao Lian, Zulfikhar A. Ali, Hyuna Kwon, and Bryan M. Wong

TL;DR
This paper introduces a laser-based method for ultrafast and efficient switching of ferroelectric polarization in materials like BaTiO3, using charge redistribution and ionic forces induced by optical pulses, avoiding complex terahertz sources.
Contribution
It proposes a novel laser-tuning mechanism for ferroelectric switching that is faster, more efficient, and compatible with existing laser technology.
Findings
Achieved polarization switching within 200 fs in BaTiO3.
Demonstrated charge redistribution reduces ferroelectric charge order.
Reversible switching enabled with standard 800 nm laser pulses.
Abstract
To enhance the efficiency of next-generation ferroelectric (FE) electronic devices, new techniques for controlling ferroelectric polarization switching are required. While most prior studies have attempted to induce polarization switching via the excitation of phonons, these experimental techniques required intricate and expensive terahertz sources and have not been completely successful. Here, we propose a new mechanism for rapidly and efficiently switching the FE polarization via laser-tuning of the underlying dynamical potential energy surface. Using time-dependent density functional calculations, we observe an ultrafast switching of the FE polarization in BaTiO3 within 200 fs. A laser pulse can induce a charge density redistribution that reduces the original FE charge order. This excitation results in both desirable and highly directional ionic forces that are always opposite to the…
Click any figure to enlarge with its caption.
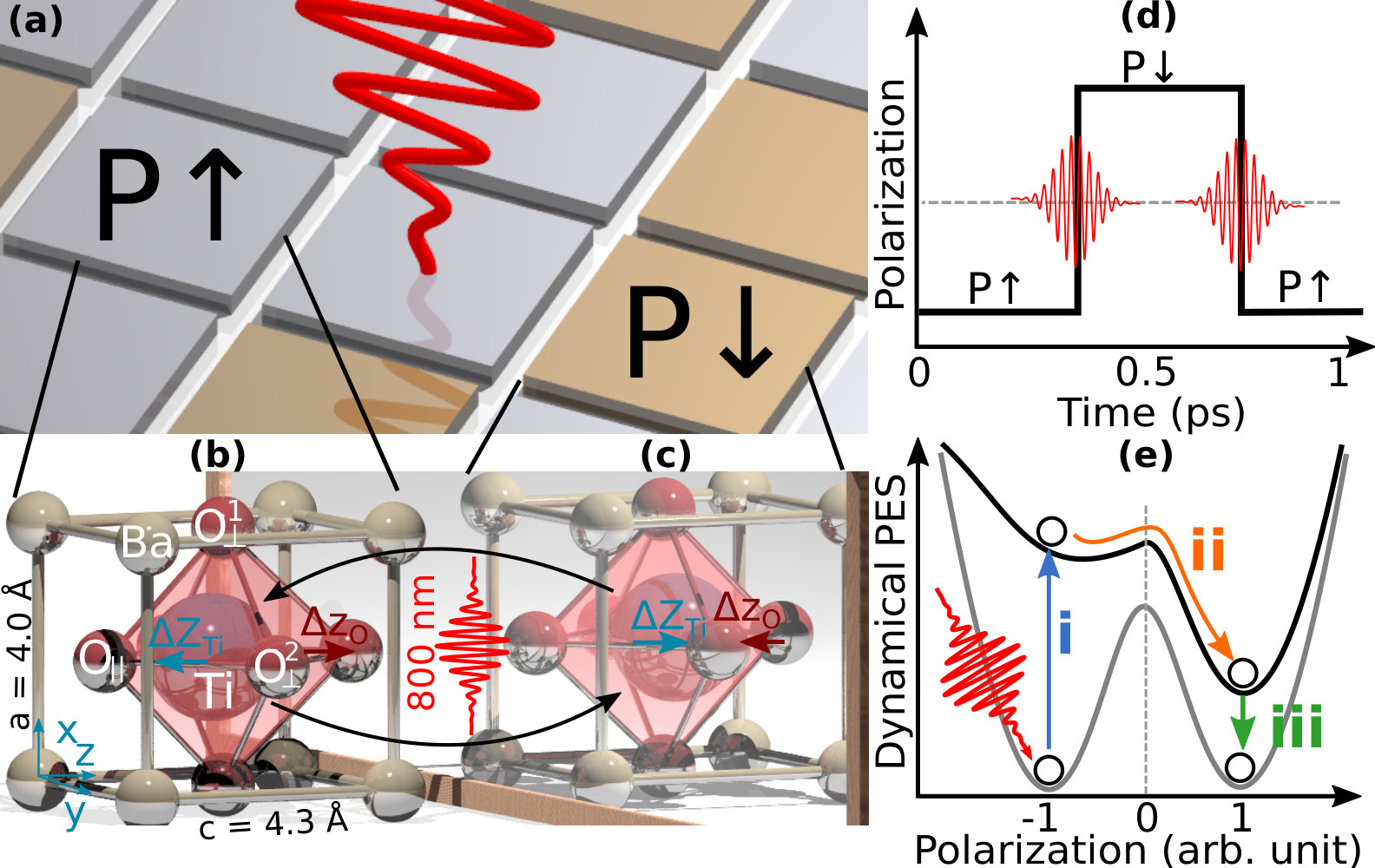 Figure 1
Figure 1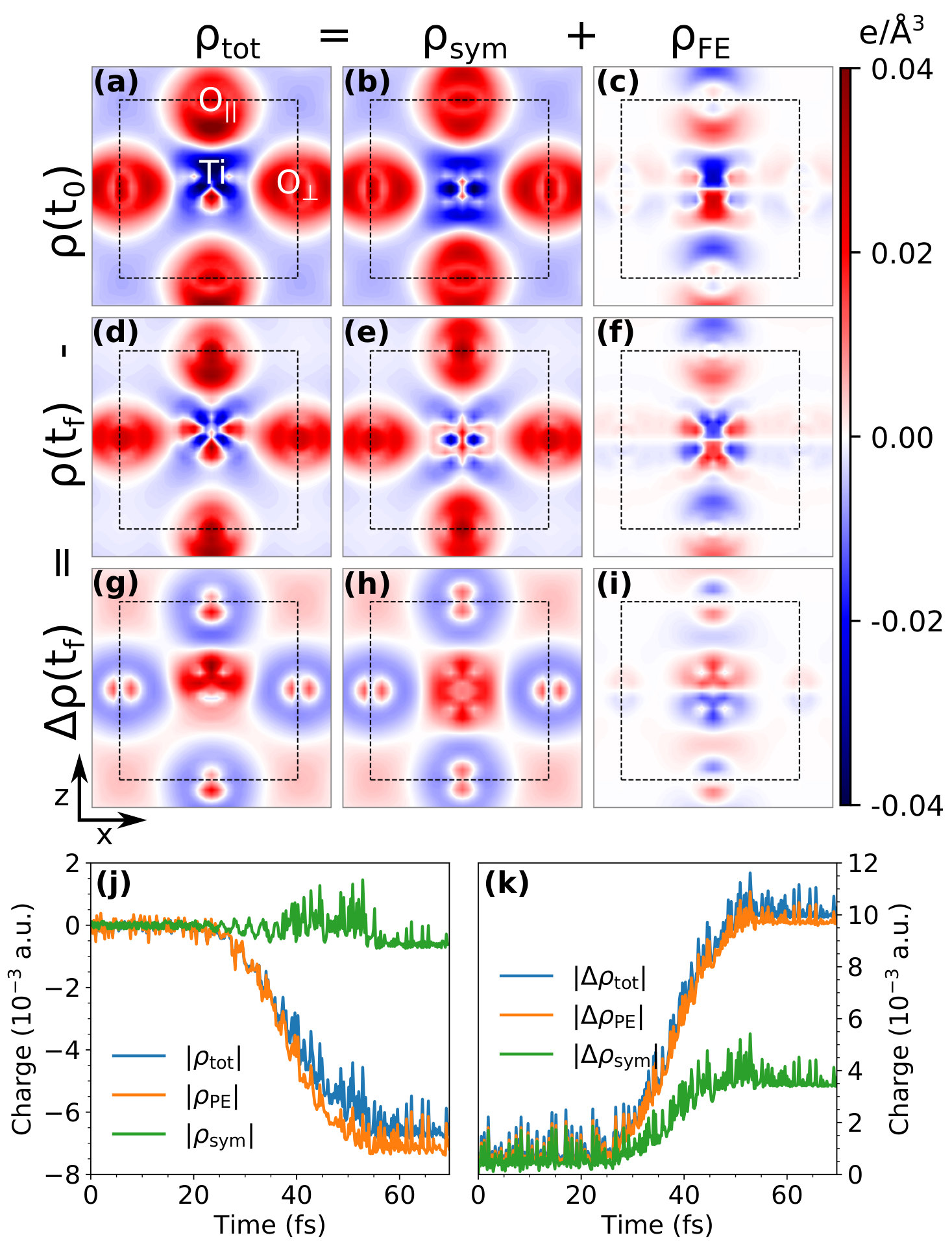 Figure 2
Figure 2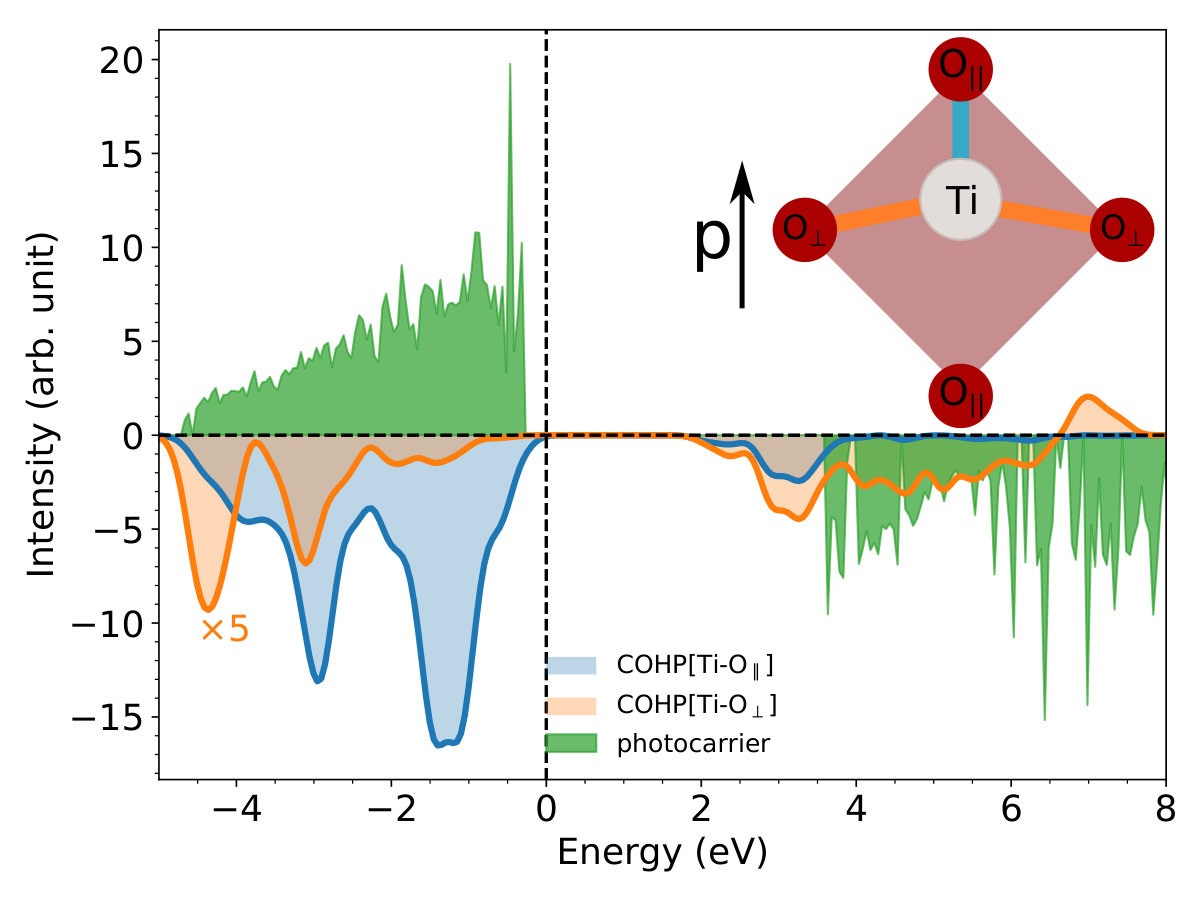 Figure 4
Figure 4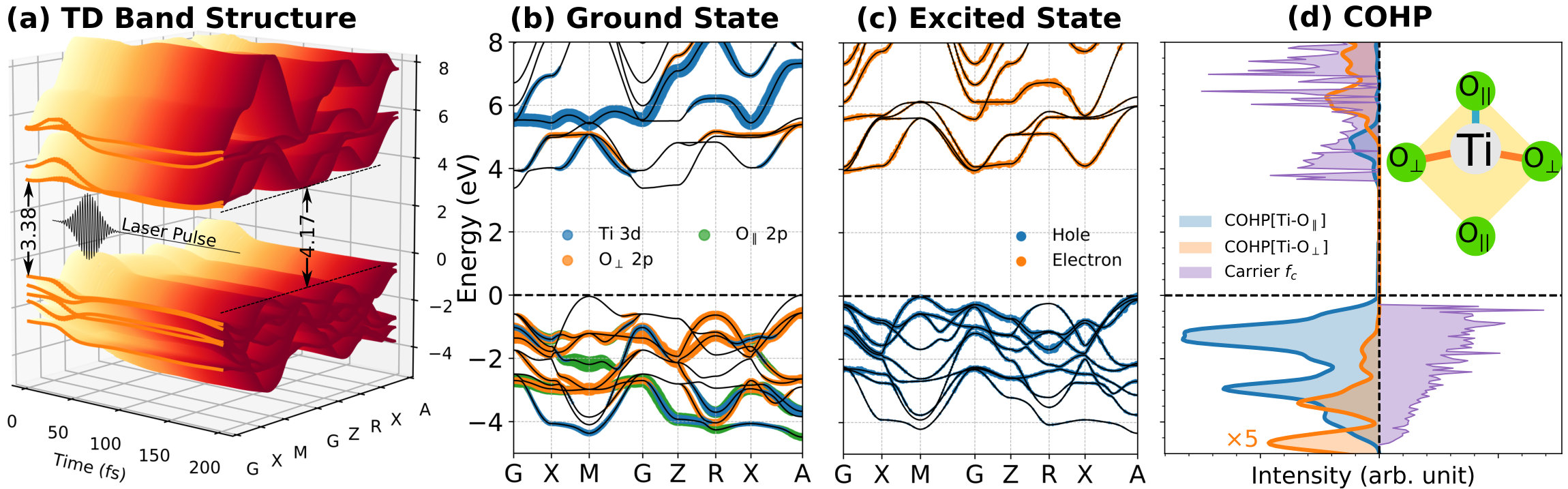 Figure 5
Figure 5 Figure 5
Figure 5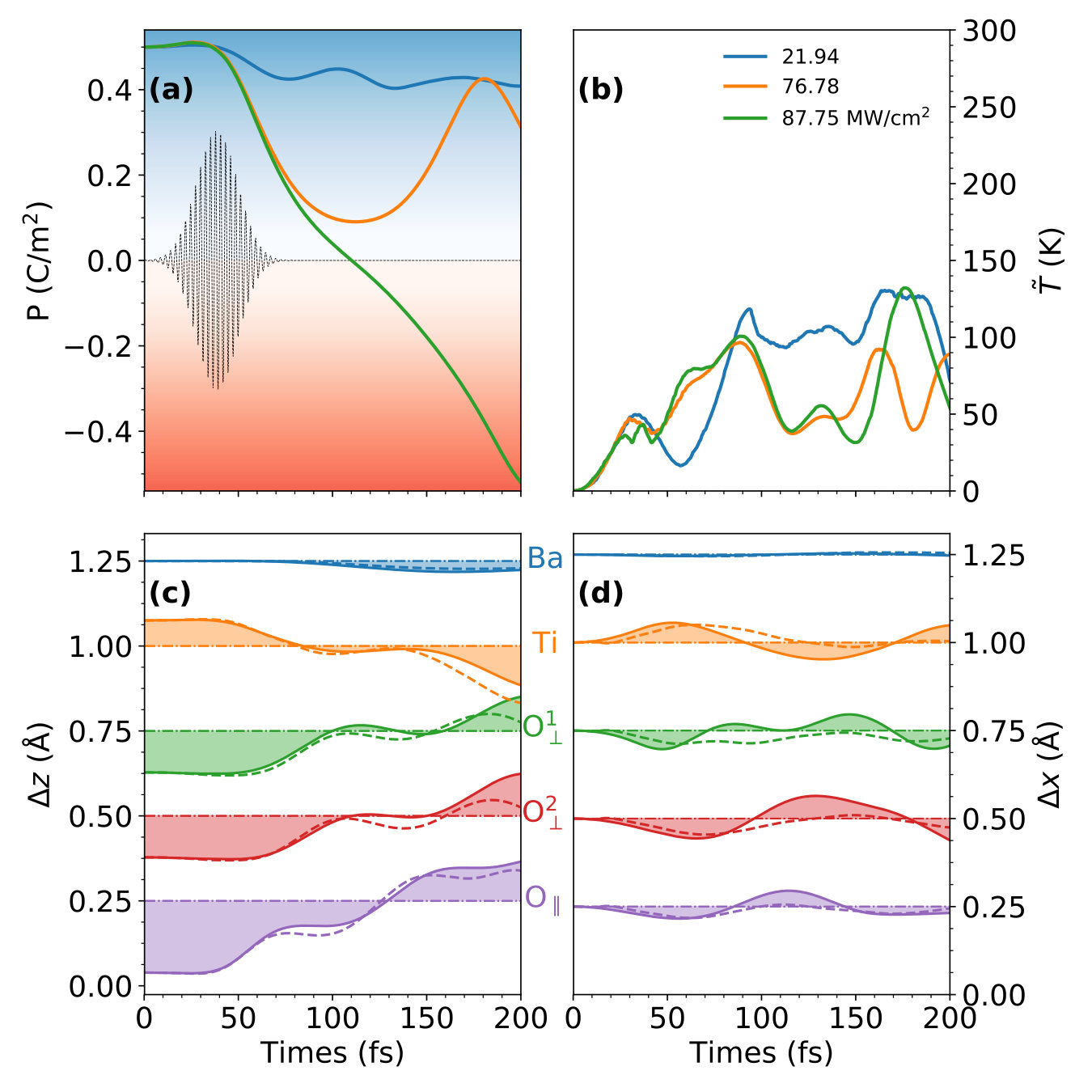 Figure 6
Figure 6| Atom | (Å) | () | ||||
|---|---|---|---|---|---|---|
| Ba | 0.0 | 0.0 | 0.0 | 0.00 | 2.73 | |
| Ti | 0.5a | 0.5a | 0.5 | 0.08 | 7.17 | |
| O | 0.0 | 0.5a | 0.5c | -0.12 | -2.02 | |
| O | 0.5a | 0.0 | 0.5c | -0.12 | -2.02 | |
| O∥ | 0.5a | 0.5a | 0.0 | 0.21 | -5.74 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Indirect but Efficient: Laser-Excited Electrons Can Drive Ultrafast Polarization Switching in Ferroelectric Materials
Chao Lian
Zulfikhar A. Ali
Hyuna Kwon
Bryan M. Wong
Department of Chemical & Environmental Engineering, Materials Science & Engineering Program, and Department of Physics & Astronomy, University of California-Riverside, Riverside, CA 92521, USA.
Abstract
To enhance the efficiency of next-generation ferroelectric (FE) electronic devices, new techniques for controlling ferroelectric polarization switching are required. While most prior studies have attempted to induce polarization switching via the excitation of phonons, these experimental techniques required intricate and expensive terahertz sources and have not been completely successful. Here, we propose a new mechanism for rapidly and efficiently switching the FE polarization via laser-tuning of the underlying dynamical potential energy surface. Using time-dependent density functional calculations, we observe an ultrafast switching of the FE polarization in BaTiO3 within 200 femtoseconds. A laser pulse can induce a charge density redistribution that reduces the original FE charge order. This excitation results in both desirable and highly directional ionic forces that are always opposite to the original FE displacements. Our new mechanism enables the reversible switching of the FE polarization with optical pulses that can be produced from existing 800-nm experimental laser sources.
Ferroelectric (FE) materials are characterized by an intrinsic spontaneous electric polarization that can be further harnessed for next-generation electronic and energy-harvesting materials. For example, tuning the FE polarization can vary the tunneling resistance over several orders of magnitude Dawber et al. (2005), enabling technological advancements such as non-volatile memory in digital electronic devices, Paz De Araujo et al. (1990) memristors, Chanthbouala et al. (2012) and integrated neuromorphic networks Jo et al. (2010); Kim and Lee (2019). In addition, by enabling a steady-state photocurrent, the use of FE polarization can substantially increase light-harvesting efficiency, particularly in hybrid organic-inorganic halide perovskite solar cells Zhou et al. (2018); Li et al. (2018); Ghosh et al. (2015); Stroppa et al. (2015); Chen et al. (2014); Wei et al. (2014); Kim et al. (2015); Tong et al. (2017); Ji et al. (2017); Jankowska et al. (2017); Jankowska and Prezhdo (2017); Li et al. (2017). Finally, the variation in FE polarization on surfaces can also dramatically change the adsorption energetics in catalytic systems and could be further harnessed to enable other polarization-dependent surface mechanisms Kakekhani and Ismail-Beigi (2015); Bal and Neyts (2019).
All of these applications are intrinsically associated with FE polarization and can, therefore, be further enhanced by tuning and controlling this intrinsic material property. Polarization switching is typically accomplished through a static electric field; however, the switching time is relatively sluggish (on the order of nanoseconds) due to the slow recrystallization time of most materials (typically hundreds of picoseconds) Merz (1954); Spierings et al. (2002). To accelerate this switching process, significant research has focused on enabling ultrafast polarization switching via optical processes Fahy and Merlin (1994); Li et al. (2004). Terahertz (THz) sources provide ultrafast pulsed electric fields that are expected to reverse the polarization via the same mechanism as a static electric field. However, even with state-of-the-art THz sources, a single THz pulse does not possess enough strength to switch this polarization Takahashi et al. (2006); Cavalleri et al. (2006); Qi et al. (2009); Hauf et al. (2018); Von Hoegen et al. (2018); Chen et al. (2016); Katayama et al. (2012). To circumvent the use of THz sources, Subedi recently proposed that activating an infrared-active phonon mode can also induce polarization switching effects Subedi (2015). Later, Mankowsky et al. Mankowsky et al. (2017) conducted experiments on LiNbO3 but only observed a temporary and partial polarization switching. While other researchers have reported related phenomena such as ultrafast domain wall movements, Daranciang et al. (2012); Lejman et al. (2014); Ahn et al. (2017); Akamatsu et al. (2018); Shinde and Singh (2018) photoinduced depolarization,Korff Schmising et al. (2007); Rana et al. (2009); Kuo et al. (2017); Rubano et al. (2018) and coherent ionic movement Först et al. (2013); Porer et al. (2018); Qi et al. (2018), ultrafast polarization switching in these FE materials have not been completely successful. Ideally, one would like a more efficient mechanism, such as that shown in Fig. 1(a) and (d), in which a readily-available laser source could be utilized to switch the polarization with one pulse and reversibly switched back with a second identical pulse.
In this Letter, we propose a new mechanism that enables this efficient switching of the polarization in FE materials. By accounting for the detailed interactions between the electrons, nuclei, and electromagnetic field, our real-time time-dependent density functional theory (RT-TDDFT) simulations show that the ultrafast FE polarization switching in BaTiO3 occurs within 200 femtoseconds (fs), after the material is excited by an 800-nm laser pulse. We find that this polarization switching commences when the laser pulse pumps electrons from 2p orbitals in oxygen to the 3d orbitals in titanium. This dynamic excitation process yields desirable and highly-directional forces on the Ti and O atoms that are (1) always opposite to the original FE displacements and (2) can be harnessed to consistently switch the polarization of the material. By laser-tuning the dynamical potential energy surface, we show that this new mechanism can switch the FE polarization in both directions using identical pulses from experimentally-available laser sources.
We use our in-house time-dependent ab initio package, (TDAP) Meng and Kaxiras (2008); Lian et al. (2018a, b), for our RT-TDDFT calculations Runge and Gross (1984); Bertsch et al. (2000); Wang et al. (2015), where the wavefunctions and charge densities are obtained from the Quantum Espresso Giannozzi et al. (2009, 2017) software package. We used the projector augmented-waves method (PAW) Blöchl (1994) and the Perdew-Burke-Ernzerhof (PBE) exchange-correlation (XC) functional Perdew et al. (1996) in both our DFT and RT-TDDFT calculations. Pseudopotentials were generated using the pslibrary Dal Corso (2014) software package. The plane-wave energy cutoff was set to 55 Ry, and the Brillouin zone was sampled using a Monkhorst-Pack scheme with an -point mesh for the unit cell and a mesh for a supercell. To reproduce the experimental band gap, a scissor correction of 1.65 eV was added to both the ground state and time-dependent calculations. The electronic timestep, , was set to fs, and the ionic timestep, , was fs. The Gaussian-type laser pulse utilized in our study is given by where is the electric field amplitude, eV is the laser frequency, and fs is the temporal location of the electric field peak. The laser pulse is linearly polarized along the direction, perpendicular to the ferroelectric polarization. The crystal orbital Hamilton population (COHP) analysis was calculated with the Lobster Dronskowski and Blöchl (1993); Deringer et al. (2011); Maintz et al. (2016) software package.
We first optimized the structure of BaTiO3, which is shown in Fig. 1(b) and Table 1. The lattice is tetragonal with Å and . The ferroelectric properties of BaTiO3 originate from the slight distortion of the Ti-O octahedron: the Ti atom deviates from the body-centered position along the direction and the O atoms deviate from the face-centered positions along the direction. These FE displacements are characterized as the displacements from the symmetric positions , where and are the positions of atom ( Ti, Ba, O⟂, and O∥) along the , , and direction in the FE and symmetric phase, respectively. We calculate the FE polarization accordingly as , where Å3 is the volume, is the ionic FE displacement, and is the Born effective charge, as shown in Fig. 1(b) and Table 1. Consistent with previous studies, the semilocal exchange-correlation functionals slightly overestimate the ratio and the static polarization, as discussed in Bilc et al. (2008); Sun et al. (2016).
We apply laser pulses having a wavelength of nm, a polarization perpendicular to the FE polarization direction, and a duration lasting from fs to fs. We characterize and monitor the laser-induced dynamics by calculating the dynamical polarization
[TABLE]
Figure 2(a) shows the results under various laser fluences 21.94, 76.78, and 87.75 MW/cm2. We identify three types of lattice dynamics: (1) at the lowest fluence of MW/cm2, slightly decreases and oscillates around . (2) when increases to a critical fluence of 76.78 MW/cm2, substantially decreases to around 0 within 100 fs, while recovers to in the following 100 fs. (3) When further increases to 87.75 MW/cm2, we observe a switching of the polarization. continuously decreases past 0 until it reaches its maximum polarization in the opposite direction.
In contrast to other experimental strategies discussed previously, this laser-induced polarization change is a non-thermal process. To clearly demonstrate this, we plot the effective temperature as a function of time, where is the kinetic energy of the ions and is the Boltzmann constant. As shown in Fig. 2(b), is much lower than room temperature throughout our entire dynamics simulations regardless of the laser fluence. In addition, is different from the thermodynamic equilibrium temperature, which is produced by the random ionic movements. Instead, in the laser-induced polarization switching process, the ionic movements are highly directional. As shown in Fig. 2(c), the laser radiation triggers a set of FE displacements, , that are nearly anti-parallel with the original values, . of the oxygen atoms and changes sign from negative to positive, while the titanium atom moves in the opposite direction from positive to negative. Considering that the oxygen and titanium atoms have effective Born charges with opposite signs (Table 1), the movements of all the atoms contribute consistently to the polarization switching. Moreover, along the directions perpendicular to the polarization, the displacement, , just slightly oscillates around the equilibrium position, as shown in Fig. 2(d). This leads to an abnormal fluence-dependence of the effective temperature , which slightly decreases as the fluence increases. Although the laser pulse induces larger ionic forces at higher fluence, the system needs to overcome the small energy barrier to reverse the polarization, as shown in Fig. 1(e), which consumes the kinetic energy and decreases the effective temperature. In comparison, with a lower influence, the ions can not overcome the energy barrier, resulting in a higher effective temperature for the oscillation. Thus, the highly directional FE movements predominantly contribute to the relatively low effective temperature , indicating a unique laser-induced non-thermal mechanism.
Coherence is another distinguishable feature of our polarization-switching mechanism in which the laser pulse drives the movement of the ions in a synchronized fashion. To verify that this coherence is not an artifact of size effects due to the use of a single unit cell, we carried out the TDDFT calculations using a supercell for comparison. We analyzed the average laser-induced displacements as a function of time, , where denotes the index of the unit cell. As shown in Fig. 2(c) and (d), in the supercell case is almost identical to in the unit cell case, which indicates that the unified switching of the FE displacements occurs over all the laser-irradiated area. Consequently, no nucleation and growth of oppositely polarized domains are needed in this polarization switching mechanism, which accelerates this switching process to finish within 200 fs.
We illustrate the atomistic mechanisms of the polarization switching by analyzing the carrier dynamics: i.e., we monitor the differential charge density as a function of time, where is the charge density, and is the superposition of the atomic charge densities. Thus, captures the spatial distribution of the bonding (+) and antibonding (–) densities. The FE polarization can be treated as the asymmetric disturbance of the symmetric phase, which is characterized by the centrosymmetric charge order. Thus, we divide the total charge into two parts, and , where , and . Obviously, is the centrosymmetric part of the total charge that characterizes the original symmetric order, whereas is the asymmetric part that characterizes the FE order.
Figure 3(a)-(i) depicts a two-dimensional contour of the charge density, (, , and ), at the Ti-O surface . Comparing Fig. 3(a)-(c), we find that is the dominant component of even when FE effects are included, whereas is localized around the Ti positions. As such, these figures show that an electron transfers from a Ti 3d orbital to an O 2p orbital, which triggers a pseudo-Jahn-Teller effect (PJTE) Polinger et al. (2015) and stabilizes the FE phase. By comparing the laser-induced charge density difference, (, , ), with the ground state charge density , we find that: (1) the laser induces an electron transfer from a bonding to an anti-bonding area, where [Fig. 3(g)] is opposite to that of the ground-state bonding charge [Fig. 3(a)]; (2) the majority of the induced charge has a centrosymmetric periodicity, i.e. [Fig. 3(h)] dominates the induced total charge [Fig. 3(g)]; (3) The most important feature is that the induced FE charge density [Fig. 3(i)] is opposite to that of the original [Fig. 3(c)], indicating a decrease in the FE order. We quantitatively evaluate the laser-induced change in the charge density by analyzing the integrated charges and , where , , and . The former characterizes the change in the bonding strength and the latter denotes the weakening of bonds, respectively. As shown in Fig. 3(j), the percentage decrease in the bond strength, , is 32.45%, 35.92%, and 10.77% for , , and , respectively. Accordingly, increases, with a ratio of , which is nearly the same as the ratio of the initial bonding charges . Thus, the laser-induced bonding-antibonding transfer is nearly homogeneous, lowering both the centrosymmetric and FE order proportionally. Since the decrease in affects all chemical bonds homogeneously, the overall effect of the decrease in is to lower the stability of the FE polarization. Thus, the laser-induced charge-density change, is always opposite to the original FE charge order and, therefore, weakens the original ionic bonding and PJTE, creating the unsymmetrical dynamical PES shown in Fig. 1(e).
We now analyze the carrier dynamics in momentum space for the inter-orbital transition. As shown in Fig. 4(a), the time-dependent band structures from our RT-TDDFT calculations show that the band gap increases from 3.38 to 4.17 eV while the band dispersions only slightly change. Since the initial and final states are degenerate at the ground state, the gap increase is attributed to the photocarrier generation. Comparing Fig. 4(b) and (c), the multi-photon excitation process produces carriers that are distributed from -4 to 6 eV, which indicates that the laser-induced electrons are mostly transfered from O2p orbitals to Ti3d orbitals. This transition is opposite to the electron flow induced by the PJTE, which introduces a highly-selective bond weakening for Ti-O*∥* over the Ti-O*⟂* bond. We can quantitatively evaluate the weakening of Ti-O bonds as
[TABLE]
where is the energy, represents either the or bond, denotes the crystal orbital Hamilton population (COHP), and is the photocarrier distribution, as shown in Fig. 4(d). From this analysis, we find that is much larger than , which indicates that the weakening of is much more significant than the weakening of the bond. Thus, the anisotropic Ti-O bonds in BaTiO3 result in a highly directional laser-induced polarization instability: the bonds parallel to the polarization are largely weakened while the bonds perpendicular to the polarization direction are barely affected.
Based on our photocarrier dynamic analysis of BaTiO3, we have uncovered an electronically-driven polarization switching mechanism that is different from the conventional ionic-driven mechanism used in existing THz experiments. As shown in Fig. 1(e), our mechanism is based on a laser-tuned dynamical potential energy surface (PES), which can be generated with the time-dependent energy and the time-dependent polarization obtained in the TDDFT calculation as . Since the dynamical PES is time-dependent, we only show the diagram to simplify the illustration. The laser radiation transforms the ground state PES into a dynamical PES by first exciting the material and automatically driving the ions to the opposite polarization. This PES-based mechanism encompasses three steps: (1) the laser pulse raises the PES of BaTiO3 by pumping electrons from the 2p orbitals of oxygen to the 3d orbitals of titanium, inducing an anti-FE charge order [Fig. 3(i)]; (2) this dynamic excitation process yields desirable and highly-directional forces on the Ti and O atoms that are always opposite to the original FE displacements, transforming the lattice into a structure with an opposite FE polarization along the non-equilibrium TD-PES within 200 fs [Fig. 2(a)]; (3) the photo-excited system relaxes to the ground state in conjunction with the recombination of the photo-carriers (which occurs beyond our simulation time). We speculate that this dynamical-PES-based polarization switching is ubiquitous in all the FE materials and, therefore, the same mechanism can be used to manipulate the polarization in other FE perovskite materials such as PbTiO3 and LiNbO3, multiferroic materials, and even recently-discovered two-dimensional SnTe-based FE materials. Chang et al. (2016) Most importantly, this new mechanism enables the switching of the FE polarization in both directions, with identical pulses that can be produced from experimentally-available 800-nm laser sources. Taken together, these findings provide a new mechanistic understanding of electronically-driven FE dynamics via laser-tuning of the dynamical potential energy surface, which can accelerate the design of more efficient, ultrafast FE devices.
C. L. and H. K. acknowledge support from the UC Riverside Collaborative Seed Grant. Z. A. A. and B. M. W. acknowledge financial support from the Office of Naval Research (Grant N00014-18-1-2740).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dawber et al. (2005) M. Dawber, K. M. Rabe, and J. F. Scott, Rev. Mod. Phys. 77 , 1083 (2005) , ar Xiv:0503372 [cond-mat] . · doi ↗
- 2Paz De Araujo et al. (1990) C. A. Paz De Araujo, L. D. Mc Millan, B. M. Melnick, J. D. Cuchiaro, and J. F. Scott, Ferroelectrics 104 , 241 (1990) . · doi ↗
- 3Chanthbouala et al. (2012) A. Chanthbouala, V. Garcia, R. O. Cherifi, K. Bouzehouane, S. Fusil, X. Moya, S. Xavier, H. Yamada, C. Deranlot, N. D. Mathur, M. Bibes, A. Barthélémy, and J. Grollier, Nat. Mater. 11 , 860 (2012) , ar Xiv:1206.3397 . · doi ↗
- 4Jo et al. (2010) S. H. Jo, T. Chang, I. Ebong, B. B. Bhadviya, P. Mazumder, and W. Lu, Nano Lett. 10 , 1297 (2010) . · doi ↗
- 5Kim and Lee (2019) M.-K. Kim and J.-S. Lee, Nano Lett. 19 , 2044 (2019) . · doi ↗
- 6Zhou et al. (2018) X. Zhou, J. Jankowska, H. Dong, and O. V. Prezhdo, J. Energy Chem. 27 , 637 (2018) . · doi ↗
- 7Li et al. (2018) W. Li, Y.-Y. Sun, L. Li, Z. Zhou, J. Tang, and O. V. Prezhdo, J. Am. Chem. Soc. 140 , 15753 (2018) . · doi ↗
- 8Ghosh et al. (2015) S. Ghosh, D. Di Sante, and A. Stroppa, J. Phys. Chem. Lett. 6 , 4553 (2015) . · doi ↗