Robust field-dressed spectra of diatomics in an optical lattice
Mariusz Pawlak, Tam\'as Szidarovszky, G\'abor J. Hal\'asz, \'Agnes, Vib\'ok

TL;DR
This study investigates the absorption spectra of Na2 molecules in an optical lattice, revealing a surprisingly robust spectrum insensitive to the dressing field, with subtle signatures of light-induced conical intersections emerging under certain conditions.
Contribution
It demonstrates the robustness of the absorption spectrum of diatomic molecules in an optical lattice despite the presence of light-induced conical intersections and explores conditions where their signatures appear.
Findings
Spectra are insensitive to dressing field intensity and wavelength.
LICI signatures are subtle and require artificial broadening to be observed.
The absorption spectrum remains robust despite nonadiabatic phenomena.
Abstract
The absorption spectra of the cold Na2 molecule dressed by a linearly polarized standing laser wave is investigated. In the studied scenario the rotational motion of the molecules is frozen while the vibrational and translational degrees of freedom are accounted for as dynamical variables. In such a situation a light-induced conical intersection (LICI) can be formed. To measure the spectra a weak field is used whose propagation direction is perpendicular to the direction of the dressing field but has identical polarization direction. Although LICIs are present in our model, the simulations demonstrate a very robust absorption spectrum, which is insensitive to the intensity and the wavelength of the dressing field and which does not reflect clear signatures of light-induced nonadiabatic phenomena related to the strong mixing between the electronic, vibration and translational motions.…
Click any figure to enlarge with its caption.
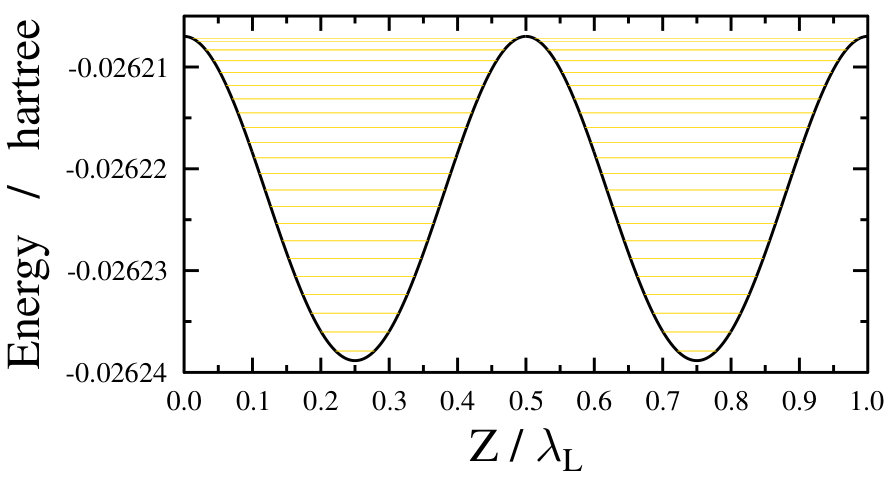 Figure 1
Figure 1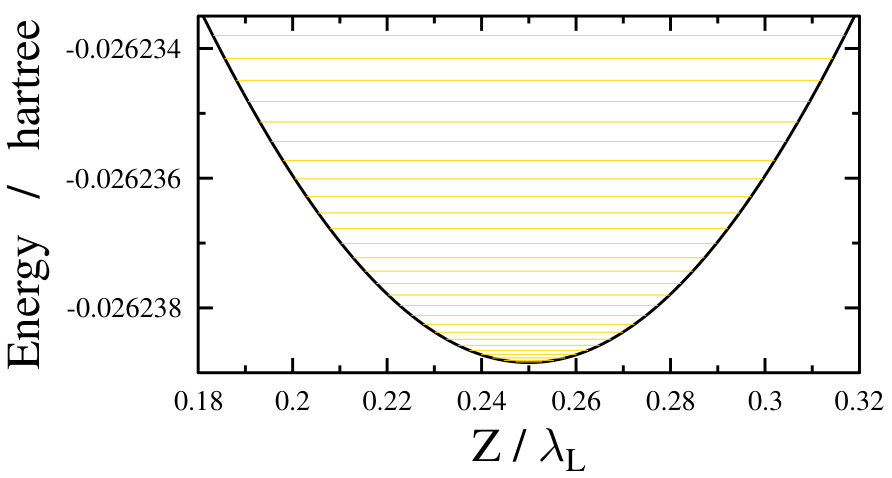 Figure 2
Figure 2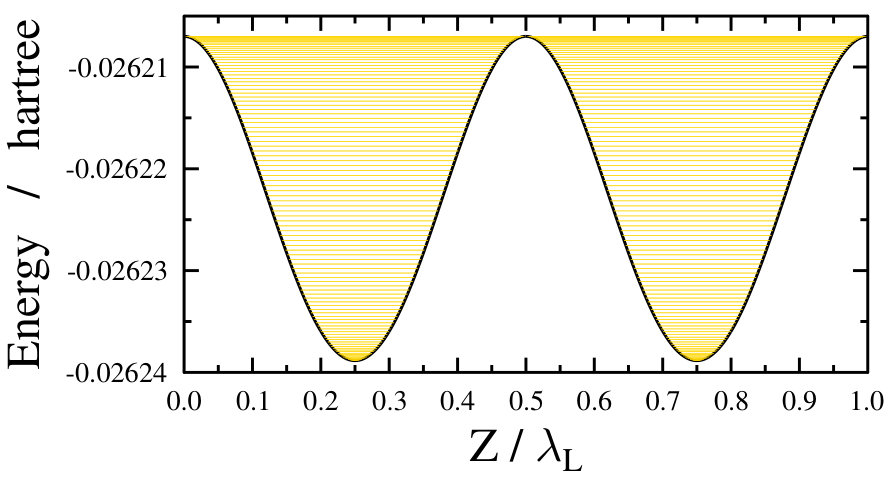 Figure 3
Figure 3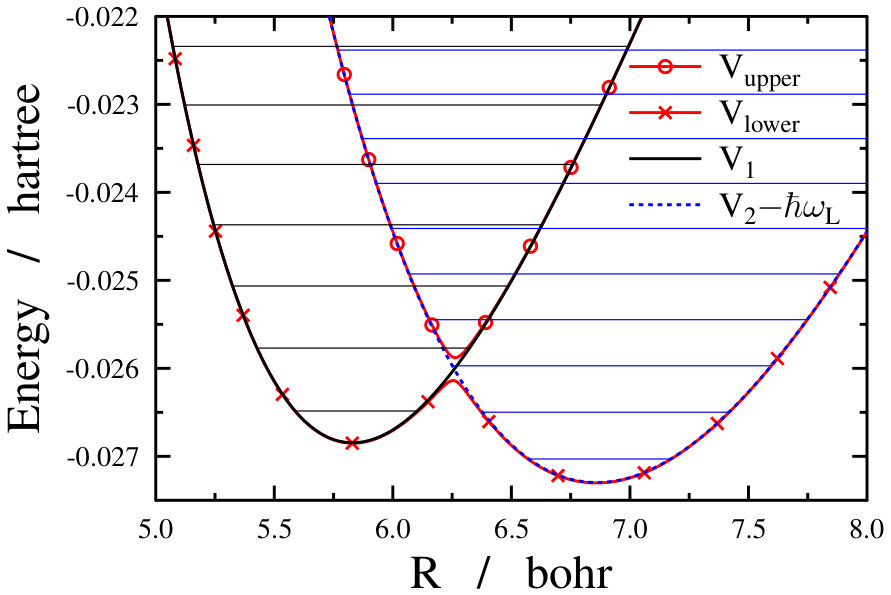 Figure 4
Figure 4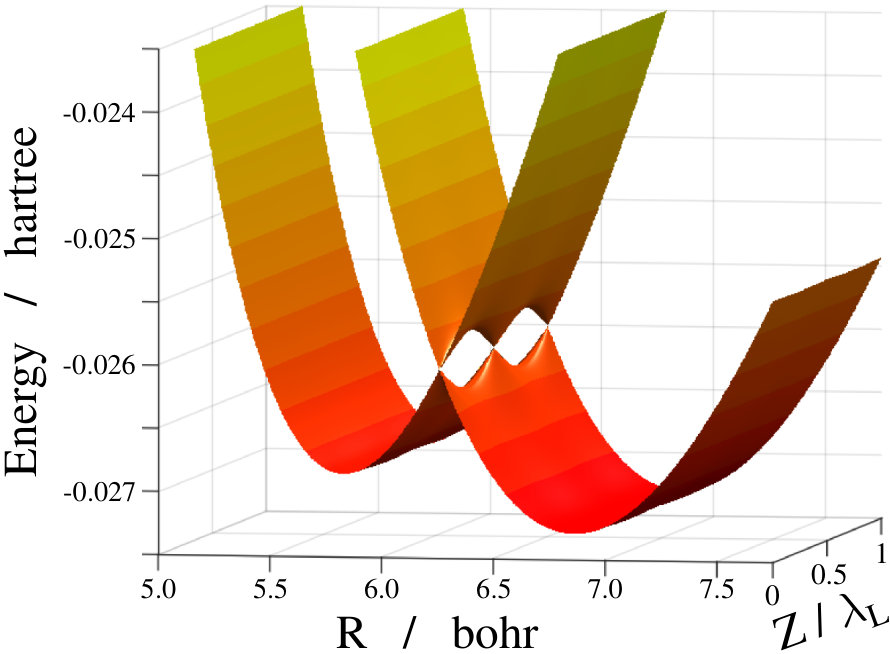 Figure 5
Figure 5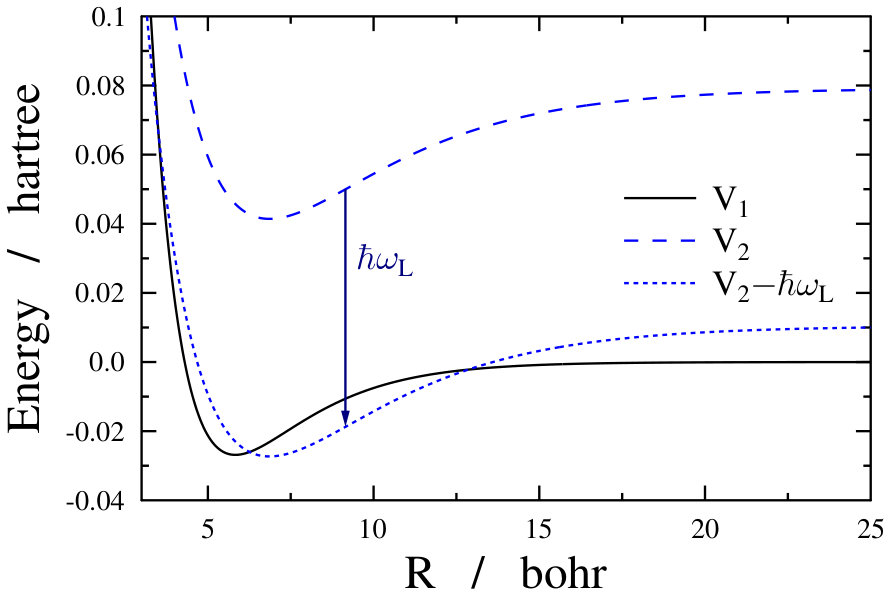 Figure 6
Figure 6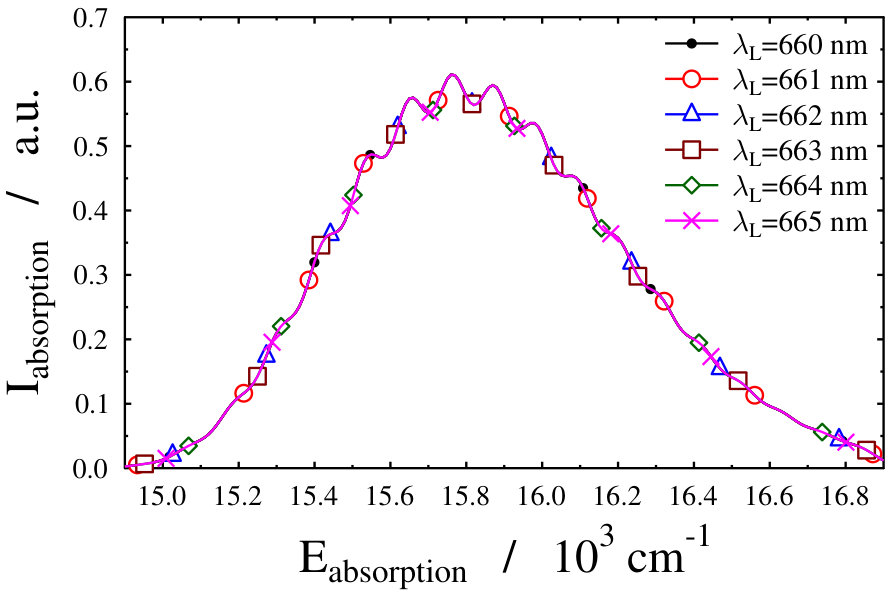 Figure 7
Figure 7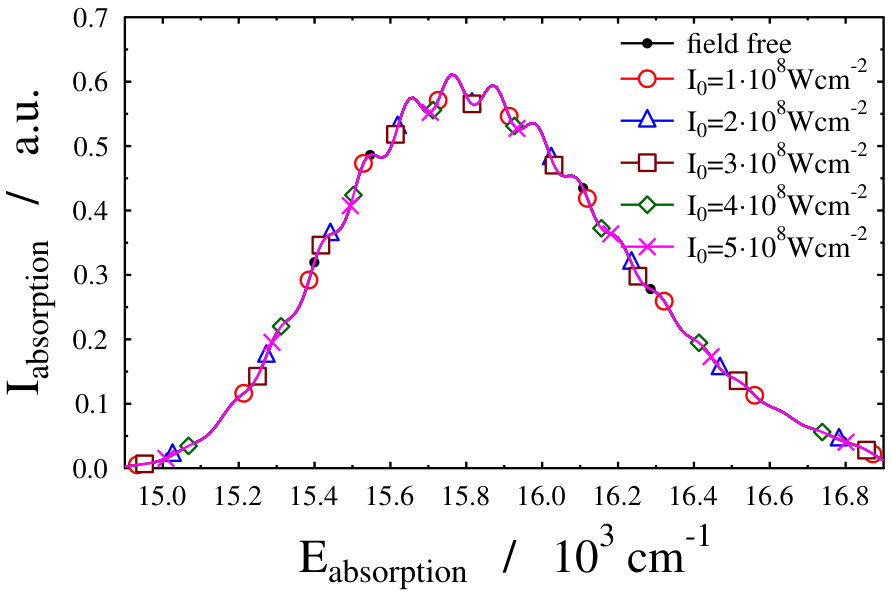 Figure 8
Figure 8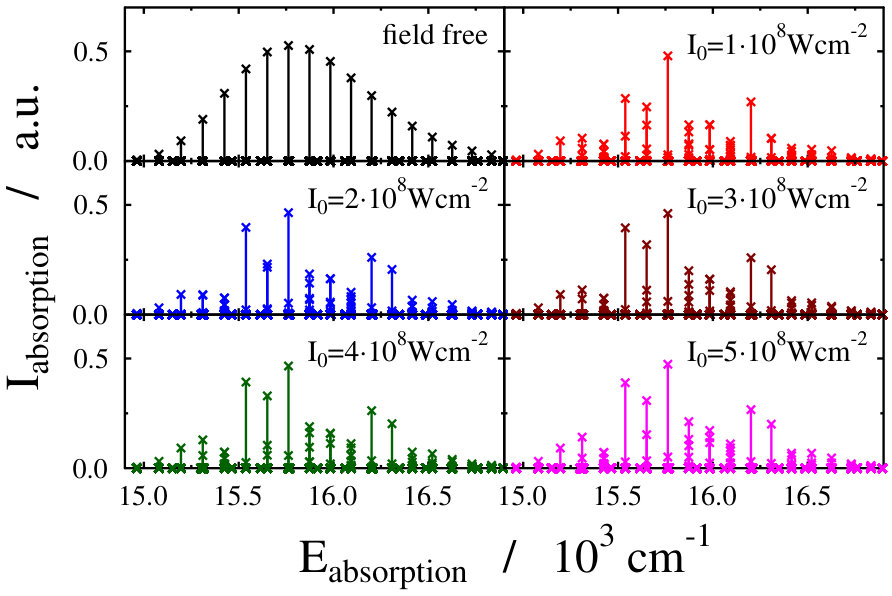 Figure 9
Figure 9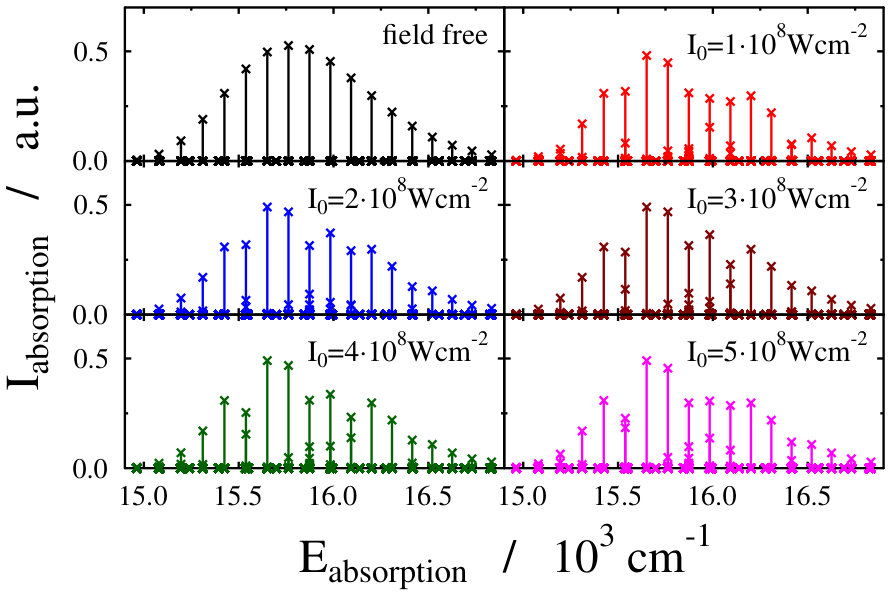 Figure 10
Figure 10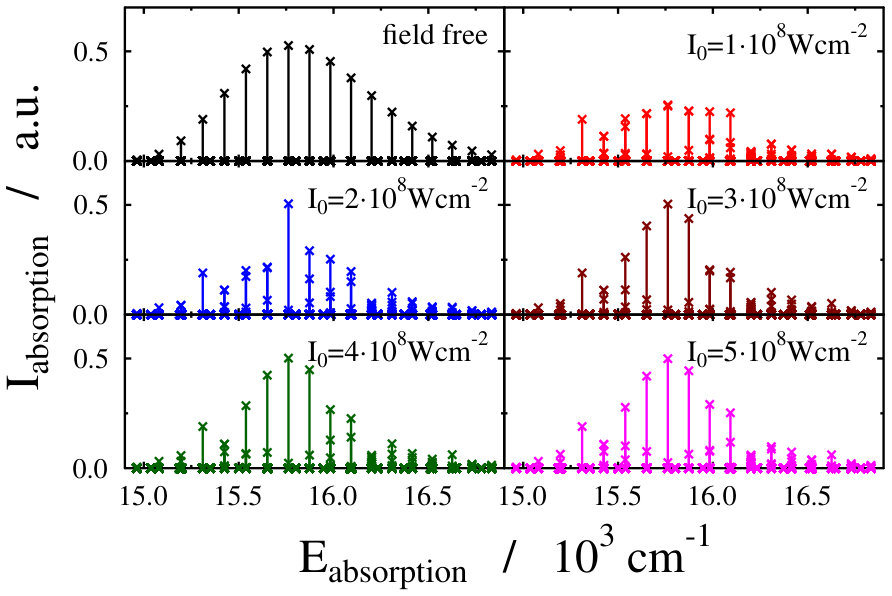 Figure 11
Figure 11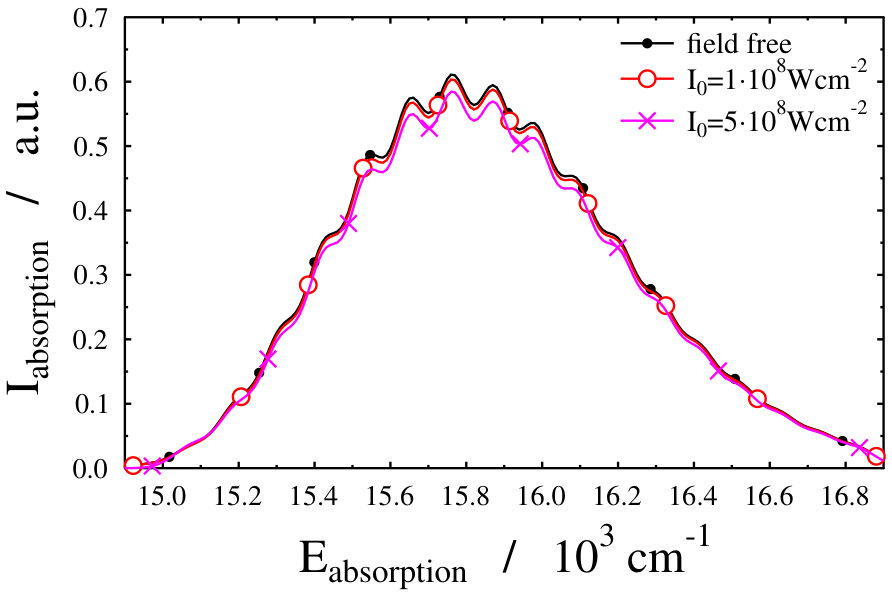 Figure 12
Figure 12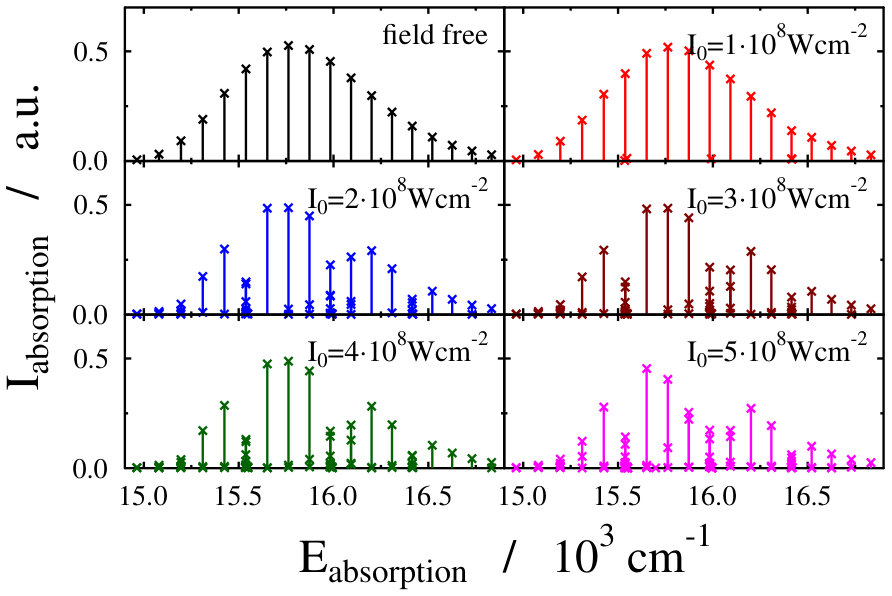 Figure 13
Figure 13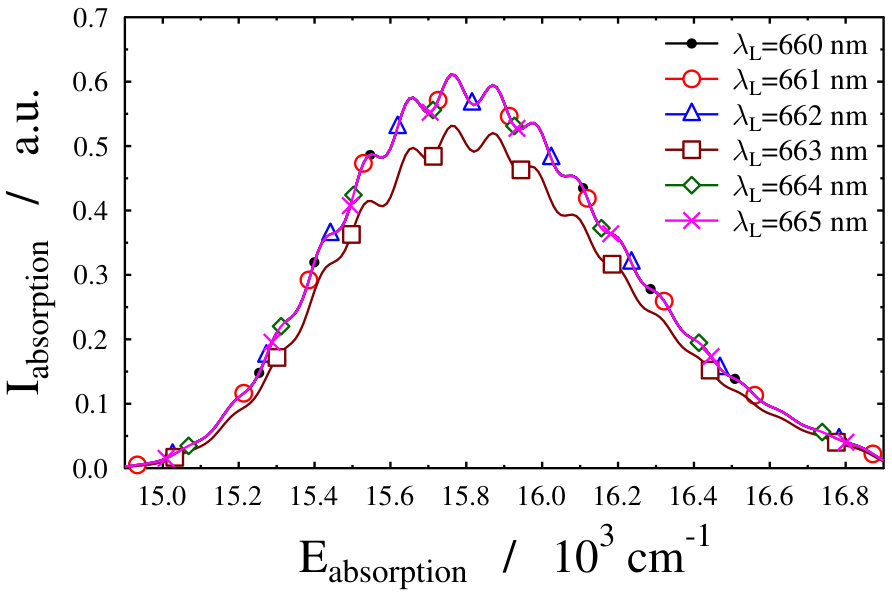 Figure 14
Figure 14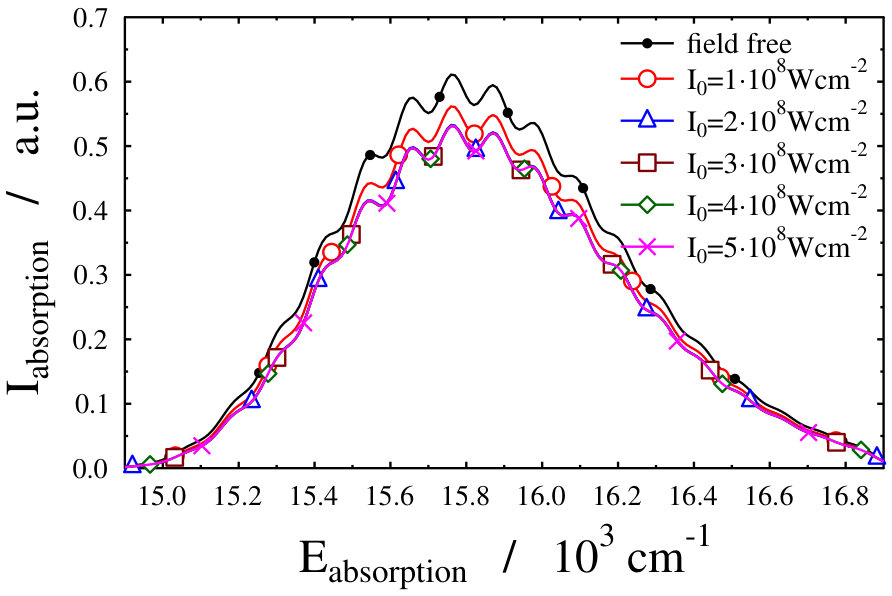 Figure 15
Figure 15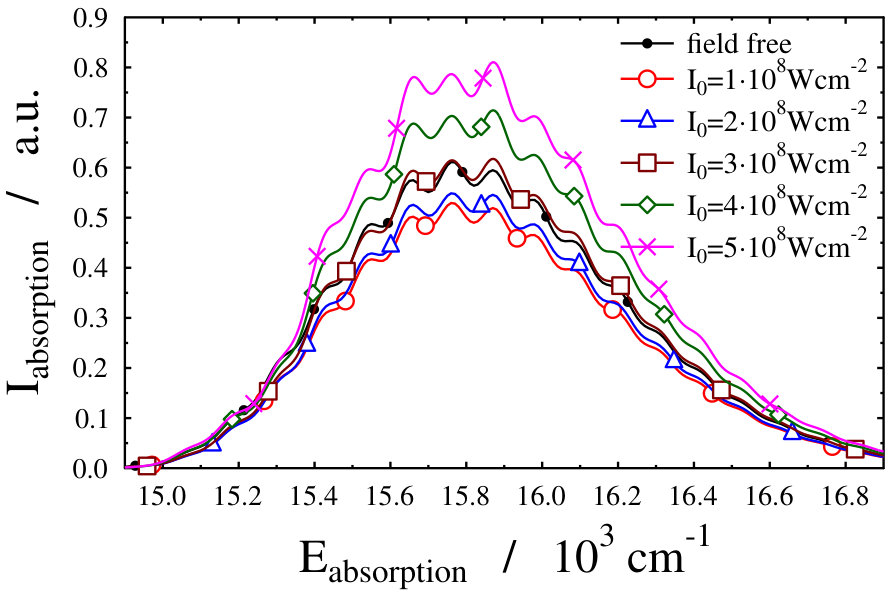 Figure 16
Figure 16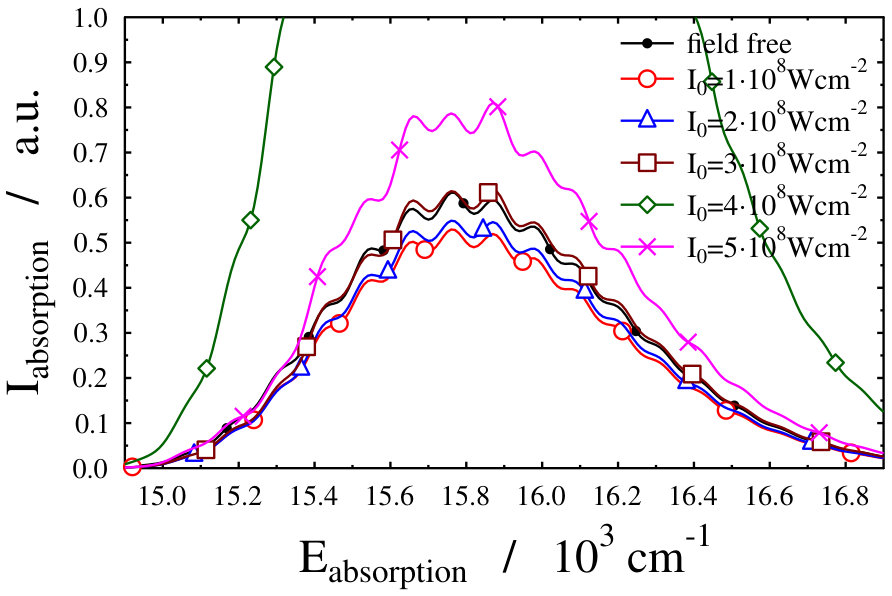 Figure 17
Figure 17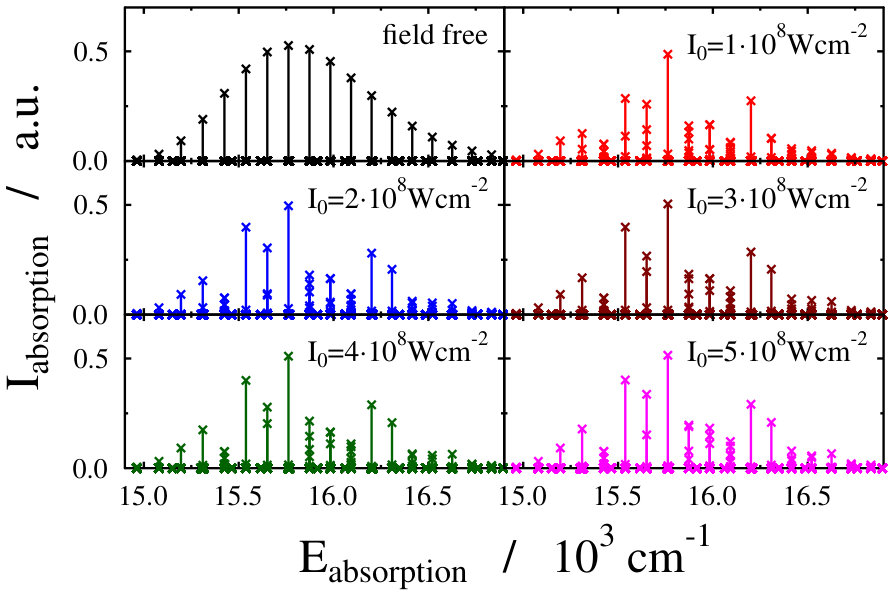 Figure 18
Figure 18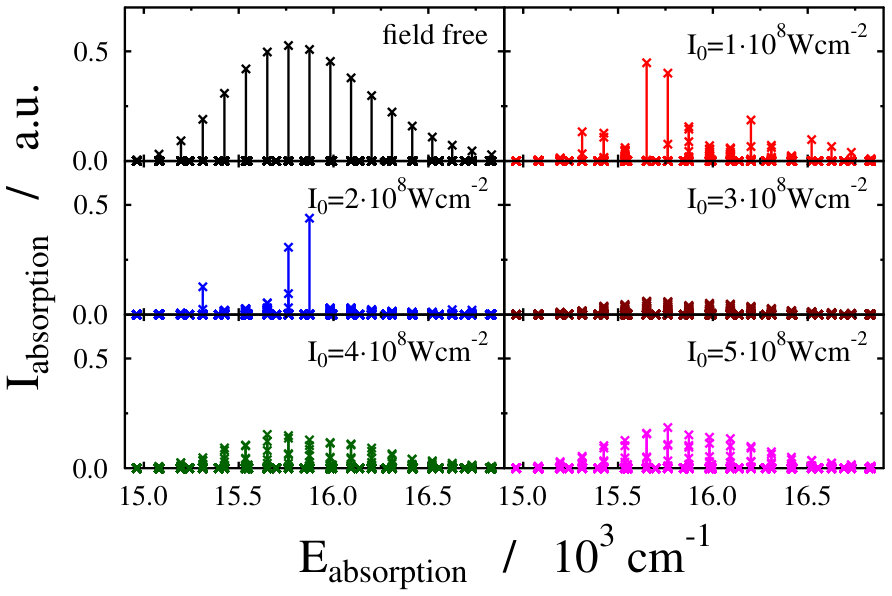 Figure 19
Figure 19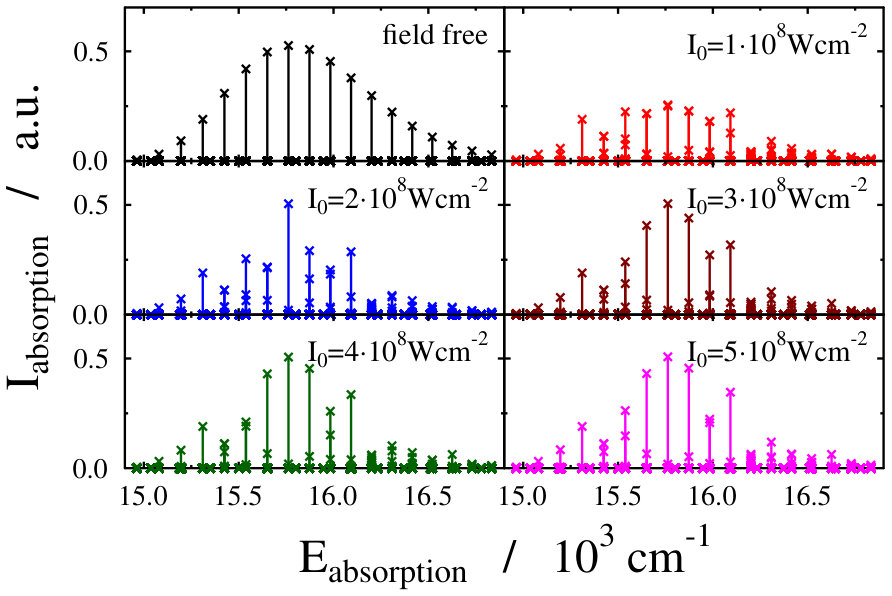 Figure 20
Figure 20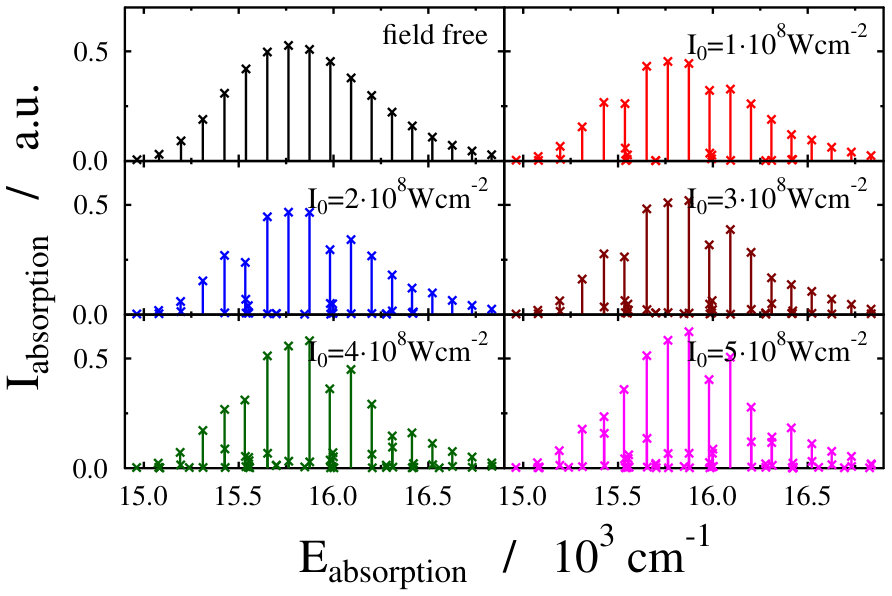 Figure 21
Figure 21Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Robust field-dressed spectra of diatomics in an optical
lattice
Mariusz Pawlak1, Tamás Szidarovszky2, Gábor J. Halász3 and Ágnes Vibók4,5
1Faculty of Chemistry, Nicolaus Copernicus University in Toruń, Gagarina 7, 87-100 Toruń, Poland
2Laboratory of Molecular Structure and Dynamics, Institute of Chemistry, ELTE Eötvös Loránd University and MTA-ELTE Complex Chemical Systems Research Group, Pázmány Péter sétány 1/A, H-1117 Budapest, Hungary
3Department of Information Technology, University of Debrecen, H-4002 Debrecen, PO Box 400, Hungary
4Department of Theoretical Physics, University of Debrecen, H-4002 Debrecen, PO Box 400, Hungary
5ELI-ALPS, ELI-HU Non-Profit Ltd, H-6720 Szeged, Dugonics tér 13, Hungary
Abstract
The absorption spectra of the cold molecule dressed by a linearly polarized standing laser wave is investigated. In the studied scenario the rotational motion of the molecules is frozen while the vibrational and translational degrees of freedom are accounted for as dynamical variables. In such a situation a light-induced conical intersection (LICI) can be formed. To measure the spectra a weak field is used whose propagation direction is perpendicular to the direction of the dressing field but has identical polarization direction. Although LICIs are present in our model, the simulations demonstrate a very robust absorption spectrum, which is insensitive to the intensity and the wavelength of the dressing field and which does not reflect clear signatures of light-induced nonadiabatic phenomena related to the strong mixing between the electronic, vibration and translational motions. However, by widening artificially the very narrow translational energy level gaps, the fingerprint of the LICI appears to some extent in the spectrum.
I Introduction
Nonadiabatic processes associated with avoided crossings (ACs) or conical intersections (CIs) Neumann ; Lenz1 ; Yarkony ; Baer1 ; Graham1 ; Domcke ; Baer2 always involve nuclear dynamics on at least two coupled potential energy surfaces (PESs) and thus can not be treated within the Born-Oppenheimer (BO) approximation Born-Oppenheimer . In this case the electronic states are coupled by the nuclear motion, and the energy exchange between the fast electrons and the slow moving nuclei may become significant. CIs between electronic PESs play a key mechanistic role in chemistry, physics and biology, most notably in photochemistry and photobiology. In several important cases like fragmentation, charge transfer, isomerization processes of polyatomics or radiationless relaxation of the excited electronic states martin ; kukura ; Hahn ; Schultz ; Lan ; Olivucci ; Tamas2 , the CIs can provide very efficient decay channel on femtosecond time scale. CIs show up in different contexts as well, such as in optical Born ; Berry ; Peleg and solid state physics Simon ; Levy . Furthermore, exceptional points – degeneracies between continuum states – can also be found in nature Lenz2 ; Lenz3 .
CIs can exist only if the molecule possesses at least two independent nuclear vibrational coordinates. In field free diatomics that have only one nuclear degree of freedom CIs are never formed. However, CIs can occur in triatomic systems and they are fairly abundant in truly large polyatomic molecules.
If the laser field is present, then due to the interaction of the diatomics with the field, the rotational nuclear coordinate comes into play and serves as an additional degree of freedom. CIs can be created both by running Lenz4 and by standing laser waves Lenz5 even in diatomic molecules. In the former situation the laser field rotates the molecule due to the interaction of the transition dipole moment of the molecule with the electric field which leads to an effective torque toward the polarization direction of the light and so called “light-induced conical intersections” (LICIs) are formed. The position of these LICIs are determined by the laser frequency while the laser intensity controls the strength of the nonadiabatic coupling. Numerous theoretical Gabi1 ; Andris1 and experimental exp1 ; exp2 studies have confirmed that the LICIs have strong impact on the dynamical Gabi1 ; Andris1 and spectroscopic Lenz4 ; Tamas1 properties of diatomics. Recently the frequency and intensity dependence of the field-dressed rovibronic spectrum of the diatomic Na2 molecule in gas phase was extensively discussed Tamas1 ; Tamas_PRA .
Light-induced nonadiabatic phenomena can also occur in optical lattices, which have been widely applied in atomic and diatomic physics so as to cool, trap and control different properties of atoms and molecules Dominic . The optical lattice is a periodic potential energy landscape that the molecules experience as a result of the standing wave pattern created by the interference of two counter propagating laser beams. The nonadiabatic phenomenon can emerge when the translational motion of the molecule strongly couples with the electronic, vibrational and rotational motions leading to a periodic array of light-induced CIs. In this case the translational motion provides the new dynamical variable which is necessary to form a branching space for diatomics. Historically, the LICIs have been predicted first in optical lattices for the molecule Lenz5 . It was demonstrated that the trapping efficiency of the molecule in its lowest electronic state can be significantly reduced due to the presence of the LICI Lenz5 . Later on the intensively studied cold weakly bound rubidium molecule Bendkowsky ; Koch has been considered which plays an important role in the cold-atom physics. As a results, due to the effect of the LICI a significant localization enhancement of the has been revealed Pawlak . Besides the theoretical studies important experimental papers have also discussed the nonadiabatic effects Tanya0 and the spectra of ultracold molecules in an optical lattice Tanya1 ; Tanya2 .
In the present work we focus again on the two-atom homonuclear cold sodium molecule and discuss the standing field-dressed spectra of this object. Our aims are two-fold. First, by investigating the frequency and intensity dependence of the spectra, we would like to explore to what extent the spectra of a non-rotating dressed by an optical lattice and a freely rotating dressed by a running laser wave are similar or different. Second, how the impact of the light-induced nonadiabatic phenomena can be visualized on the absorption spectra of the standing field-dressed non-rotating molecule.
The article is arranged as follows. In the next section, we present a brief outline of the theory and the computation details. In the third section results are shown and analyzed. Finally, conclusions are given in the last section.
II Theory and computation details
In the present paper a two step numerical pump-probe simulation is derived to study the weak-field absorption spectrum of the field-dressed homonuclear molecule in an optical lattice.
*First, we specify the field-dressed states of the system by applying a medium intensity “pump” standing laser wave which mixes the field-free eigenstates of the sodium dimer. *The applied standing laser wave can be written as the sum of two counter propagating linearly polarized running waves nimrod :
[TABLE]
where is the amplitude, is the frequency and is the wave vector (k_{L}=$$\omega_{L}/c=2\pi/\lambda_{L}) of the laser field.* *
Second, we determine the dipole transition amplitudes between the field-dressed states in the framework of first-order time-dependent perturbation theory by using a second weak probe pulse.* *
The two relevant electronic states of the molecule which are involved in the numerical calculations are the ground and the first excited states (further denoted as and , respectively). These are characterized by the and potential energy curves (see Fig. 1).
We represent the time-dependent Hamiltonian in the Floquet form Floquet .
Assuming dipole approximation and only one net photon absorption the well known () static dressed state form can be obtained for the Hamiltonian 111In the dressed state representation the interaction between the molecule and the electromagnetic field is obtained by shifting the energy of the appropriate electronic states by (n=1,2,...$$\infty). As one net photon absorption is assumed, :
[TABLE]
Here and are the kinetic energy operators of the molecular rovibrational motion and of the translational motion of the center of mass of the molecule, respectively. These are written in the following form:
[TABLE]
[TABLE]
where is the reduced and is the total mass of the two atoms, respectively, is the squared angular momentum operator associated with diatomic rotations, is the molecular vibrational coordinate, and is the transition dipole moment which is parallel to the molecular axis. denotes the angle between the polarization direction and the direction of the transition dipole and thus one of the angles of rotation of the molecule. The light is propagated along the direction. The potentials and were taken from Ref. Magnier , whereas the transition dipole moment from Ref. Zemke .
The form of the Hamiltonian demonstrated in Eq. (4) helps to understand the essence of the light-induced conical intersection phenomenon. In this representation the laser light shifts the excited potential curve downwards by (see the upper panel of Fig. 1) and a crossing is formed between the diabatic ground and the () shifted diabatic excited potential energy curves (see the middle panel of Fig. 1). After diagonalizing the potential energy matrix the adiabatic potential curves and can be obtained (middle panel of Fig. 1). These two curves can cross each other, giving rise to a light-induced conical intersection whenever the following two conditions are simultaneously fulfilled: (i) ; () ( is an integer) or . The lower panel of Fig. 1 visualizes CIs induced by a laser light for the sodium dimer.
Now, we reduce the three-dimensional problem to a two-dimensional one by assuming that the initial ground electronic and rotational state is coupled with only the first excited electronic and rotational state , . Then, we can obtain the field-dressed (FD) eigenstates and the corresponding quasi-energies by solving the matrix eigenvalue problem with the Hamiltonian of Eq. (4). Therefore, let us build up the Hamiltonian matrix by using basis functions which are the products of plain wave functions describing the translational motion of the system and field-free molecular vibrational eigenfunctions of the ground and first excited electronic states:
[TABLE]
Here
[TABLE]
is the th vibrational eigenfunction of the th electronic state, where and denote the and electronic states, respectively. The functions in Eq. (11) stand for the basis function used for expanding the vibrational states. Moreover
[TABLE]
describes the plain wave functions moving along direction, where is the imaginary unit, , and .
The resulting field-dressed states in the Floquet framework can be obtained as the linear combination of the products of our basis functions, given spherical harmonics, and the Fourier vectors of the Floquet states, namely
[TABLE]
where are the expansion coefficients gained by diagonalizing the Hamiltonian of Eq. (4) after representing it in the basis set of Eq. (10).
Using the field-dressed states of Eq. (LABEL:eq:field-dressed) one can determine the spectrum of the field-dressed molecule. We assume a weak probe pulse whose propagation direction is perpendicular to the direction of the pump pulse but has identical polarization direction. Furthermore, we assume that the transitions induced by this probe pulse can be described by one-photon processes. In this case the transition amplitudes in dipole approximation can be computed in the framework of first-order time-dependent perturbation theory (TDPT) which is a standard procedure of computational molecular spectroscopy Spect1 . Then, the transition amplitude from a initial state to a final state reads
[TABLE]
Here, the unit vector defines the polarization direction of the dressing pump and probe pulses. The first term of the expression stands for absorption while the second term represents the simulated emission spectra. From now on we will focus only on the absorption, therefore the first term will serve as our working formula.
III Results and discussion
We discuss the field-dressed absorption spectrum of the molecule in an optical lattice obtained from an initial state chosen to be a single field-dressed state.
III.1 Adiabatic approximation
Let us first choose the field-dressed state according to the so-called “adiabatic approximation” adiabatic_theorem . Within this framework the field-dressed state chosen is the one populated adiabatically from the field-free ground electronic and vibrational molecular state by slowly switching on the dressing field. This approach has been used previously in a similar context Tamas1 and provides a reasonable approximation as long as the dressing field is turned on much slower than the characteristic timescales of the system. Because we can not suggest a specific value (see Eq. (12)) for the field-free initial state of an optical lattice experiment, we identified the adiabatically populated field-dressed state as that having the largest coefficient, see Eq. (LABEL:eq:field-dressed), for a single but any value. In Figs. 2 and 3 the field-dressed absorption spectra are presented for several different dressing-light intensities and wavelengths from to W/cm2 and to nm. Inspecting Fig. 2 we see that for the particular dressing-field wavelength of nm, the presence of the dressing field leads to a decrease in the absorption of the molecule with respect to the field-free case. In addition, tiny discrepancies between the different field-dressed absorption envelopes can be realized. Similarly, the absorption envelope obtained by a nm dressing wavelength (see on Fig. 3) also differs from the other wavelength dressing fields.
These findings are not surprising, because such and even more pronounced deviations have already been visualized on the spectra of freely rotating molecules dressed by running wave laser lights Tamas1 . For the sake of comparison we present in Fig. 4 some field-dressed absorption spectra of the freely rotating for several different light intensities of a nm wavelength dressing-field 222For the calculation of the spectra the first term of the last line in Eq. (3) of Tamas1 is applied.. It can clearly be seen that the field-dressed absorption envelopes are undergoing some remarkable changes via modifying the dressing-field intensities. They can move either up or down relative to the position of the field-free envelope.
The lower panels of Figs. 2 and 4, showing the individual spectral lines of the respective field-dressed spectra, demonstrate significant differences between a non-rotating Na2 dressed by an optical lattice and a freely rotating Na2 dressed by a running wave laser fields. In both cases Autler–Townes-type splittings AutlerTownes1 of the field-free peaks can be observed, but the splittings are much more pronounced in the optical lattice case. This is probably related to the fact that the density of translational states in the optical lattice is much higher than the density of rotational states for the freely-rotating Na2, therefore, the mixing of the different field-free translational states can occur more efficiently than the mixing of the different rotational states during the dressing process.
On the other hand, the spectrum envelope for the freely-rotating Na2 is more sensitive to the dressing field intensity (see upper panels of Figs. 2 and 4). The changes in the overall spectral peak heights resulting from couplings (induced by the dressing field) between eigenstates of a zeroth-order (field-free) Hamiltonian is referred to as intensity-borrowing Lenz1 ; Spect1 . It is known that in classical vibronic molecular spectroscopy the intensity-borrowing effect is a clear fingerprint of the presence of a strong nonadibatic mixing between the different degrees of freedom in the close vicinity of an intrinsic conical intersection. It has been shown that similar situation holds for the dressed state spectra of a freely rotating molecule when running laser waves are used for the dressing resulting in the formation of a light-induced conical intersection Tamas1 . In the optical lattice, the different degree of intesity borrowing is most probably related to the fact that while the transition amplitude induced by the probe pulse is a function of the rotational coordinate, it is not a function of the translational coordinate, see Eq. (3) in Ref. Tamas1 and Eq. (14). Therefore, the mixing of the different translational states in the light-dressed states can lead to slight peak splittings, but do not lead to significant changes in the summed overall transition amplitudes of Eq. (14). Therefore, the impact of the light-induced conical intersection is not so pronounced.
As already stated before, during the above simulations an adiabatic turn-on of the dressing field was assumed, i.e., the field-free ground state was assumed to change into a single light-dressed state in an adiabatic fashion. This light-dressed state was used as the initial state when computing the field-dressed spectra. However, such an adiabatic approximation is only valid if the turn-on time of the dressing field is much longer than the characteristic timescales of the system under study. For the freely rotating Na2 this can be a reasonable approximation, as the smallest rotational transition energy of approximately 0.3 cm*-1* stands for a timescale of around 100 ps. On the other hand, the timescale corresponding to the very dense, nearly continuous energy levels of the translational motion in an optical lattice (see Fig. 5) is very long, around a few seconds. Such a slow turn-on time for the dressing field is not realistic, thus the adiabatic approximation seems to be not so adequate for the optical lattice case.
III.2 Second adiabatic approximation
Another possible approach to select the initial light-dressed state is to choose the one in which the sum of the field-free basis coefficients is the largest, i.e. the field-dressed eigenvector with the largest contribution from the field-free is selected (see Eq. (LABEL:eq:field-dressed)). Here k numbers the basis functions related to the translational motion (see Eq. (12)). With such a selection process, we aim to represent a physical scenario in which the turn-on time is adiabatic with respect to the timescale of the vibrational and electronic degrees of freedom, but instantaneous with respect to the timescale of the translational degree of freedom. This selection criteria was applied during the investigation of the molecular localization in an optical lattice as well Pawlak (we shortly refer to it as “second adiabatic approach”). Curves of the field-dressed absorption spectra for several different light intensity and wavelength values obtained by this framework are displayed in Figs. 6 and 7. The most striking result is the robustness of the spectra. We obtained a significantly different picture than for the situation of the previously studied freely-rotating Na2 molecule. The absorption envelopes are almost identical, not only at different intensities but also when compared with the field-free one. In contrast, the height of the peaks of the stick spectra are substantially different and each of which differ prominently from the height of the field-free peaks. In other words, new transitions appear due to the standing laser field formed dressed states. Here again, the densely packed small pikes can be understood as the fingerprint of the Autler–Townes effect. The presence of the huge number of small spikes results in a rich structure in the line spectra, despite the fact that the envelopes do not change their shape. The latter fact, however, allows us to suggest that the intensity borrowing effect – which can be seen as the clear fingerprint of the light-induced conical intersection – is missing now. Investigating in detail the initial field-dressed states obtained with the "adiabatic approximation" or the "second adiabatic approximation" reveals that while in the "adiabatic approximation" the overall contribution of the state is around 91.63% (for Wcm*-2* and nm), it is almost 99.98% for the "second adiabatic approximaition". This suggests, that for the intensity borrowing effect to be seen in the optical lattice setup represented in our simulations, the initially prepared field-dressed state should be somewhat contaminated by the excited electronic state manifold. This can be rationalized by the previously mentioned fact that the mixing of only translational states does not lead to significant changes in the transition amplitudes induced by the probe pulse.
III.3 Model system
By scaling artificially the total mass (denoted by in Eq. 9) of the Na2 molecule in an optical lattice without changing the reduce mass () related to the vibrational motion, an increased separation of the translational energy levels can be achieved. Fig. 8 shows the translational energy levels of such a model system wherein the actual mass is obtained by reducing the mass of the Na2 molecule to its 20000 portion. The resulting object shows a close similarity with the previously studied rotating Na2 molecule in the sense that the rotational energy levels of the molecule are much further apart from each other than the very dense, almost continuous energy levels belonging to the unscaled translation motion in the lattice. Studying the field-dressed spectra of this model system with the scaled total mass, it can be realized that similarly to the field-dressed spectra of the freely-rotating Na2 molecule (see in Fig. 4) the absorption envelopes, even if not so significantly, but differ from each other. Due to some numerical problems that emerge during the construction of the model, we could manage a reduction of the mass of the Na2 molecule only to its 20000 portion, but even in this case it is clearly visible that the envelopes belonging to different intensity values noticeably differ from each other. The robustness of the spectrum disappears, the absorption envelopes are moving, although these modifications are much less sensitive to the change of the wavelength and intensity of the dressing field than those of the freely-rotating Na2. Even though the “second adiabatic approach” was applied for the selection of the field-dressed state - which keeps the absorption envelopes of the unscaled Na2 in an optical lattice fixed – the envelopes of the absorption spectra are still changed as a result of the artificial widening of the translational energy level gaps. This means that besides the always existsing Autler–Townes-type splittings the intensity borrowing slowly appears as well, representing the signature of the light-induced nonadibatic phenomena. This effect can be traced back once again to the contamination of the initial field-dressed state with the excited electronic state manifold, contribution of the state is 98.69% (for Wcm*-2* and nm).
Obtained results demonstrate that irrespective of the evaluation algorithm used for the selection of the field-dressed molecular states, the very dense energy level structure of the translational motion taking place in an optical lattice play an essential role in the robustness of the field-dressed spectrum.
IV Conclusions
Assuming a weak probe pulse, we simulated the standing laser field-dressed absorption spectra of non-rotating Na2 molecules located in an optical lattice. The direction of the applied probe pulse was set to be perpendicular to the direction of the dressing pulse and the polarization direction of the two fields were set the same. In this case the dependence of the translational coordinate Z in the probe pulse does not appear. Applying the proposed algorithm of Pawlak for the selection of a single dressed state which is the most similar to the field-free ground electronic and vibrational state of the molecule, a robust field-dressed absorption spectrum for the Na2 molecule was obtained. Although, the present model of the optical lattice provides almost identical envelopes of the absorption spectra in the studied wavelength and intensity ranges, it seems that this robustness can be influenced by changing the density of the states and the method of selecting the initial field-dressed state, which is of course, not a trivial decision.
We think that the present results are likely to change if the probe pulse is applied in a different direction, because changes may appear in the picture due to the emerging translational coordinate (Z) dependence of the probe pulse. Furthermore, by envolving an additional degree of freedom in the probe stage might allow us to visualize the signature of the light-induced nonadibatic phenomena in the standing field-dressed spectra, which is hardly present in this model. A more accurate method for the standing laser field-dressed spectroscopy can be constructed if rotations are included or if one considers spectra obtained from not a single field-dressed state, but from the combination of several of them. To continue, we plan to go in this direction. However, it is a great challenge, as one has to solve the time-dependent dynamical Schrödinger equation for the pump stage so as to determine the contributions of the different field-dressed states composing the initial wave function measured by the proceeding probe process.
Acknowledgements.
This research was supported by the EU-funded Hungarian grant EFOP-3.6.2-16-2017-00005. The authors are grateful to NKFIH for support (Grant K128396 and PD124623).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1(1) J. von Neumann and E. P. Wigner, Phys. Z. 30 , 467 (1929).
- 2(2) H. Köppel, W. Domcke, and L. S. Cederbaum, Adv. Chem. Phys. 57 , 59 (1984).
- 3(3) D. R. Yarkony, Rev. Mod. Phys. 68 , 985 (1996).
- 4(4) M. Baer, Phys. Rep. 358 , 75 (2002).
- 5(5) G. A. Worth and L. S. Cederbaum, Ann. Rev. Phys. Chem. 55 , 127 (2004).
- 6(6) W. Domcke, D. R. Yarkony, and H. Köppel, Conical Intersections: Electronic Structure, Dynamics and Spectroscopy , World Scientific, Singapore, 2004.
- 7(7) M. Baer, Beyond Born Oppenheimer: Electronic Non-Adiabatic Coupling Terms and Conical Intersections , Wiley, Hoboken, NJ, 2006.
- 8(8) M. Born and R. Oppenheimer, Ann. Phys. 84 , 457 (1927).