Theoretical rotation-vibration spectroscopy of {\it cis}- and {\it trans}-diphosphene (P$_2$H$_2$) and the deuterated species P$_2$HD
Alec Owens, Sergei N. Yurchenko

TL;DR
This paper provides the first comprehensive rotation-vibration line lists for cis- and trans-diphosphene (P$_2$H$_2$) and its deuterated form, aiding astronomical detection and spectral analysis of these P-bearing molecules.
Contribution
It introduces the first-ever theoretical line lists for P$_2$H$_2$, computed using high-level ab initio methods, for the first time enabling detailed spectroscopic studies of these molecules.
Findings
First line lists for P$_2$H$_2$ provided
Transitions considered up to 8000 cm$^{-1}$ and J ≤ 25
Analysis of deuterated species P$_2$HD spectrum included
Abstract
Growing astronomical interest in phosphorous (P) chemistry is stimulating the search for new interstellar P-bearing molecules; a task requiring detailed knowledge of the microwave and infrared molecular spectrum. In this work, we present comprehensive rotation-vibration line lists of the \cis- and \trans-isomers of diphosphene (PH). The line lists have been generated using robust, first-principles methodologies based on newly computed, high-level \ai\ potential energy and dipole moment surfaces. Transitions are considered between states with energies up to ~cm and total angular momentum . These are the first-ever line lists to be reported for PH and they should significantly facilitate future spectroscopic characterization of this system. The deuterated species \trans-PHD and the effect of its dynamic dipole moment on the rovibrational spectrum…
Click any figure to enlarge with its caption.
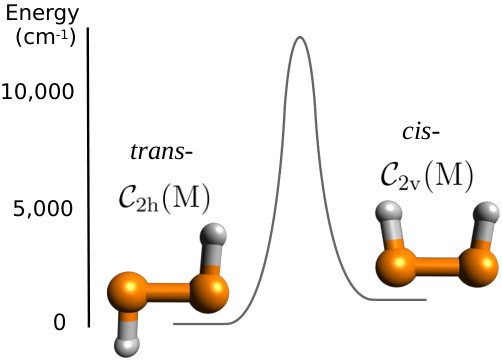 Figure 1
Figure 1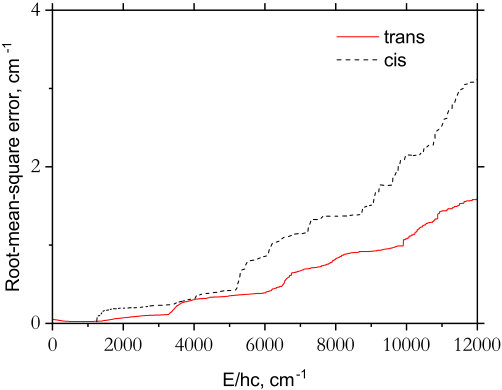 Figure 2
Figure 2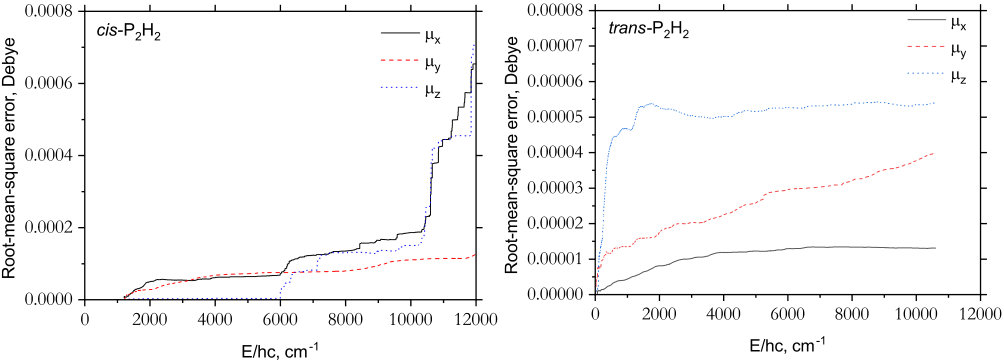 Figure 3
Figure 3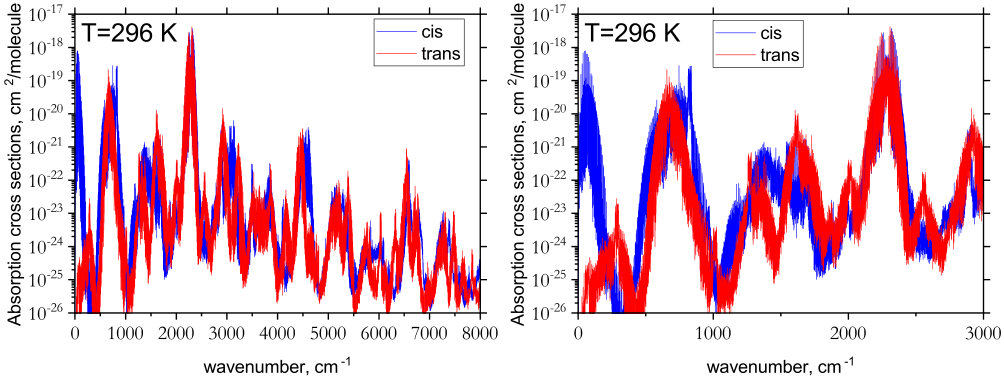 Figure 4
Figure 4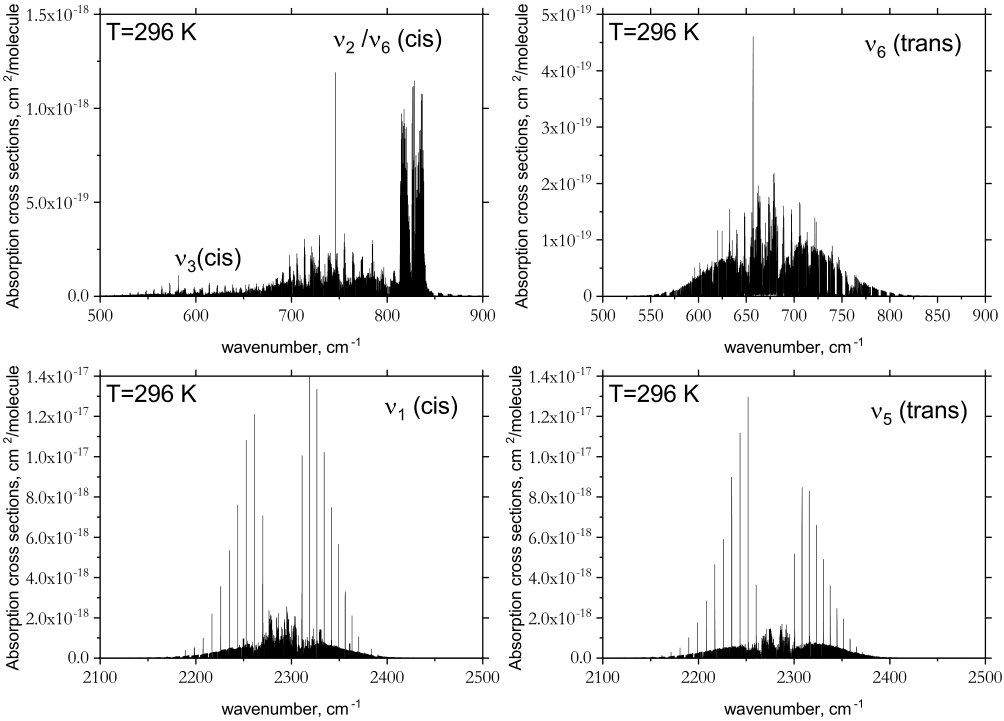 Figure 5
Figure 5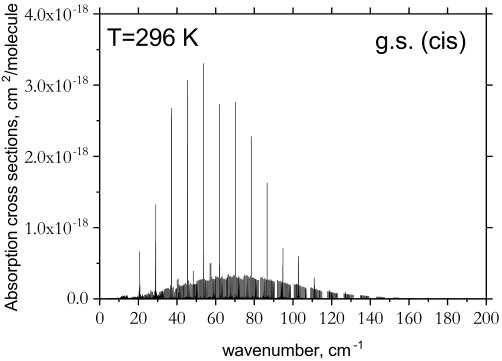 Figure 6
Figure 6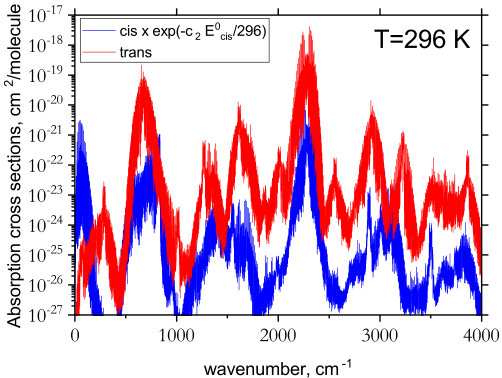 Figure 7
Figure 7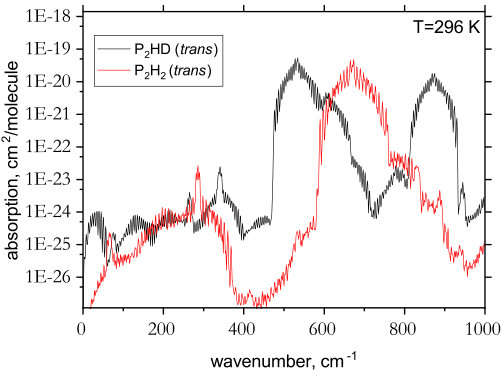 Figure 8
Figure 8| Mode | Sym. | Description | Calculated |
|---|---|---|---|
| Symmetric PH stretching | 2290.93 | ||
| Symmetric bending | 733.68 | ||
| PP stretching | 593.87 | ||
| Torsional motion | 678.21 | ||
| Asymmetric PH stretching | 2285.13 | ||
| Asymmetric bending | 825.70 |
| Mode | Sym. | Description | Calculated |
|---|---|---|---|
| Symmetric PH stretching | 2263.88 | ||
| Symmetric bending | 954.73 | ||
| PP stretching | 604.52 | ||
| Torsional motion | 753.94 | ||
| Asymmetric PH stretching | 2280.89 | ||
| Asymmetric bending | 669.07 |
| Mode | Sym. | Description | Calculated |
|---|---|---|---|
| PH stretching | 2233.04 | ||
| PD stretching | 1652.41 | ||
| PPH bending | 871.69 | ||
| PP stretching | 607.28 | ||
| PPD bending | 530.90 | ||
| Torsional motion | 658.04 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Theoretical rotation-vibration spectroscopy of cis- and trans-diphosphene (P2H2) and the deuterated species P2HD
Alec Owens
Department of Physics and Astronomy, University College London, Gower Street, WC1E 6BT London, United Kingdom
Sergei N. Yurchenko
Department of Physics and Astronomy, University College London, Gower Street, WC1E 6BT London, United Kingdom
Abstract
Growing astronomical interest in phosphorous (P) chemistry is stimulating the search for new interstellar P-bearing molecules; a task requiring detailed knowledge of the microwave and infrared molecular spectrum. In this work, we present comprehensive rotation-vibration line lists of the cis- and trans-isomers of diphosphene (P2H2). The line lists have been generated using robust, first-principles methodologies based on newly computed, high-level ab initio potential energy and dipole moment surfaces. Transitions are considered between states with energies up to cm*-1* and total angular momentum . These are the first-ever line lists to be reported for P2H2 and they should significantly facilitate future spectroscopic characterization of this system. The deuterated species trans-P2HD and the effect of its dynamic dipole moment on the rovibrational spectrum is also discussed.
I Introduction
Phosphorous (P) chemistry is of increasing astronomical interest (Jiménez-Serra et al., 2018), notably because of the essential role phosphorous plays in biochemical processes. To date, only a handful of P-bearing molecules (CP (Guélin et al., 1990), PN (Turner and Bally, 1987; Ziurys, 1987), PO (Tenenbaum, Woolf, and Ziurys, 2007), C2P (Halfen, Clouthier, and Ziurys, 2008), PH3 (Agúndez et al., 2014), HCP (Agúndez, Cernicharo, and Guélin, 2007), and the tentative identification of NCCP (Agúndez, Cernicharo, and Guélin, 2014)) have been detected in interstellar and circumstellar environments, and such observations require detailed knowledge of the microwave and infrared molecular spectrum. Despite its relatively low cosmic abundance, the fact that phosphorous is highly reactive means that other P-bearing species are likely to be observed in the future.
One potential system is diphosphene (P2H2), which structurally can exist in either the cis- or trans-isomeric form. Diphosphene has been detected in the pyrolysis of diphosphine (P2H4) Fehlner (1966), and is also a potential intermediate in the photolysis of phosphine (PH3) to phosphorus and hydrogen Ferris and Benson (1980). The phosphorus-phosphorus double bond in P2H2, which is an unusual property in phosphorous chemistry, has led to interest in this molecule and a number of theoretical studies have been reported Yoshifuji et al. (1983); Ha, Nguyen, and Ruelle (1984); Allen et al. (1985); Schmidt and Gordon (1986); Ito and Nagase (1986); Allen et al. (1986); Nguyen and Ha (1989); Fueno and Akagi (1995); Matus, Nguyen, and Dixon (2007); Lu et al. (2009, 2010); Vogt-Geisse and Schaefer III (2012). However, they have largely been concerned with the calculation of isomerization transition states and the respective equilibrium structures. As such, spectroscopic data on this system is in very short supply. For example, we are unaware of any known values for the fundamental vibrational frequencies of the P2H2 isomers. We thus find it worthwhile to investigate the rovibrational spectrum of cis- and trans-P2H2 using rigorous, first-principles methodologies.
Nowadays, the rovibrational spectra of small, closed-shell polyatomic molecules can be accurately calculated using high-level ab initio methods in conjunction with a variational treatment of nuclear motion (Tennyson, 2016). Purely ab initio potential energy surfaces (PESs) can predict vibrational energy levels to within 1 cm*-1* (Polyansky et al., 2003; Schwenke, 2002; Yachmenev et al., 2011; Malinyszek and Koput, 2012; Owens et al., 2015a, b, 2016a, 2019), while ab initio dipole moment surfaces (DMSs) are capable of producing transition intensities comparable to, if not more reliable in some instances, than experiment Yurchenko (2014); Tennyson (2014). Such approaches have considerable predictive power and are becoming commonplace in a range of applications, for example, the ExoMol database (Tennyson and Yurchenko, 2012; Tennyson et al., 2016) is constructing molecular line lists to aid the atmospheric characterization of exoplanets and other hot bodies. It is within the ExoMol computational framework (Tennyson and Yurchenko, 2017) that we treat diphosphene. As shown in Fig. 1, the potential energy barrier between cis- and trans-P2H2 is large enough (approximately cm*-1* (Lu et al., 2009), where is the Planck constant and is the speed of light) that we can consider them as separate molecules with different molecular symmetry.
The paper is organized as follows: The electronic structure calculations and analytic representation of the PESs and DMSs are described in Sec. II and Sec. III, respectively. The variational nuclear motion computations and intensity simulations are detailed in Sec. IV. The line lists, which consider transitions up to in the 0–8000 cm*-1* region, are presented in Sec. V as well as a discussion of the deuterated species P2HD and the effect of its dynamic dipole moment on the rovibrational spectrum. Concluding remarks are offered in Sec. VI.
II Potential Energy Surface
II.1 Electronic structure calculations
The cis and trans-isomers of P2H2 are known to have little multireference character (Lu et al., 2009), so their electronic structure can be accurately described using coupled cluster methods. Similar to our previous work on ab initio PESs (Owens et al., 2015a, b, 2016a, 2019), we employ focal-point analysis (Császár, Allen, and Schaefer III, 1998) to represent the total electronic energy as
[TABLE]
The energy at the complete basis set (CBS) limit was determined using the explicitly correlated coupled cluster method CCSD(T)-F12b (Ref. Adler, Knizia, and Werner, 2007) with the F12-optimized correlation consistent basis sets, cc-pVTZ-F12 and cc-pVQZ-F12 Peterson, Adler, and Werner (2008). A parameterized, two-point formula Hill et al. (2009)
[TABLE]
was used to extrapolate to the CBS limit. The coefficient is unique to the CCSD-F12b and (T) component of the total correlation energy and assumed a value of and , respectively (Hill et al., 2009). No extrapolation was applied to the Hartree-Fock (HF) energy, instead the HF+CABS (complementary auxiliary basis set) singles correction Adler, Knizia, and Werner (2007) computed in the quadruple zeta basis set was used. Calculations employed the frozen core approximation and the diagonal fixed amplitude ansatz 3C(FIX) Ten-No (2004) with a Slater geminal exponent value of Hill et al. (2009). For the auxiliary basis sets (ABS), the resolution of the identity OptRI Yousaf and Peterson (2008) basis was employed, and the cc-pV5Z/JKFIT Weigend (2002) and aug-cc-pwCV5Z/MP2FIT Hättig (2005) basis sets for density fitting. Calculations were performed with MOLPRO2015 Werner et al. (2012) unless stated otherwise.
Core-valence (CV) electron correlation was accounted for at the CCSD(T)-F12b/cc-pCVTZ-F12 (Ref. Hill, Mazumder, and Peterson, 2010) level of theory with the same ansatz and ABS as listed above but with a Slater geminal exponent value of . The (1s) orbital of the phosphorous atoms was frozen in all-electron calculations since basis sets cannot describe this orbital adequately.
Scalar relativistic (SR) effects were computed using the second-order Douglas-Kroll-Hess approach Douglas and Kroll (1974); Heß (1986) at the CCSD(T)/cc-pVQZ-DK (Ref. de Jong, Harrison, and Dixon, 2001) level of theory in the frozen core approximation. After the completion of this study it was brought to our attention that the electronic energies that include the Douglas-Kroll one-electron integrals lose accuracy when using a basis set above triple-zeta quality, see e.g. Ref. Huang and Lee, 2008. Tests were performed on some select geometries to investigate this degradation but the results were inconclusive. Given that the size of the SR correction ranged from cm*-1*, we do not expect this effect to significantly change the shape of the PESs and the subsequent accuracy of our variational calculations.
Higher-order (HO) correlation effects were computed using the hierarchy of coupled cluster methods CCSD(T), CCSDT, and CCSDT(Q), such that . Here, the full triples contribution is , and the perturbative quadruples contribution is . Calculations in the frozen core approximation employed the general coupled cluster approach Kállay and Gauss (2005, 2008) as implemented in the MRCC code MRCC interfaced to CFOUR CFOUR . The correlation consistent basis sets cc-pVTZ(+d for P) and cc-pVDZ(+d for P) Dunning Jr. (1989) were utilized for the full triples and perturbative quadruples contributions, respectively.
The terms in Eq. (1) were determined on a uniformly-spaced grid of 68,686 nuclear geometries with energies up to cm*-1*. The grid was constructed in terms of six internal coordinates: the P–P bond length , two P–H bond lengths , two interbond angles , and the dihedral angle between the planes containing the P–P–H1 and P–P–H2 nuclei. Geometries with were classified as cis-P2H2, while those with were treated as trans-P2H2. This separation is consistent with the value of the dihedral angle corresponding to the isomerization transition state linking the cis- and trans-isomers (Lu et al., 2009). All ab initio points computed for the PESs are provided as supplementary material.
We mention that the energy difference between the equilibrium structures of cis- and trans-P2H2 (considering CBS extrapolation and all HL corrections) was computed to be 1229.7 cm*-1*, slightly higher than the trans-cis energy gap of cm*-1* determined in the theoretical study of Ref. Lu et al., 2009.
II.2 Analytic representation
The potential energy function of the cis- and trans-isomers was represented using the following expansion,
[TABLE]
with maximum expansion order . This analytic representation is essentially a sextic force field in an appropriate coordinate system with proper limiting behaviour; an important factor in variational calculations. The coordinates,
[TABLE]
where , , , , , , and the deviations are taken relative to the respective reference equilibrium values , , , and or 180*∘* for the cis and trans configurations, respectively. The stretching Morse oscillator parameters and were optimized in the fittings and had slightly different values for the cis and trans PESs, see the supplementary material.
The expansion parameters were established through least-squares fittings to the ab initio data and were subject to the permutation condition,
[TABLE]
i.e., for or . The trans-PES required 737 parameters which were determined from fitting to 36,609 points, while the cis-PES needed 733 parameters fitted to 32,722 points. Weight factors of the form suggested by Partridge and Schwenke (1997)
[TABLE]
were employed in the fit to favour energies below cm*-1*. Here, where is the potential energy at the th geometry above equilibrium and the normalization constant (all values in cm*-1*). Watson’s robust fitting scheme Watson (2003) was also utilized to reduce the weights of outliers and further improve the description at lower energies.
The results of the PES fittings are shown in Fig. 2 where it can be seen that the errors for the cis-PES are systematically larger than the trans-PES. Because the cis-isomer resides at higher energy, this would explain the increased fitting error compared to trans-P2H2. In both cases, the quality of the fit is excellent around the equilibrium region but gradually deteriorates as we go to higher energies. The isomerization transition state between cis- and trans-P2H2 lies at around cm*-1* and requires a multireference description (Lu et al., 2009). As such, the computed coupled cluster energies will be unreliable as we sample the PES in the region of the isomerization transition state, and this will contribute to the degradation in the fittings, particularly for the cis-isomer. However, this should not substantially affect our rovibrational computations as we are treating the cis- and trans-isomers as separate molecules with different PESs. The expansion parameters of the PESs are provided as supplementary material along with a program to construct the analytic representation.
III Dipole Moment Surface
III.1 Electronic structure calculations
The DMSs were generated on the same grid of nuclear geometries as the cis- and trans- PESs. Defining a Cartesian laboratory-fixed coordinate system with origin at one of the P nuclei, an external electric field with components a.u. was applied along each coordinate axis and the respective dipole moment component for determined through central finite differences. Calculations were performed at the CCSD(T)/aug-cc-pVTZ(+d for P) level of theory in the frozen core approximation using MOLPRO2015 Werner et al. (2012).
III.2 Analytic representation
To represent the DMSs, it is beneficial to transform to a suitable molecule-fixed axis system before fitting an analytic representation to the ab initio data. For P2H2, the following was used: The axis is aligned along the P–P bond, while the axis is perpendicular and lies in the plane bisecting the two P–P–H planes (i.e., the planes containing the P–P and P–H bonds). The axis is oriented such that the axis system is right-handed. This representation is similar to that used for hydrogen peroxide (HOOH) Al-Refaie et al. (2015).
For trans-P2H2, the DMS spans the (M) symmetry group with the , and components of the dipole moment transforming according to the , , and irreducible representations, respectively. For cis-P2H2, the DMS spans the (M) symmetry group and the , and components transform according to the , , and representations, respectively. In the molecule-fixed axis system, the symmetry-adapted projections of the electronically averaged Cartesian dipole moment components and are given as analytic representations, where each component is expanded in Taylor series around the equilibrium configuration in terms of internal coordinates, that is,
[TABLE]
The coordinates are defined as,
[TABLE]
and the expansion parameters of the , and dipole moment components obey the following permutation rules,
[TABLE]
which corresponds to permuting the two hydrogen atoms. Thus, when for any value of the index.
The expansion coefficients for were determined simultaneously through a least-squares fitting to the ab initio data. Watson’s robust fitting scheme (Watson, 2003) and the same weight factors as shown in Eq. (11) were used. Similarly, the reference equilibrium parameters assumed similar values to those employed in the PES fittings, see the supplementary material. For the cis-isomer, the three dipole moment surfaces, , , and , required 378, 242 and 404 parameters, respectively, whilst the trans dipole components used 496, 420 and 421 parameters. The results of the DMS fittings are illustrated in Fig. 3. The component of the cis-isomer (which has a permanent electric dipole) has the largest magnitude and hence the largest errors, otherwise the behaviour of the DMS fittings is as expected. The expansion parameters of the DMSs are given as supplementary material along with a program to construct the analytic representation.
IV Variational calculations
The computer program TROVE (Yurchenko, Thiel, and Jensen, 2007) was used for all nuclear motion computations. Since its procedures and methodology are well documented Yurchenko, Thiel, and Jensen (2007); Yurchenko et al. (2009); Yachmenev and Yurchenko (2015); Yurchenko, Yachmenev, and Ovsyannikov (2017), we summarize only the key aspects relevant for this work. Calculations are based on the assumption that the two isomers are separated by an impenetrable barrier ( cm*-1*) with zero tunneling. The cis- and trans-isomers are treated as separate molecules with different molecular symmetry.
The rovibrational Hamiltonian was constructed numerically with both the kinetic and potential energy operators truncated at 6th order (see Refs. Yurchenko, Thiel, and Jensen, 2007; Yachmenev and Yurchenko, 2015 for a discussion of the associated errors of such a scheme). The Hamiltonian was represented in the Eckart frame as a power series expansion around the equilibrium geometry in terms of the six coordinates introduced in Eqs. (4)–(9), except in the case of the kinetic energy operator which used linear displacement variables for the stretching terms. The symmetrized rovibrational basis set was constructed using a general, multi-step contraction scheme Yurchenko, Yachmenev, and Ovsyannikov (2017). The polyad number, defined as
[TABLE]
was used to control the size of the primitive and contracted basis sets. Here, the quantum numbers for correspond to primitive basis functions , which are determined by solving one-dimensional Schrödinger equations for each th vibrational mode. Products of are used to build the vibrational basis set. Initially, reduced symmetry sub-spaces are set up by coupling equivalent modes and solving the reduced-mode Schrödinger equations; for example all stretching degrees of freedom are treated together. The resulting eigenfunctions are then combined and used as a basis set to solve the full-dimensional problem. The final basis sets were truncated with and , resulting in 8892 and 8701 basis functions (containing all symmetries) for cis- and trans-P2H2, respectively. All primitive basis functions were generated using the Numerov-Cooley approach Noumerov (1924); Cooley (1961) on grids of 1000 points and then symmetrized using the TROVE symmetry-adaptation procedure Yurchenko, Yachmenev, and Ovsyannikov (2017). The eigenfunctions were multiplied with symmetrized rigid-rotor functions to produce the final basis set for calculations. Nuclear masses were employed in TROVE calculations.
The trans-isomer is of (M) symmetry with transitions obeying the dipole selection rules . The nuclear spin statistical weights are for states of symmetry , respectively. In calculations, the Hamiltonian was expanded around equilibrium values of , , and . The zero-point energy (ZPE) was computed to be 3865.759 cm*-1*.
The cis-isomer is of (M) symmetry with transitions obeying the dipole selection rules . The nuclear spin statistical weights are for states of symmetry , respectively. In calculations, the Hamiltonian was expanded around equilibrium values of , , and . The ZPE was computed to be 5025.816 cm*-1*, i.e., 1160.061 cm*-1* above that of the trans-isomer.
All transitions and Einstein coefficients involving states with rotational excitation up to have been computed using the MPI-GAIN program Al-Refaie, Yurchenko, and Tennyson (2017) for the 0–8000 cm*-1* region (above the ZPE of each isomer). The lower and upper state energy thresholds were chosen to be 4000 cm*-1* and 8000 cm*-1*, respectively. Spectral simulations employed the ExoCross code Yurchenko, Al-Refaie, and Tennyson (2018) and were carried out at a temperature of K with a partition function value of and for cis- and trans-P2H2, respectively. Transitions obeyed the symmetry selection rules defined above and the standard rotational selection rules , where ′ and ′′ denote the upper and lower state, respectively. Overall, the trans-P2H2 line list contained 10,667,208,951 transitions between 5,881,876 states, while the cis-P2H2 line list consisted of 11,020,092,365 transitions between 6,009,302 states.
As an aside, we have found it worthwhile to investigate the effects of isotopic substitution on the spectra of diphosphene, in particular the rotational spectrum of the trans-isomer. To this end, variational calculations for the deuterated species trans-P2HD have been performed utilizing the trans-P2H2 PES and DMS. Since this is an asymmetric species, calculations employed the (M) molecular symmetry group which consists of only two irreducible representations, and , therefore leading to larger symmetry-adapted Hamiltonian matrices when solving the respective Schrödinger equations with TROVE. The contracted vibrational basis set used a polyad truncation number of resulting in 17 312 functions, significantly larger than those of the P2H2 isomers. Due to this increased size, our study of P2HD was restricted to states with . The final line list contained 2,499,185,732 transitions between 895,884 states in the 0–8000 cm*-1* region. Note that the nuclear spin statistical weight for P2HD.
V Results
V.1 Vibrational energies
The computed fundamental frequencies of cis- and trans-P2H2 are listed in Table 1 and 2, respectively. We are unaware of any known values to compare against, however, our computed “anharmonic” values are in sensible agreement with accurate ab initio harmonic frequencies computed using coupled cluster and multireference methods in conjunction with large Gaussian basis sets (Lu et al., 2009). Based on our previous experience of generating high-level ab initio PESs of closed-shell molecules (Owens et al., 2015a, b, 2016a, 2019), which often utilized the same focal-point approach and levels of theory, the combined rms error of the computed fundamentals when compared to experiment has been well within cm*-1*. The fundamentals of P2HD are listed in Table 3.
Convergence tests utilizing larger vibrational basis sets ( in Eq. (24)) were performed for cis- and trans-P2H2. The convergence error for states below 3000 cm*-1* (used in the line lists) showed a mean absolute deviation of 0.14 cm*-1* for cis-P2H2, and 0.02 cm*-1* for trans-P2H2, with the fundamentals exhibiting orders-of-magnitude better convergence than other states. The improved convergence for trans-P2H2 is due to the larger polyad number in variational calculations. Naturally, the convergence errors increase at higher energies, particularly for highly excited modes which are the most difficult to converge. In the worst instances this error can be of the order of tens of wavenumbers. Some caution should therefore be exercised when using the line lists for transitions above 4000 cm*-1* (as a conservative estimate). A list of energies up to 8000 cm*-1* (above the ZPE of each isomer) with symmetry and TROVE quantum number labelling (determined from the contribution of the primitive basis functions) is provided as supplementary material.
V.2 Line lists
An overview of the rotation-vibration spectrum of cis- and trans-P2H2 is shown in Fig. 4. The strongest intensity features in both isomers occur for the PH stretching modes around cm*-1*. A closer inspection of these bands along with the other fundamentals is shown in Fig. 5. The most noticeable difference between the isomers occurs below 500 cm*-1*. Because trans-P2H2 has no permanent dipole moment it possesses a very weak rotational spectrum. The cis-isomer, however, has a large permanent dipole moment (computed in this study to be 1.033 D which is consistent with values determined in Ref. Lu et al., 2009) and a well pronounced rotational structure, evident in Fig. 6.
The cis- and trans-isomers are relatively stable and the rate for isomerization is as low as s*-1* (corresponding to a half-life of s) at K (Lu et al., 2009). Since the cis-isomer has a higher ZPE, if both species were to exist in a region at local thermodynamic equilibrium, the spectrum of cis-P2H2 would appear slightly weaker in comparison to trans-P2H2. In Fig. 7, we show the relative temperature effects on the spectrum of cis-P2H2 at K using the Boltzmann scaling factor where cm*-1*. Promisingly, the rotational band of cis-P2H2 appears strong enough to be detected. Note that no Boltzmann scaling was applied to the spectra in Fig. 4.
In Fig. 8, the spectrum of trans-P2HD is plotted alongside that of trans-P2H2. The effect of isotopic substitution is clearly seen in the shifted band centers but more noticeably in the rotational spectrum below 200 cm*-1*. Despite the fact that P2HD has no permanent ab initio dipole moment, the interaction of the rotational and vibrational degrees of freedom in this asymmetric isomer leads to a non-zero effective dipole moment, thus increasing the intensity of the pure rotational lines by about three orders of magnitude. Although fairly weak the rotational spectrum of P2HD should be detectable, at least in the laboratory. It is worth mentioning that recent work has investigated the effects of H D isotopic substitution on phosphine spectra Viglaska et al. (2018).
The computed line lists of these three species are available in the ExoMol format (Tennyson et al., 2016) from the ExoMol database at www.exomol.com. If combining the separate cis- and trans-P2H2 line lists into one diphosphene spectrum, it is necessary to shift the ZPE of the cis energy levels by 1160.061 cm*-1*.
Commenting on the accuracy of the line lists, we expect band centers of the fundamentals to be accurate to within 3 cm*-1* (as a conservative estimate) but this could be in the 1 cm*-1* range for certain bands. Line intensities should on average be accurate to within 5–10% of experiment. These estimates are based on our previous experience generating high-level ab initio PESs and DMSs for closed-shell systems using similar levels of theory (Owens et al., 2015a, b, 2016a, 2019, 2016b). That said, the isomerization transition state between cis- and trans-P2H2 is located at around cm*-1* so the ab initio points computed at geometries near this will be unreliable due to its multireference character. Also, because we have neglected the double-well nature of the PES of P2H2, computed higher rovibrational energies, i.e. those above cm*-1* (particularly for the cis-isomer), will possess larger errors. Transitions involving these states will have very weak intensities so their inclusion is not expected to significantly impact the computed spectra. However, we would recommend some caution when using the line lists for transitions above 4000 cm*-1*.
VI Conclusions
The rotation-vibration spectrum of diphosphene has been investigated using accurate, first-principles methodologies. High-level ab initio theory was used to construct new six-dimensional PESs and DMSs which were then utilized in variational nuclear motion computations. Line lists were generated for the cis- and trans-isomers of P2H2 which considered transitions between states with energies up to cm*-1* (above the ZPE of each isomer) and total angular momentum . Comparisons of the fundamental frequencies, their relative intensities, and the ground-state electric dipole moment of cis-P2H2 are consistent with previous calculations Lu et al. (2009), giving us confidence in the validity of the presented work. There is, however, a pressing need for quantitative spectral data on the diphosphene molecule as this would enable future benchmarking and the opportunity to improve our theoretical spectroscopic model through empirical refinement of the PESs.
Regarding future astronomical detection of P2H2, e.g., in the interstellar medium (ISM), the rotational spectrum of cis-P2H2 is dipole allowed and appears strong enough at colder temperatures, e.g., at K the intensities of the rotational band are of the order cm/molecule. That said, the presented line list may not be accurate enough in the microwave region and experimentally determined frequencies or more sophisticated ab initio calculations could be required Puzzarini, Stanton, and Gauss (2010). For the deuterated trans-P2HD system, which has a non-zero dynamic dipole moment induced by centrifugal distortion, the rotational intensities (of the order of cm/molecule) should be detectable in laboratory studies.
Supplementary Material
See the supplementary material for a list of computed energy levels, ab initio data points, and the expansion parameters and Fortran routines to construct the PESs and DMSs of diphosphene.
Acknowledgements.
This work was supported by the STFC Projects No. ST/M001334/1 and ST/R000476/1, and by the COST action MOLIM (CM1405). The authors acknowledge the use of the UCL Legion High Performance Computing Facility (Legion@UCL) and associated support services in the completion of this work, along with the Cambridge Service for Data Driven Discovery (CSD3), part of which is operated by the University of Cambridge Research Computing on behalf of the STFC DiRAC HPC Facility (www.dirac.ac.uk). The DiRAC component of CSD3 was funded by BEIS capital funding via STFC capital grants ST/P002307/1 and ST/R002452/1 and STFC operations grant ST/R00689X/1. DiRAC is part of the National e-Infrastructure.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jiménez-Serra et al. (2018) I. Jiménez-Serra, S. Viti, D. Quénard, and J. Holdship, Astrophys. J. 862 , 128 (2018) . · doi ↗
- 2Guélin et al. (1990) M. Guélin, J. Cernicharo, G. Paubert, and B. E. Turner, Astron. Astrophys. 230 , L 9 (1990).
- 3Turner and Bally (1987) B. E. Turner and J. Bally, Astrophys. J. 321 , L 75 (1987) . · doi ↗
- 4Ziurys (1987) L. M. Ziurys, Astrophys. J. 321 , L 81 (1987) . · doi ↗
- 5Tenenbaum, Woolf, and Ziurys (2007) E. D. Tenenbaum, N. J. Woolf, and L. M. Ziurys, Astrophys. J. 666 , L 29 (2007) . · doi ↗
- 6Halfen, Clouthier, and Ziurys (2008) D. T. Halfen, D. J. Clouthier, and L. M. Ziurys, Astrophys. J. 677 , L 101 (2008) . · doi ↗
- 7Agúndez et al. (2014) M. Agúndez, J. Cernicharo, L. Decin, P. Encrenaz, and D. Teyssier, Astrophys. J. Lett. 790 , L 27 (2014) . · doi ↗
- 8Agúndez, Cernicharo, and Guélin (2007) M. Agúndez, J. Cernicharo, and M. Guélin, Astrophys. J. Lett. 662 , L 91 (2007) . · doi ↗