Laser induced persistent orientation of chiral molecules
Ilia Tutunnikov, Johannes Flo{\ss}, Erez Gershnabel, Paul Brumer and, Ilya Sh. Averbukh

TL;DR
This paper demonstrates a novel laser-induced persistent orientation of chiral molecules, which remains long after excitation and can be used for chiral detection and separation.
Contribution
It introduces a new method for enantioselective molecular orientation using twisted laser fields, with long-lasting effects demonstrated computationally.
Findings
Persistent orientation lasts long after laser pulses.
Orientation is controllable and unique to chiral molecules.
Potential applications in chiral detection and separation.
Abstract
We show, both classically and quantum mechanically, enantioselective orientation of gas phase chiral molecules excited by laser fields with twisted polarization. Counterintuitively, the induced orientation, whose direction is laser controllable, does not disappear after the excitation, but stays approximately constant long after the end of the laser pulses, behavior unique to chiral systems. We computationally demonstrate this long-lasting orientation using propylene oxide molecules (, or PPO) as an example, and consider two kinds of fields with twisted polarization: a pair of delayed cross-polarized pulses, and an optical centrifuge. This novel chiral effect opens new avenues for detecting molecular chirality, measuring enantiomeric excess and separating enantiomers with the help of inhomogeneous external fields.
Click any figure to enlarge with its caption.
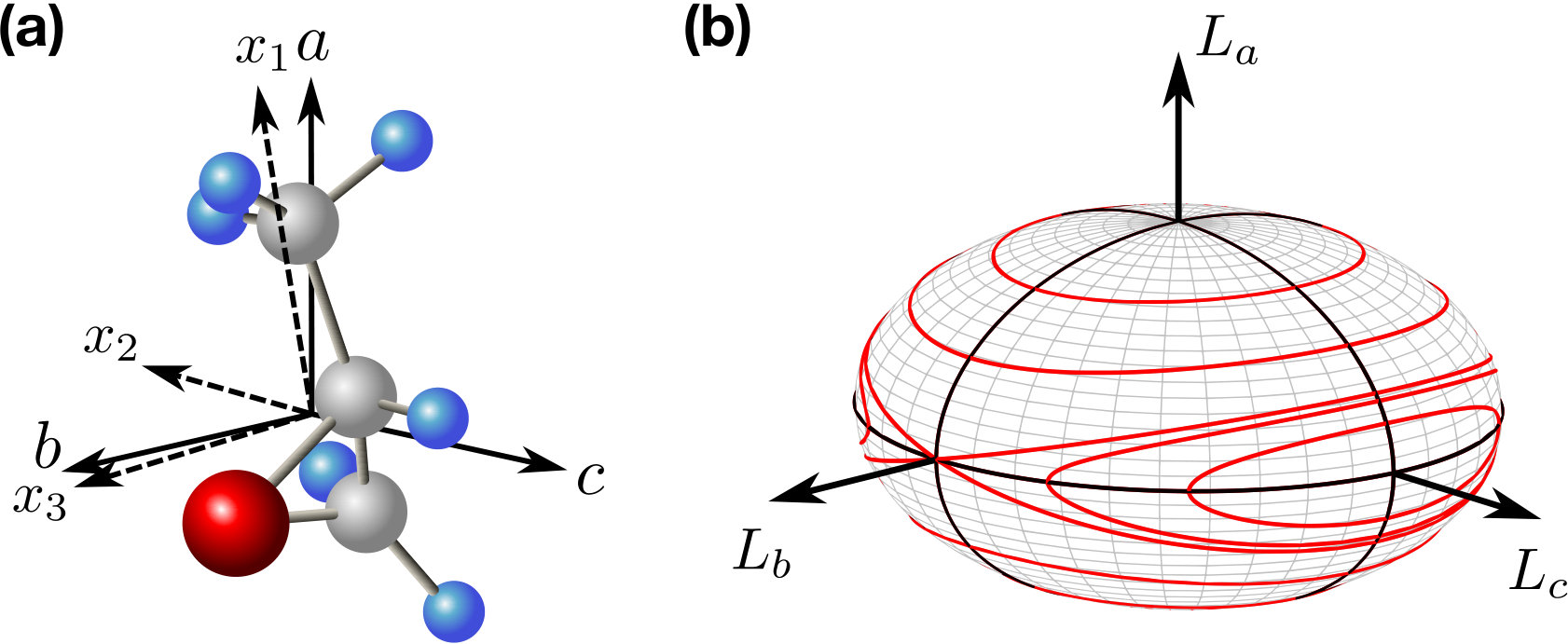 Figure 1
Figure 1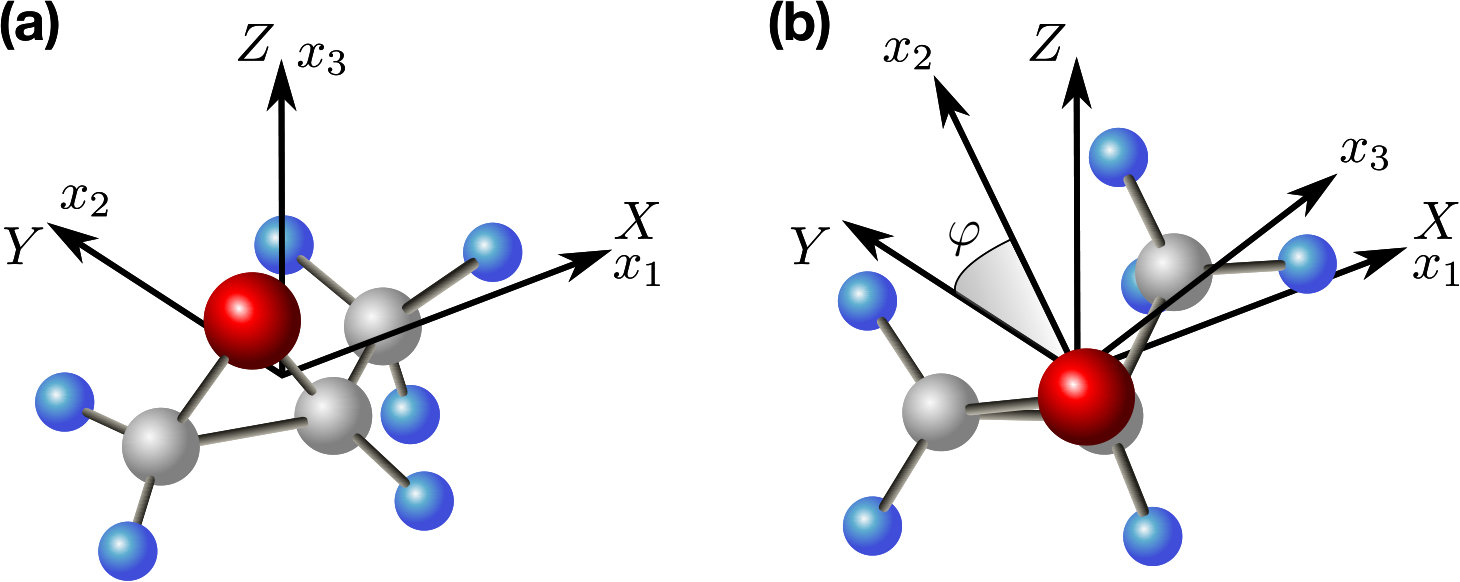 Figure 2
Figure 2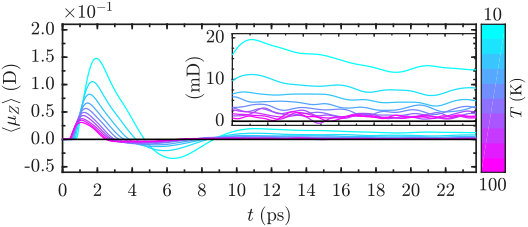 Figure 3
Figure 3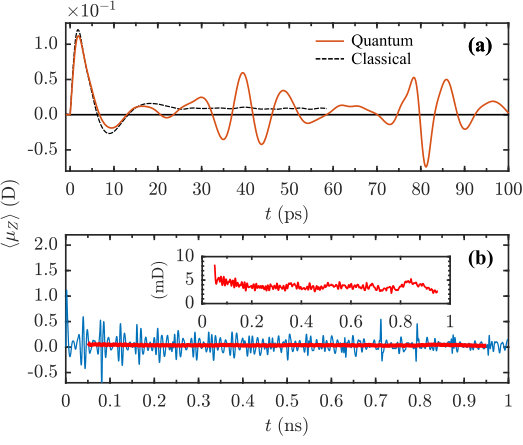 Figure 4
Figure 4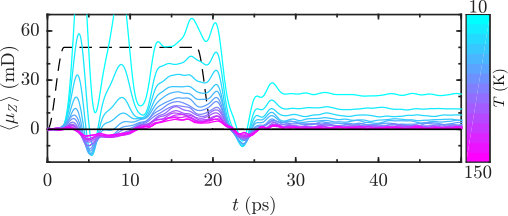 Figure 5
Figure 5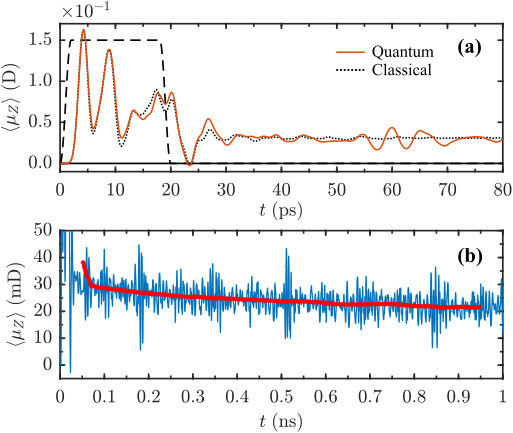 Figure 6
Figure 6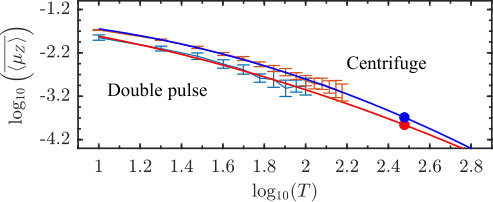 Figure 7
Figure 7 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
††thanks: I. T. and J. F. contributed equally to this work.††thanks: I. T. and J. F. contributed equally to this work.††thanks: [email protected]††thanks: [email protected]
Laser induced persistent orientation of chiral molecules
Ilia Tutunnikov
AMOS and Department of Chemical and Biological Physics, Weizmann Institute of Science, Rehovot 7610001, Israel
Johannes Floß
Chemical Physics Theory Group, Department of Chemistry, and Center for Quantum Information and Quantum Control, University of Toronto, Toronto, Ontario M5S 3H6, Canada
Erez Gershnabel
AMOS and Department of Chemical and Biological Physics, Weizmann Institute of Science, Rehovot 7610001, Israel
Paul Brumer
Chemical Physics Theory Group, Department of Chemistry, and Center for Quantum Information and Quantum Control, University of Toronto, Toronto, Ontario M5S 3H6, Canada
Ilya Sh. Averbukh
AMOS and Department of Chemical and Biological Physics, Weizmann Institute of Science, Rehovot 7610001, Israel
Abstract
We show, both classically and quantum mechanically, enantioselective orientation of gas phase chiral molecules excited by laser fields with twisted polarization. Counterintuitively, the induced orientation, whose direction is laser controllable, does not disappear after the excitation, but stays approximately constant long after the end of the laser pulses, behavior unique to chiral systems. We computationally demonstrate this long-lasting orientation using propylene oxide molecules (, or PPO) as an example, and consider two kinds of fields with twisted polarization: a pair of delayed cross-polarized pulses, and an optical centrifuge. This novel chiral effect opens new avenues for detecting molecular chirality, measuring enantiomeric excess and separating enantiomers with the help of inhomogeneous external fields.
I Introduction
Chiral molecules exist in two enantiomeric forms. The two enantiomers are mirror images of each other, and they are nonsuperimposable by translation and rotation (Cotton, ). Molecular chirality is an omnipresent natural phenomenon of extreme importance in physics, chemistry and biology (UniversalChirality, ). The ability to discriminate and separate mixtures of enantiomers is important, for example, in drug synthesis as different enantiomers of chiral drugs may exhibit strikingly different biological activity.
The related studies of gas phase chiral molecules focus on the measurements of enantiomeric excess, handedness of a given compound, and on devising techniques for manipulating mixtures containing both enantiomers (ShapiroBrumer, ; Brumer2002, ; Lux2012, ; Patterson2013, ; Pitzer2013, ; Herwig2013, ; Lehmann2013, ; Janssen2014, ; Patterson2014, ; Steinbacher2015, ; Lux2015, ; Christensen2015, ; Kastner2016, ; Beaulieu2018, ; Pitzer2018, ; Fehre2019, ; Neufeld2019, ; Rozen2019, ; Leibscher2019, ). In addition, over the years more ambitious directions were considered theoretically - laser assisted asymmetric synthesis, enantiomeric interconversion and purification (e.g. (Shapiro2000, ; ShapiroBrumer, ) and references therein).
Recently, a pair of non-resonant delayed cross-polarized laser pulses was proposed as a new tool for discrimination of chiral molecules (Yachmenev2016, ; Gershnabel2018, ; Tutunnikov2018, ) and the underlying classical enantioselective molecular orientation mechanism was exposed (Gershnabel2018, ; Tutunnikov2018, ). The approach was extended to general fields with time-dependent polarization twisting in a plane. Optical fields with fixed linear polarization are unable to induce molecular orientation because of the symmetry of light interaction with the induced dipole. Polarization twisting in a certain plane breaks that symmetry and defines a preferred spatial direction perpendicular to that plane that depends on the sense of polarization rotation. When interacting with molecules, the twisted field has a two-fold effect. First, it induces unidirectional rotation (UDR) of the most polarizable molecular axis in the plane of polarization twisting (Fleischer2009, ; Kitano2009, ; Khodorkovsky2011, ; Korech2013, ; Mizuse2015, ; Lin2015, ), thereby orienting the averaged angular momentum vector perpendicular to the plane. In addition, in the case of chiral molecules, the twisted field induces an orienting torque along the most polarizable axis, thus orienting the molecule itself perpendicular to the above plane (Gershnabel2018, ; Tutunnikov2018, ). The direction of orientation depends on both the sense of polarization twisting and the handedness of the molecule. A pair of delayed cross-polarized laser pulses (Fleischer2009, ; Kitano2009, ) provides the simplest example of the field with twisted polarization, but there are also more complex fields, such as chiral pulse trains (Zhdanovich2011, ; Bloomquist2012, ; Floss2012, ), polarization-shaped pulses (Karras2015, ; Prost2017, ; Prost2018, ) and optical centrifuge (Karczmarek1999, ; Villeneuve2000, ; Yuan2011, ; Korobenko2014, ; Korobenko2018, ). Most recently, the orientation of chiral propylene oxide (PPO) molecules by means of an optical centrifuge was experimentally achieved in (Milner2019, ), thus providing the first demonstration of enantioselective laser control over molecular rotation.
In this paper, we substantiate the enantioselective molecular orientation by twisted fields in two significant ways. First, we provide a fully quantum treatment of this behavior and, second, we show that the orientation in chiral molecules is long-lived. Fully quantum studies on laser-driven PPO are provided and compared to classical results. Two implementations of the laser fields with twisted polarization are considered - a pair of delayed cross-polarized pulses, and an optical centrifuge. The laser-induced orientation is shown to be robust against normally detrimental temperature effects.
The paper is organized as follows. In Section II, the conditions needed for long-lasting field-free orientation in a pulse-excited molecular ensemble are discussed, and we show that chiral molecules excited by a laser field with twisted polarization satisfy these conditions. In Section III, the results of classical and fully quantum simulations of the enantio-selective orientation at thermal conditions are provided and compared with one another. Section IV concludes the paper.
These results substantiate, quantum mechanically, the classical expectation (Gershnabel2018, ; Tutunnikov2018, ) that the orientation does not disappear after the excitation, but stays at an approximately constant level long after the end of the laser field.
II Classical Analysis
In this section we use classical mechanics to analyze conditions leading to the existence of permanent orientation in a gas of field-free rotating asymmetric-top molecules. We show that excitation of an isotropic ensemble of chiral molecules by twisted fields satisfies these conditions.
Background. The Binet construction (Goldstein, ; LandauLifshitzMechanics, ) allows one to classify the trajectories traversed by the angular momentum vector in the molecule-fixed frame defined by the three principal axes of moment of inertia tensor. Figure 1(a) shows one of the enantiomers (right handed, (R))of our example molecule, propylene oxide (, or PPO) with two sets of axes: principal axes of moment of inertia and polarizability tensors. Hereafter, the letters , , refer to the inertia principal frame, whereas the numbers 1, 2, 3 refer to the polarizability principal frame.
We briefly describe the construction. Free motion of an asymmetric-top rotor has four constants of motion, energy and the three components of angular momentum expressed in an inertial frame (laboratory fixed frame). In the frame of principal axes of the inertia tensor [see Fig. 1(a)], the allowed trajectories of the angular momentum vector satisfy the following equations
[TABLE]
where are the moments of inertia, are the components of the angular momentum vector , is the rotational energy and is the magnitude of the angular momentum vector. The moments of inertia are ordered according to . The first expression defines an ellipsoid with semi-axes , and ; these coincide with the principal axes of inertia tensor. The second expression defines a spherical shell with radius . The angular momentum vector tip moves on the ellipsoid-spherical shell intersections. In addition to the six stationary rotations about each of the three inertia tensor principal axes, the allowed trajectories can be divided into sets of closed curves, as shown in Fig. 1(b).
For and , the trajectories are divided into two sets of curves () enclosing the poles on and axes, respectively [see Fig. 1(b)]. The sign of is conserved on these trajectories, and depending on the enclosed pole (either on positive or negative side of the axis), we denote the corresponding sets by . Although the Binet construction provides a qualitative picture of trajectories as seen from the molecule fixed frame, it allows to deduce some valuable information about the motion in the laboratory frame, as well. In case of a single top, we can assume that the conserved vector of angular momentum, points along the laboratory axis. For definiteness, we assume trajectory, meaning that moves on a “taco-shaped” curve around one of the poles on the axis [see Fig. 1(b)]. This implies that in the laboratory frame the axis “precesses” around while the sign of the projection remains unchanged, which means preferred orientation of the axis in the course of time. Notably, even in case of an ensemble of asymmetric tops, i.e. when initially angular momenta vectors point in various directions, the permanent orientation is still possible. In that case permanent orientation corresponds to the ensemble-averaged quantities or having a constant sign. This may be achieved by, first, orienting the averaged angular momentum vector (e.g. along axis) and, second, breaking the symmetry between vs trajectories.
**Particular case of twisted polarization. **Here we consider a specific example of an ensemble of chiral molecules excited by pair of delayed cross-polarized laser pulses (Fleischer2009, ; Kitano2009, ; Khodorkovsky2011, ), which constitutes the simplest implementation of a field with twisted polarization. We show that such an excitation leads to the conditions discussed above. Here, the first pulse is polarized along the laboratory axis, while the polarization of the second one is in the plane at to the axis. The first pulse induces alignment of the most polarizable molecular axis. For the sake of simplicity of the qualitative analysis, we assume that after the first pulse all the molecules are perfectly aligned along the axis, and are stationary (Gershnabel2018, ). Initially, we consider only half of all molecules, in which the most polarizable axis points along [see Fig. 2(a)]. Angle is the angle between the axis and the axis. lying in the plane [see Fig. 2(b)]. The aligned molecules are uniformly distributed in . The interaction potential, and torque, induced by a non-resonant optical field are given by
[TABLE]
where the angle brackets denote time averaging over the optical cycle, is the induced dipole, is the polarizability tensor, and is the vector of the electric field. The duration of the laser pulses is assumed to be short as compared to the typical rotational periods of the chiral molecules, therefore the effect of the second pulse is considered in the impulsive approximation, .
The second pulse (twisted with respect to the first one) induces unidirectional rotation in the plane resulting in orientation of along , which constitutes the first criterion for the long-lasting orientation. In addition, there is an orienting torque acting on the aligned most polarizable axis, . To check whether the second criterion for the long-lasting orientation is satisfied, we evaluate the torques along the molecular and axes, ()
[TABLE]
where , , , , and . Here, are the elements of the orthogonal rotation matrix relating the principal polarizability and inertia frames. (See Supplementary Materials for details.) For chiral molecule, the two frames do not align [see Fig. 1(a)] and the off-diagonal elements of differ from zero. In this case, the subdomains of positive and negative in the interval are not equal, which is precisely the required vs asymmetry. When the two frames do align (as in non-chiral molecules), becomes diagonal resulting in torques and . In this case, the vs symmetry is preserved.
It may be shown that Eq. 1 remains the same for molecules with the most polarizable axis, pointing along . For the complimentary enantiomer ((S)-PPO), signs of some of the elements of are reversed, resulting in the opposite orientation direction.
III Numerical Simulations
In this section, we present the results of numerical simulations of the laser driven orientation dynamics of (R)-PPO molecule. We consider two implementations of laser fields with twisted polarization, a pair of delayed cross-polarized pulses (Fleischer2009, ; Kitano2009, ; Khodorkovsky2011, ) and an optical centrifuge (Karczmarek1999, ; Villeneuve2000, ; Yuan2011, ; Korobenko2014, ; Korobenko2018, ). The behavior of an ensemble of molecules is investigated using both classical and quantum mechanical tools. The chiral molecule is modeled as a rigid asymmetric top having anisotropic polarizablity and a dipole moment. Supplementary Table 1 summarizes the molecular properties used.
For the classical simulations, the behavior of a thermal ensemble was simulated using the Monte Carlo approach. Our numerical scheme relies on solving the Euler equations for angular velocities and parametrizing the rotations by quaternions (Art-of-Molecular-Simulation, ). The details of the scheme may be found in (Tutunnikov2018, ). Before the excitation, the molecules are isotropically distributed, and the angular velocities are assigned according to the Boltzmann distribution for a given temperature.
For the quantum simulations, we use the symmetric top wavefunctions as a basis set (Zare, ). Here is the total angular momentum (in Section II we used the label for the modulus of the classical angular momentum), is its projection on the molecule-fixed axis, and is its projection onto the laboratory fixed axis. In this basis, the matrix representing the kinetic energy Hamiltonian has a tridiagonal form, in which the states of different ’s are coupled with states (see Sup. Eq. 8). The interaction potential is expressed in terms of Wigner D-matrices and its matrix elements are evaluated in the basis. The explicit expressions used may be found in the Supplementary Material. Then, a unitary transformation is applied transforming the matrices to the asymmetric-top basis, in which the kinetic energy Hamiltonian becomes diagonal. All the thermally populated (for a given temperature) eigenstates of the chiral molecule are propagated in time separately, and thermal averaging is performed to obtain the observables of interest. The explicit expressions for matrix elements of the observables are presented in the Supplementary Material, as well.
Double pulse excitation. Here we consider excitation of an ensemble of (R)-PPO molecules by a pair of delayed cross-polarized laser pulses. The first pulse is polarized along the laboratory axis, while the polarization of the second one is in the plane at angle to the axis. The electric field of the pulses is given by , where is the maximal amplitude of the electric field of the pulse, FWHM is the full width at half maximum of the intensity profile, is the carrier frequency of the pulse, and is a unit vector along the polarization direction (). The maximal amplitude of the electric field is related to the peak intensity by , where is the permittivity of vacuum and is the speed of light in vacuum.
The simulated classical results span the range of temperatures K with a step of 10 K. The first pulse leads to the alignment of the most polarizable molecular axis along , and the second pulse is applied when this alignment reaches the maximal value. Since this value as well as the time required to reach it are temperature dependent, the moment of application of the second pulse was optimized for each temperature.
Figure 3 shows the ensemble-averaged projection of the molecular dipole on the axis as a function of time, resulting from excitation by both pulses. As may be seen, the second pulse results in a sharp transient orientation of the ensemble-averaged dipole reaching an approximately constant value in the long term. It is important to emphasize that the orientation direction is perpendicular to the plane of twisting and its sign depends on both the sense of twisting and the handedness of the molecule. The sign of orientation is opposite for the second enantiomer (S)-PPO. The classical mechanism explaining the induced enantioselective transient orientation is described elsewhere (Gershnabel2018, ; Tutunnikov2018, ). The maximal amplitudes of both the transient and the persistent dipole are temperature dependent and become lower with increasing the temperature. The long-lasting dipole signals shown in Figure 3 exhibit small-amplitude beats about the asymptotically constant values. These beats stem from the statistical nature of the simulations, and they are inversely proportional to .
To access the influence of the quantum effects on the permanent orientation, we carried out fully quantum mechanical simulations of the dynamics of (R)-PPO molecules kicked by the two pulses. The pulse envelope used in the quantum simulations, is given by Sup. Eq. 6. The difference between this envelope and the Gaussian one used in the classical simulation is insignificant, as the pulses are short and the difference in their integrals is negligible (). The rotational temperature was set to K. The delay of the second pulse was adjusted to the moment of maximal alignment of the most polarizable axis towards the axis, as measured using the alignment factor (see Sup. Eq. 23). Depending on the initial conditions, the pulses excite the molecules to the states with up to , with the mean value of about , which is above the average thermal value of . The short-time dipole signal (see Sup. Eq. 18) is shown in Fig. 4(a) with its classical counterpart for comparison. Shortly after the pulse, the polarization peaks at about . In contrast to the classically predicted permanent steady-state dipole signal, the quantum curve exhibits beats, spaced by ps (the first few). These quantum beats are a well-known phenomenon for quantum rotors (Felker1992, ). However, the long-term time-averaged signal differs from zero, see Figure 4(b). The inset of the figure shows the time average, as defined in the figure’s caption.
Optical centrifuge excitation. Optical centrifuge is a laser pulse, whose linear polarization undergoes an accelerated rotation around its propagation direction (Karczmarek1999, ; Villeneuve2000, ; Yuan2011, ; Korobenko2014, ; Korobenko2018, ). We model the electric field of such a pulse by
[TABLE]
where is the angular acceleration and is the envelope (dashed curve, Fig. 5 and 6), for explicit expression see Sup. Eq. 7. As in the previous case, we start with the results of the classical simulations. Figure 5 shows the ensemble-averaged projection of the molecular dipole on the axis as a function of time, resulting from excitation by the optical centrifuge. The temperature changes in steps of 10 K in the range of K.
Akin to the double pulse scheme, excitation by the optical centrifuge results in enantioselective dipole signal. The signal drops down when the centrifuge is switched off after 20 ps. However, the orientation signal does not vanish completely, but levels off at a non-zero value. Although the amplitude of the transient signal is higher in the case of the double pulse excitation, the amplitudes of the long-time signals are higher in this scenario, making the optical centrifuge more efficient in inducing the permanent dipole orientation. The amplitudes of the signals, both during the driven and field-free dynamics periods are temperature dependent and decrease with temperature.
To investigate the long-lasting dynamics of the induced dipole moment, we carried out a fully quantum mechanical simulation of the centrifuge-driven (R)-PPO rotational dynamics. Centrifuge pulse had the same parameters as shown in the caption to Fig. (5), while the rotational temperature was set to K. Such a pulse excites angular momenta of up to , with the mean excitation of about , which is higher as compared to the previous double-pulse excitation example, and significantly above the average thermal value of . Higher angular momentum results in a better quantum-classical agreement, as compared to the double pulse excitation. The short-time dipole signal is shown in Fig. 6(a). During the pulse, the polarisation peaks at about , and it stays practically constant at the level of after the end of the pulse, with a small-amplitude beating structure present at long times. For comparison, the classical signal obtained under the same conditions is plotted and the correspondence with the quantum results is very good up to ps. On the nanosecond time-scale [Fig. 6(b)], one can observe multiple beats, however the coarse-grained time-averaged quantum signal remains positive, and almost constant as the classically predicted one.
Dynamical Tunneling. Due to the coupling of different -states by the rotational Hamiltonian (see Sup. Eq. 8), the quantum mechanical rigid asymmetric top does not have eigenstates with the angular momentum being oriented within the molecular frame. As a consequence, any state that is internally oriented at some point in time can not be an eigenstate and will oscillate between being oriented and anti-oriented, an effect known as dynamical tunneling (Keshavamurthy2011, ). In other words, a clockwise internal rotation would eventually become counter-clockwise, and vice versa. This leads to a breakdown of quantum-classical analogy, as the classical model introduced in Sec. II assumes a strict separation of clockwise and counter-clockwise rotation (no interchange between and ). Therefore, one would expect no permanent orientation after turn-off of all external fields for a quantum mechanical chiral rotor.
Yet, in spite of the dynamical tunneling, our simulations show a significant long-time orientation of the quantum rotors, equal in magnitude to the classical model. The reason is that the dynamical tunneling time grows very fast with the angular momentum (Harter1984, ), being larger the more the angular momentum is (anti-)oriented along the or axis. Indeed, already for , the tunneling time may exceed microseconds (Harter1984, ), explaining why orientation was observed in the quantum simulations on a nanosecond time-scale. As a matter of fact, the slight decrease of the base line visible in Fig. 6b (thick red line) is possibly a signature of the dynamical tunneling.
At this point one should also note that whilst dynamical tunneling would be of greater importance on longer time-scales than the ones presented here, other effects (neglected here), like rovibrational couplings, hyperfine structure (Thomas2018, ) and intermolecular collisions are likely of higher importance and would probably bury any signs of the dynamical tunneling in an actual experiment.
**Extrapolation to room temperature. **Due to the statistical nature of our classical simulations on one hand and basis size limitations of the quantum simulation on the other, direct simulations at room temperature are highly numerically demanding, therefore we resort to extrapolation. Figure 7 shows the average long term value of the classically calculated , denoted by as a function of temperature on the double logarithmic scale. The error bars for are obtained by the standard error propagation formula (Ku1966, ) based on the variance of . The points are fit by a polynomial and each point is assigned a weight equal to inverse of its variance. The fit allows to extrapolate the results to temperatures beyond those simulated. The predicted value of the permanent dipole at room temperature is and , for the double pulse and centrifuge excitation schemes, respectively (see Fig. 7). Because of the relatively high vapor pressure of propylene oxide at room temperature (vapor, ), even such modest values of the mean molecular dipole moment result in a sizable macroscopic polarization in the gas sample, and cause a collective electric field of about 0.05 V/m in the focal region of a laser beam focused to a spot of and having the Rayleigh range of about . The magnitude and longevity of this field (and the corresponding light-induced voltage) favor its detection using high-speed nanosecond electronics.
The appearance of long-time orientation in a gas sample can be detected by means of Coulomb explosion (Pitzer2013, ; Herwig2013, ; Christensen2015, ; Pitzer2018, ; Fehre2019, ; Milner2019, ) and nonlinear optics through high harmonic generation of various orders (Kamta_2005, ; Frumker2012, ; Frumker2012b, ; Kraus2014, ), especially by means of the second harmonic generation. Another approach to detecting the induced orientation is by measuring THz emission due to free induction decay of coherently oscillating molecular dipoles (Harde1991, ; Fleischer2011, ; Babilotte2016, ).
IV Conclusions
Summarizing, we have demonstrated long-lived orientation of chiral molecules by laser fields with twisted polarization using both classical mechanics and fully quantum-mechanical treatments. The problem was analyzed using propylene oxide molecules, as an example, and considering two different implementations of the twisted optical field: (i) a pair of delayed cross-polarized laser pulses, and (ii) an optical centrifuge. We found very good agreement between the classical and quantum approaches over a wide range of experimentally relevant parameters, and over an extended time range. Significantly, we demonstrated the novel chiral phenomenon of persistent molecular orientation lasting long after the end of the exciting pulses. This counterintuitive effect was first conjectured in our previous papers (Gershnabel2018, ; Tutunnikov2018, ) utilizing a classical approach, but its validity is now verified on a much longer time scale (several orders of magnitude longer) where the use of quantum treatment is unavoidable. The sign of the oriented dipole moment depends on both the sense of polarization twisting and the handedness of the molecule. This long-lasting orientation provides new modalities for detecting molecular chirality with the help of optical harmonics generation, or direct high-speed measurements of electric fields caused by laser-induced macroscopic polarization of the gas. Moreover, it is well known that optical pre-orientation of molecules affects their deflection by inhomogeneous fields (see e.g. (Gershnabel2011a, ; Gershnabel2011b, ; Yachmenev2019, ), references therein, and recent reviews (Fleischer2012, ; Lemeshko2013, ; Chang2015, )). Enantioselective orientation effects considered in this paper may open up new avenues for separation of molecular enantiomers.
We note that ongoing experiments in Milner’s group show initial signals of the long-time enantioselective orientation, supporting our theoretical predictions. A report on these results and the corresponding theoretical analysis is in preparation (LongChiralExp_tobepub, ).
Acknowledgments
The authors appreciate useful discussions with A. A. Milner and V. Milner. This work was supported by the Israel Science Foundation (Grant No. 746/15), the ISF-NSFC joint research program (Grant No. 2520/17), and by Natural Sciences and Engineering Research Council of Canada grant to PB. IA acknowledges support as the Patricia Elman Bildner Professorial Chair, and thanks the UBC Department of Physics & Astronomy for hospitality extended to him during his sabbatical stay. This research was made possible in part by the historic generosity of the Harold Perlman Family.
References
- [1]
F. A. Cotton.
Chemical Applications of Group Theory.
John Wiley & Sons, Hoboken, NJ, USA, 3rd edition, 1990.
- [2]
G. H. Wagnière.
On Chirality and the Universal Asymmetry: Reflections on Image and Mirror Image.
Wiley-VCH, 2007.
- [3]
M. Shapiro and P. Brumer.
Quantum Control of Molecular Processes.
Wiley-VCH, 2nd edition, 2012.
- [4]
P. Brumer, E. Frishman, and M. Shapiro.
Principles of electric-dipole-allowed optical control of molecular chirality.
Phys. Rev. A, 65:015401, 2002.
- [5]
C. Lux, M. Wollenhaupt, T. Bolze, Q. Liang, J. Köhler, C. Sarpe, and T. Baumert.
Circular dichroism in the photoelectron angular distributions of camphor and fenchone from multiphoton ionization with femtosecond laser pulses.
Angew. Chem, 51(20):5001–5005, 2012.
- [6]
D. Patterson, M. Schnell, and J. M. Doyle.
Enantiomer-specific detection of chiral molecules via microwave spectroscopy.
Nature, 497(7450):475–477, 2013.
- [7]
M. Pitzer, M. Kunitski, A. S. Johnson, T. Jahnke, H. Sann, F. Sturm, L. Ph. H. Schmidt, H. Schmidt-Böcking, R. Dörner, J. Stohner, J. Kiedrowski, M. Reggelin, S. Marquardt, A. Schießer, R. Berger, and M. S. Schöffler.
Direct determination of absolute molecular stereochemistry in gas phase by Coulomb explosion imaging.
Science, 341(6150):1096–1100, 2013.
- [8]
P. Herwig, K. Zawatzky, M. Grieser, O. Heber, B. Jordon-Thaden, C. Krantz, O. Novotný, R. Repnow, V. Schurig, D. Schwalm, Z. Vager, A. Wolf, O. Trapp, and H. Kreckel.
Imaging the absolute configuration of a chiral epoxide in the gas phase.
Science, 342(6162):1084–1086, 2013.
- [9]
C. S. Lehmann, N. B. Ram, I. Powis, and M. H. M. Janssen.
Imaging photoelectron circular dichroism of chiral molecules by femtosecond multiphoton coincidence detection.
J. Chem. Phys, 139(23):234307, 2013.
- [10]
M. H. M. Janssen and I. Powis.
Detecting chirality in molecules by imaging photoelectron circular dichroism.
Phys. Chem. Chem. Phys., 16:856–871, 2014.
- [11]
D. Patterson and M. Schnell.
New studies on molecular chirality in the gas phase: Enantiomer differentiation and determination of enantiomeric excess.
Phys. Chem. Chem. Phys., 16:11114–11123, 2014.
- [12]
A. Steinbacher, P. Nuernberger, and T. Brixner.
Optical discrimination of racemic from achiral solutions.
Phys. Chem. Chem. Phys., 17:6340–6346, 2015.
- [13]
C. Lux, A. Senftleben, C. Sarpe, M. Wollenhaupt, and T. Baumert.
Photoelectron circular dichroism observed in the above-threshold ionization signal from chiral molecules with femtosecond laser pulses.
J. Phys. B, 49(2):02LT01, 2015.
- [14]
L. Christensen, J. H. Nielsen, C. S. Slater, A. Lauer, M. Brouard, and H. Stapelfeldt.
Using laser-induced Coulomb explosion of aligned chiral molecules to determine their absolute configuration.
Phys. Rev. A, 92:033411, 2015.
- [15]
A. Kastner, C. Lux, T. Ring, S. Züllighoven, C. Sarpe, A. Senftleben, and T. Baumert.
Enantiomeric excess sensitivity to below one percent by using femtosecond photoelectron circular dichroism.
ChemPhysChem, 17(8):1119–1122, 2016.
- [16]
S. Beaulieu, A. Comby, D. Descamps, B. Fabre, G. A. Garcia, R. Géneaux, A. G. Harvey, F. Légaré, Z. Mašín, L. Nahon, A. F. Ordonez, S. Petit, B. Pons, Y. Mairesse, O. Smirnova, and V. Blanchet.
Photoexcitation circular dichroism in chiral molecules.
Nat. Phys., 14(5):484–489, 2018.
- [17]
M. Pitzer, R. Berger, J. Stohner, R. Dörner, and M. Schöffler.
Investigating absolute stereochemical configuration with Coulomb explosion imaging.
Chimia, 72(6):384–388, 2018.
- [18]
K. Fehre, S. Eckart, M. Kunitski, M. Pitzer, S. Zeller, C. Janke, D. Trabert, J. Rist, M. Weller, A. Hartung, L. Ph. H. Schmidt, T. Jahnke, R. Berger, R. Dörner, and M. S. Schöffler.
Enantioselective fragmentation of an achiral molecule in a strong laser field.
Sci. Adv., 5(3), 2019.
- [19]
O. Neufeld, D. Ayuso, P. Decleva, M. Y. Ivanov, O. Smirnova, and O. Cohen.
Ultrasensitive chiral spectroscopy by dynamical symmetry breaking in high harmonic generation.
Phys. Rev. X, 9:031002, 2019.
- [20]
S. Rozen, A. Comby, E. Bloch, S. Beauvarlet, D. Descamps, B. Fabre, S. Petit, V. Blanchet, B. Pons, N. Dudovich, and Y. Mairesse.
Controlling subcycle optical chirality in the photoionization of chiral molecules.
Phys. Rev. X, 9:031004, 2019.
- [21]
M. Leibscher, T. F. Giesen, and C. P. Koch.
Principles of enantio-selective excitation in three-wave mixing spectroscopy of chiral molecules.
J. Chem. Phys, 151(1):014302, 2019.
- [22]
M. Shapiro, E. Frishman, and P. Brumer.
Coherently controlled asymmetric synthesis with achiral light.
Phys. Rev. Lett., 84:1669–1672, 2000.
- [23]
A. Yachmenev and S. N. Yurchenko.
Detecting chirality in molecules by linearly polarized laser fields.
Phys. Rev. Lett., 117:033001, 2016.
- [24]
E. Gershnabel and I. Sh. Averbukh.
Orienting asymmetric molecules by laser fields with twisted polarization.
Phys. Rev. Lett., 120:083204, 2018.
- [25]
I. Tutunnikov, E. Gershnabel, S. Gold, and I. Sh. Averbukh.
Selective orientation of chiral molecules by laser fields with twisted polarization.
J. Phys. Chem. Lett, 9(5):1105–1111, 2018.
- [26]
S. Fleischer, Y. Khodorkovsky, Y. Prior, and I. Sh. Averbukh.
Controlling the sense of molecular rotation.
New J. Phys., 11(10):105039, 2009.
- [27]
K. Kitano, H. Hasegawa, and Y. Ohshima.
Ultrafast angular momentum orientation by linearly polarized laser fields.
Phys. Rev. Lett., 103:223002, 2009.
- [28]
Y. Khodorkovsky, K. Kitano, H. Hasegawa, Y. Ohshima, and I. Sh. Averbukh.
Controlling the sense of molecular rotation: Classical versus quantum analysis.
Phys. Rev. A, 83:023423, 2011.
- [29]
O. Korech, U. Steinitz, R. J. Gordon, I. Sh. Averbukh, and Y. Prior.
Observing molecular spinning via the rotational Doppler effect.
Nat. Photonics, 7(9):711–714, 2013.
- [30]
K. Mizuse, K. Kitano, H. Hasegawa, and Y. Ohshima.
Quantum unidirectional rotation directly imaged with molecules.
Sci. Adv., 1(6), 2015.
- [31]
K. Lin, Q. Song, X. Gong, Q. Ji, H. Pan, J. Ding, H. Zeng, and J. Wu.
Visualizing molecular unidirectional rotation.
Phys. Rev. A, 92:013410, 2015.
- [32]
S. Zhdanovich, A. A. Milner, C. Bloomquist, J. Floß, I. Sh. Averbukh, J. W. Hepburn, and V. Milner.
Control of molecular rotation with a chiral train of ultrashort pulses.
Phys. Rev. Lett., 107:243004, 2011.
- [33]
C. Bloomquist, S. Zhdanovich, A. A. Milner, and V. Milner.
Directional spinning of molecules with sequences of femtosecond pulses.
Phys. Rev. A, 86:063413, 2012.
- [34]
Johannes Floß and Ilya Sh. Averbukh.
Molecular spinning by a chiral train of short laser pulses.
Phys. Rev. A, 86:063414, Dec 2012.
- [35]
G. Karras, M. Ndong, E. Hertz, D. Sugny, F. Billard, B. Lavorel, and O. Faucher.
Polarization shaping for unidirectional rotational motion of molecules.
Phys. Rev. Lett., 114:103001, Mar 2015.
- [36]
E. Prost, H. Zhang, E. Hertz, F. Billard, B. Lavorel, P. Bejot, Joseph Zyss, Ilya Sh. Averbukh, and O. Faucher.
Third-order-harmonic generation in coherently spinning molecules.
Phys. Rev. A, 96:043418, Oct 2017.
- [37]
E. Prost, E. Hertz, F. Billard, B. Lavorel, and O. Faucher.
Polarization-based tachometer for measuring spinning rotors.
Opt. Express, 26(24):31839–31849, Nov 2018.
- [38]
J. Karczmarek, J. Wright, P. Corkum, and M. Ivanov.
Optical centrifuge for molecules.
Phys. Rev. Lett., 82:3420–3423, 1999.
- [39]
D. M. Villeneuve, S. A. Aseyev, P. Dietrich, M. Spanner, M. Yu. Ivanov, and P. B. Corkum.
Forced molecular rotation in an optical centrifuge.
Phys. Rev. Lett., 85:542–545, 2000.
- [40]
L. Yuan, S. W. Teitelbaum, A. Robinson, and A. S. Mullin.
Dynamics of molecules in extreme rotational states.
Proc. Natl. Acad. Sci. U. S. A., 108(17):6872–6877, 2011.
- [41]
A. Korobenko, A. A. Milner, and V. Milner.
Direct observation, study, and control of molecular superrotors.
Phys. Rev. Lett., 112:113004, 2014.
- [42]
A. Korobenko.
Control of molecular rotation with an optical centrifuge.
J. Phys. B, 51(20):203001, 2018.
- [43]
A. A. Milner, J. A. M. Fordyce, I. MacPhail-Bartley, W. Wasserman, V. Milner, I. Tutunnikov, and I. Sh. Averbukh.
Controlled enantioselective orientation of chiral molecules with an optical centrifuge.
Phys. Rev. Lett., 122:223201, 2019.
- [44]
H. Goldstein.
Classical mechanics.
Addison Wesley, San Francisco, 2002.
- [45]
L. D. Landau and E. M. Lifshitz.
Mechanics.
Butterworth-Heinemann, Oxford, 3rd edition, 1976.
- [46]
D. C. Rapaport.
The Art of Molecular Dynamics Simulation.
Cambridge University Press, 2nd edition, 2004.
- [47]
R. Zare.
Angular momentum : understanding spatial aspects in chemistry and physics.
Wiley, New York, 1988.
- [48]
P. M. Felker.
Rotational coherence spectroscopy: studies of the geometries of large gas-phase species by picosecond time-domain methods.
J. Phys. Chem., 96(20):7844–7857, 1992.
- [49]
S. Keshavamurthy and P. Schlagheck.
Dynamical tunneling: theory and experiment.
CRC Press, Boca Raton, 2011.
- [50]
W. G. Harter and C. W. Patterson.
Rotational energy surfaces and high- eigenvalue structure of polyatomic molecules.
J. Chem. Phys., 80:4241, 1984.
- [51]
E. F. Thomas, A. A. Søndergaard, B. Shepperson, N. E. Henriksen, and H. Stapelfeldt.
Hyperfine-structure-induced depolarization of impulsively aligned molecules.
Phys. Rev. Lett., 120:163202, Apr 2018.
- [52]
H. H. Ku.
Notes on the use of propagation of error formulas.
J. Res. Nat. Bur. Stand. Sec. C: Eng. Inst., 70C(4):263, 1966.
- [53]
T. R. Bott and H. N. Sadler.
Vapor pressure of propylene oxide.
J. Chem. Eng. Data, 11(1):25–25, 1966.
- [54]
G. L. Kamta, A. D. Bandrauk, and P. B. Corkum.
Asymmetry in the harmonic generation from nonsymmetric molecules.
J. Phys. B, 38(20):L339–L346, sep 2005.
- [55]
E. Frumker, C. T. Hebeisen, N. Kajumba, J. B. Bertrand, H. J. Wörner, M. Spanner, D. M. Villeneuve, A. Naumov, and P. B. Corkum.
Oriented rotational wave-packet dynamics studies via high harmonic generation.
Phys. Rev. Lett., 109:113901, Sep 2012.
- [56]
E. Frumker, N. Kajumba, J. B. Bertrand, H. J. Wörner, C. T. Hebeisen, P. Hockett, M. Spanner, S. Patchkovskii, G. G. Paulus, D. M. Villeneuve, A. Naumov, and P. B. Corkum.
Probing polar molecules with high harmonic spectroscopy.
Phys. Rev. Lett., 109:233904, Dec 2012.
- [57]
P. M. Kraus, D. Baykusheva, and H. J. Wörner.
Two-pulse field-free orientation reveals anisotropy of molecular shape resonance.
Phys. Rev. Lett., 113:023001, Jul 2014.
- [58]
H. Harde, S. Keiding, and D. Grischkowsky.
THz commensurate echoes: Periodic rephasing of molecular transitions in free-induction decay.
Phys. Rev. Lett., 66:1834–1837, 1991.
- [59]
S. Fleischer, Y. Zhou, R. W. Field, and K. A. Nelson.
Molecular orientation and alignment by intense single-cycle THz pulses.
Phys. Rev. Lett., 107:163603, Oct 2011.
- [60]
P. Babilotte, K. Hamraoui, F. Billard, E. Hertz, B. Lavorel, O. Faucher, and D. Sugny.
Observation of the field-free orientation of a symmetric-top molecule by terahertz laser pulses at high temperature.
Phys. Rev. A, 94:043403, Oct 2016.
- [61]
E. Gershnabel and I. Sh. Averbukh.
Electric deflection of rotating molecules.
J. Chem. Phys., 134(5):054304, 2011.
- [62]
E. Gershnabel and I. Sh. Averbukh.
Deflection of rotating symmetric top molecules by inhomogeneous fields.
J. Chem. Phys., 135(8):084307, 2011.
- [63]
A. Yachmenev, J. Onvlee, E. Zak, A. Owens, and J. Küpper.
Field-induced diastereomers for chiral separation.
arXiv 1905.07166, 2019.
- [64]
S. Fleischer, Y. Khodorkovsky, E. Gershnabel, Y. Prior, and I. Sh. Averbukh.
Molecular alignment induced by ultrashort laser pulses and its impact on molecular motion.
Isr. J. Chem., 52(5):414–437, 2012.
- [65]
M. Lemeshko, R. V. Krems, J. M. Doyle, and S. Kais.
Manipulation of molecules with electromagnetic fields.
Mol. Phys., 111(12-13):1648–1682, 2013.
- [66]
Y.-P. Chang, D. A. Horke, S. Trippel, and J. Küpper.
Spatially-controlled complex molecules and their applications.
Int. Rev. Phys. Chem., 34(4):557–590, 2015.
- [67]
I. Tutunnikov, J. Floß, E. Gershnabel, P. Brumer, I. Sh. Averbukh, A. A. Milner, and V. Milner.
(to be published).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1[1] F. A. Cotton. Chemical Applications of Group Theory . John Wiley & Sons, Hoboken, NJ, USA, 3 rd edition, 1990.
- 2[2] G. H. Wagnière. On Chirality and the Universal Asymmetry: Reflections on Image and Mirror Image . Wiley-VCH, 2007.
- 3[3] M. Shapiro and P. Brumer. Quantum Control of Molecular Processes . Wiley-VCH, 2 nd edition, 2012.
- 4[4] P. Brumer, E. Frishman, and M. Shapiro. Principles of electric-dipole-allowed optical control of molecular chirality. Phys. Rev. A , 65:015401, 2002.
- 5[5] C. Lux, M. Wollenhaupt, T. Bolze, Q. Liang, J. Köhler, C. Sarpe, and T. Baumert. Circular dichroism in the photoelectron angular distributions of camphor and fenchone from multiphoton ionization with femtosecond laser pulses. Angew. Chem , 51(20):5001–5005, 2012.
- 6[6] D. Patterson, M. Schnell, and J. M. Doyle. Enantiomer-specific detection of chiral molecules via microwave spectroscopy. Nature , 497(7450):475–477, 2013.
- 7[7] M. Pitzer, M. Kunitski, A. S. Johnson, T. Jahnke, H. Sann, F. Sturm, L. Ph. H. Schmidt, H. Schmidt-Böcking, R. Dörner, J. Stohner, J. Kiedrowski, M. Reggelin, S. Marquardt, A. Schießer, R. Berger, and M. S. Schöffler. Direct determination of absolute molecular stereochemistry in gas phase by Coulomb explosion imaging. Science , 341(6150):1096–1100, 2013.
- 8[8] P. Herwig, K. Zawatzky, M. Grieser, O. Heber, B. Jordon-Thaden, C. Krantz, O. Novotný, R. Repnow, V. Schurig, D. Schwalm, Z. Vager, A. Wolf, O. Trapp, and H. Kreckel. Imaging the absolute configuration of a chiral epoxide in the gas phase. Science , 342(6162):1084–1086, 2013.