First Principles Study of Electronic Structure and Fermi Surface in Rare-earth Filled Skutterudites RPt4Ge12
Gheorghe Lucian Pascut, Michael Widom, Kristjan Haule, and Khandker F., Quader

TL;DR
This study uses first-principles calculations to analyze the electronic structure and Fermi surfaces of rare-earth filled skutterudites RPt4Ge12, revealing insights into their superconducting, thermoelectric, and correlated electron properties.
Contribution
It provides a comprehensive theoretical analysis of electronic structures, Fermi surfaces, and the effects of f-orbital occupancy and Hubbard U in RPt4Ge12 skutterudites, advancing understanding of their properties.
Findings
Fermi surfaces suggest multi-band superconductivity potential
Density of states explains photoemission experiments
Presence of pseudogaps may influence thermoelectric behavior
Abstract
Experiments on rare-earth filled skutterudites demonstrate an intriguing array of thermodynamic, transport and superconducting properties, and bring to fore theoretical challenges posed by f-electron systems. First principle calculations based density functional theory and its extensions for strongly correlated systems such as the Hubbard U correction, provide valuable information about electronic structure that can be used to understand experiments. We present a comprehensive study of the electronic structure and Fermi surface of a series of rare earth filled skutterudites, RPt4Ge12 (where R = La, Ce, Pr), aimed at shedding light on: consequences of progressive increase of f-orbital occupancy in the series; the effects of the Hubbard parameter U; the Fermi surfaces, band structures and densities of states. The calculated Fermi surfaces may be relevant to the question of multi-band…
Click any figure to enlarge with its caption.
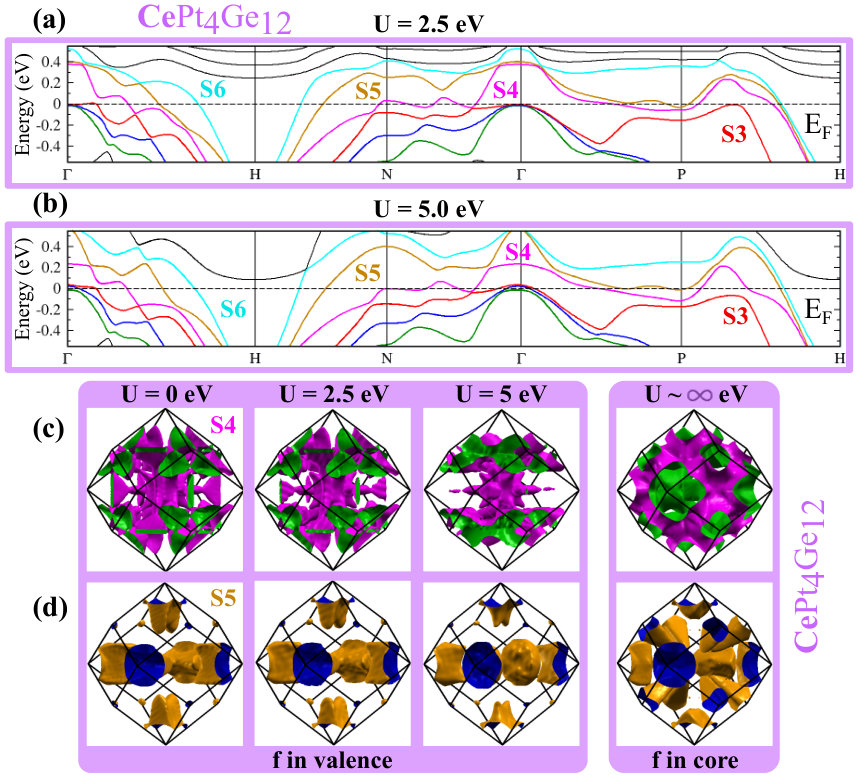 Figure 10
Figure 10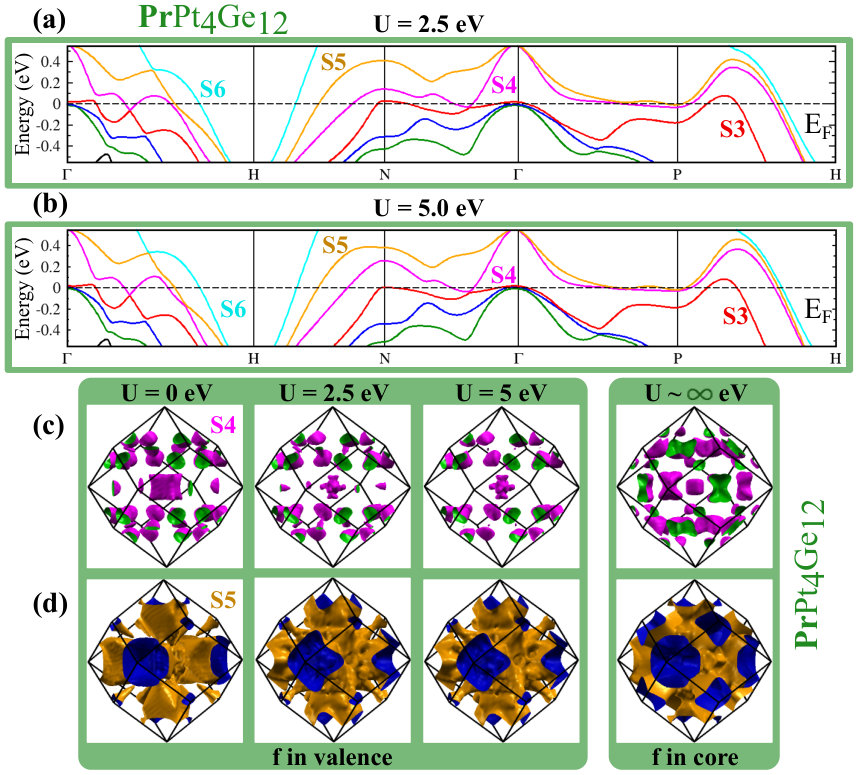 Figure 11
Figure 11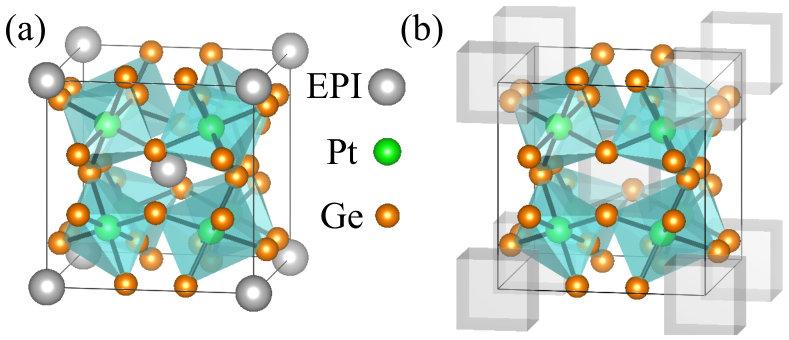 Figure 1
Figure 1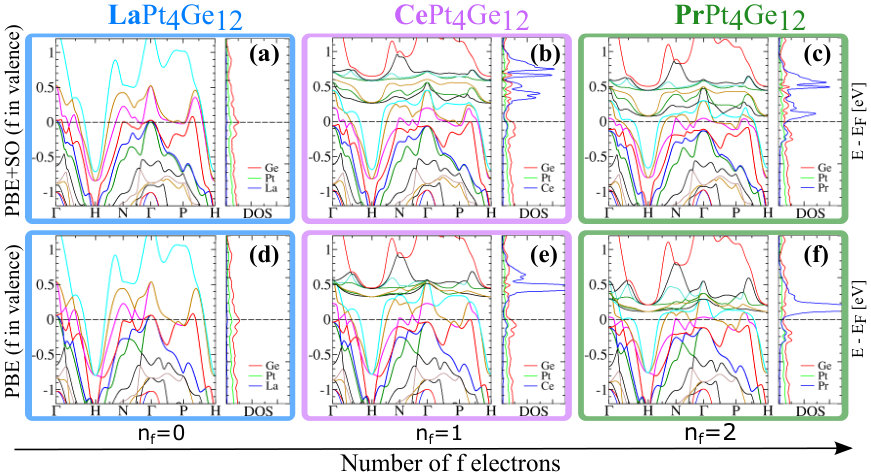 Figure 2
Figure 2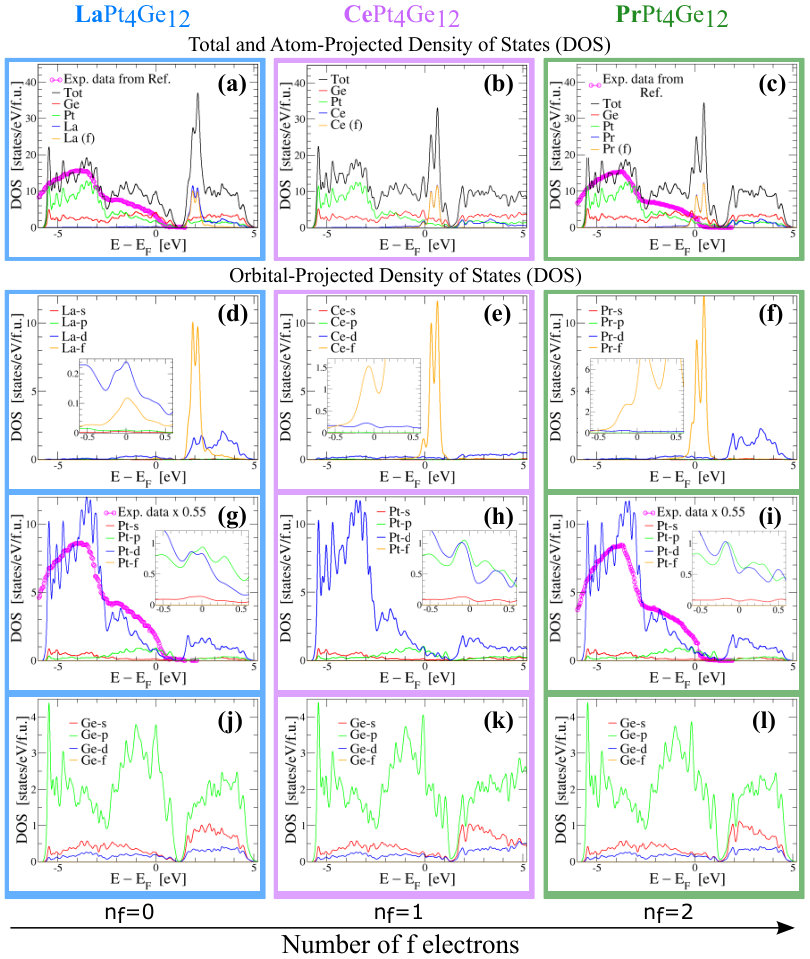 Figure 3
Figure 3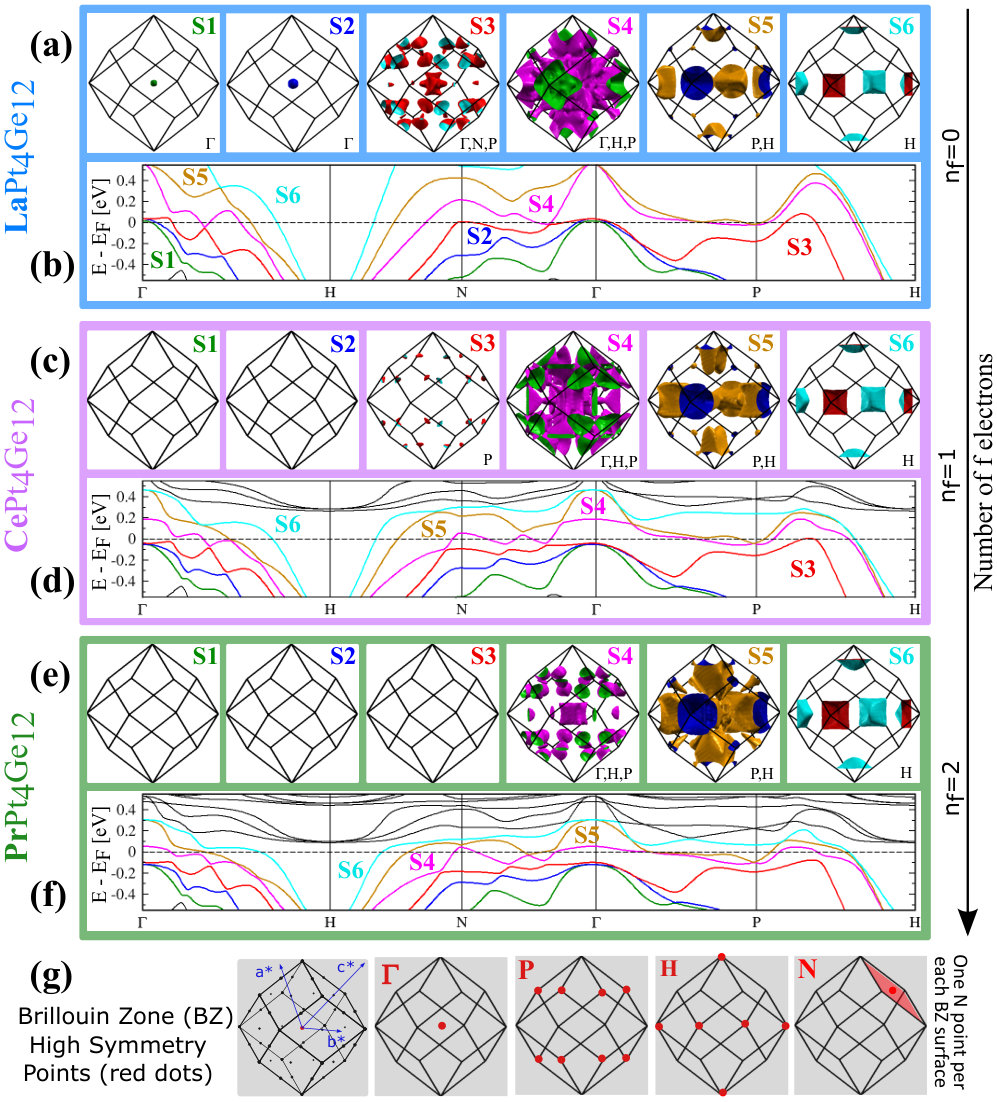 Figure 4
Figure 4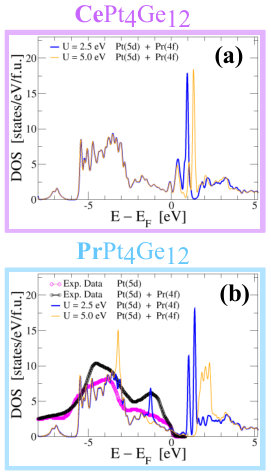 Figure 5
Figure 5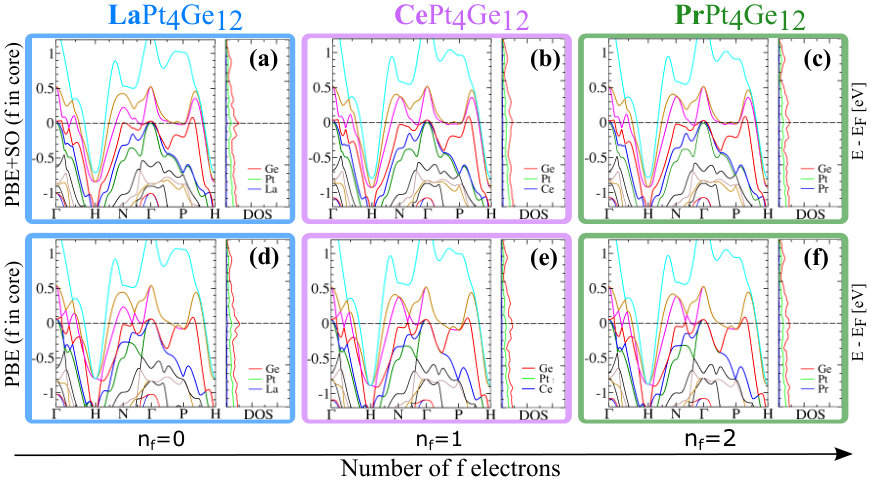 Figure 6
Figure 6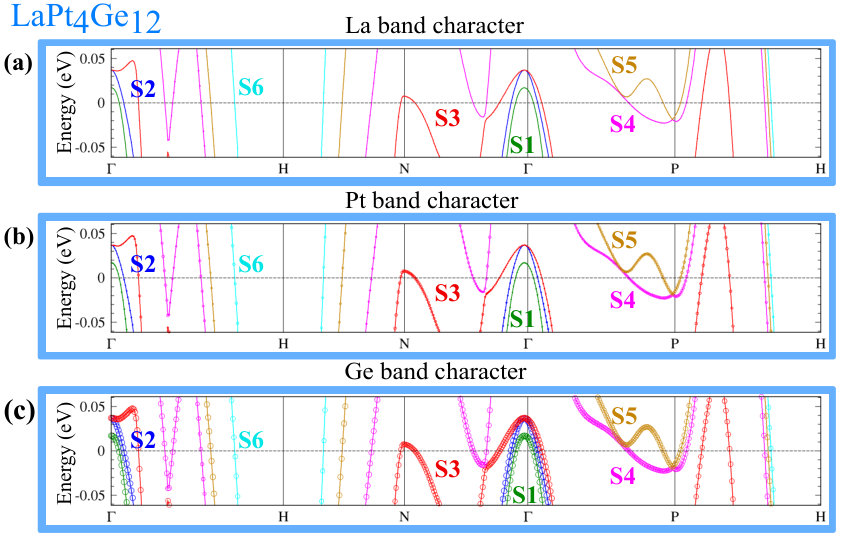 Figure 7
Figure 7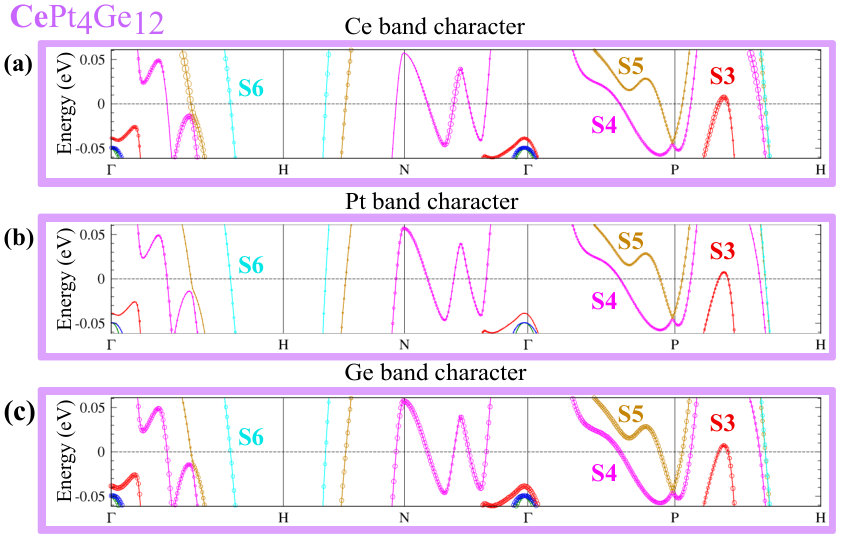 Figure 8
Figure 8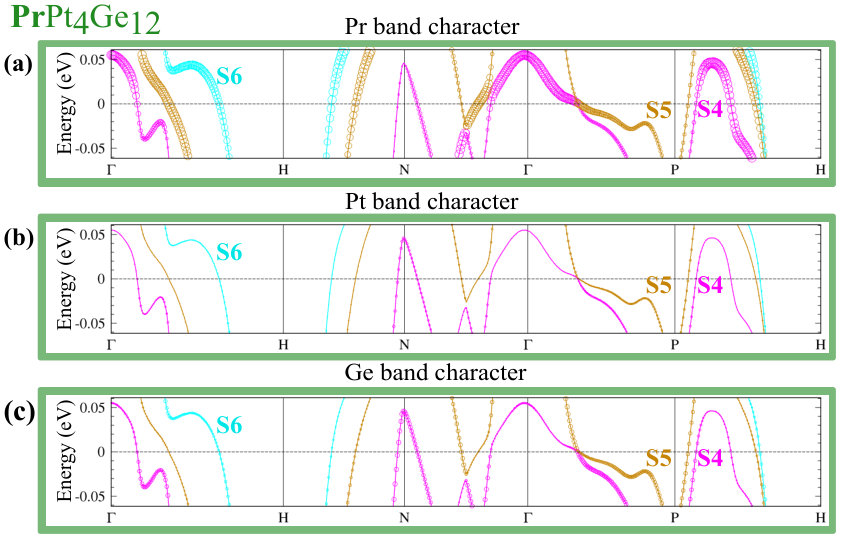 Figure 9
Figure 9| DFT scenarios | D()[states/eV/f.u.] - calc | - estimate | ||||
| La | Ce | Pr | Laa,b | Cec | Prd,e,f | |
| PBE (f in valence) | 11.9 | 10.9 | 14.7 | 2.7/2.0 | 4.1 | 2.5/2.1/2.0 |
| PBE + SOC | 10.0 | 12.2 | 16.4 | 3.2/2.4 | 3.7 | 2.3/1.9/1.8 |
| PBE + SOC + U ( = 5) | – | 10.0 | 11.3 | – | 4.5 | 3.3/2.8/2.6 |
| PBE (f in core) | – | 9.6 | 9.6 | – | 4.7 | 3.9/3.3/3.1 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
First Principles Study of Electronic Structure and Fermi Surface
in Rare-earth Filled Skutterudites Pt4Ge12
Gheorghe Lucian Pascut
Department of Physics & Astronomy, Rutgers University, Piscataway, NJ O8854, USA
MANSiD Research Center and Faculty of Forestry, Stefan Cel Mare University (USV), Suceava 720229, Romania
Michael Widom
Department of Physics, Carnegie Mellon University, Pittsburgh, PA 15213, USA
Kristjan Haule
Department of Physics & Astronomy, Rutgers University, Piscataway, NJ O8854, USA
Khandker F. Quader
Department of Physics, Kent State University, Kent , OH 44242, USA
Abstract
Experiments on rare-earth filled skutterudites demonstrate an intriguing array of thermodynamic, transport and superconducting properties, and bring to fore theoretical challenges posed by f-electron systems. First principle calculations based density functional theory and its extensions for strongly correlated systems such as the Hubbard U correction, provide valuable information about electronic structure that can be used to understand experiments. We present a comprehensive study of the electronic structure and Fermi surface of a series of rare earth filled skutterudites, RPt4Ge12 (where R = La, Ce, Pr), aimed at shedding light on: consequences of progressive increase of f-orbital occupancy in the series; the effects of the Hubbard parameter U; the Fermi surfaces, band structures and densities of states. The calculated Fermi surfaces may be relevant to the question of multi-band versus single-band superconductivity. Computed densities of states qualitatively explain the available resonant photoemission spectroscopy experiments, and (together with available specific heat measurements) provide estimates of the effective masses. We also show the existence of pseudogaps in the total density of states which may be relevant for the thermoelectric properties of these systems.
I Introduction
Filled skutterudites are a class of materials that exhibit complex crystal structures Jeitschko and Braun (1977); Gumeniuk et al. (2010a), and correspondingly complex electronic ground states, such as conventional BCS-type B. Maple et al. (2008) and unconventional superconductivity Aoki et al. (2007); Maisuradze et al. (2009, 2010), non-Fermi liquid behavior Takeda and Ishikawa (2000); Yamamoto et al. (2006), anomalous metal-to-insulator transition Sekine et al. (1997), multipolar ordering Yoshizawa et al. (2005), topological insulator state Yan et al. (2012); Smith et al. (2011), Kondo lattice behavior B. Maple et al. (2008), valence fluctuations Masatoshi et al. (2008), heavy fermion behavior Bauer et al. (2002); B. Maple et al. (2002); Maple et al. (2005), various magnetically ordered states Maple et al. (2007); Sato et al. (2009); Jeon et al. (2016); Yuhasz et al. (2006), etc. They are also promising materials for next generation thermoelectric applications. Morelli and Meisner (1995); Sales et al. (1997); Nolas et al. (1998); Sales et al. (1996)
In this paper, using first principles density functional theory (DFT) and its extensions for strongly correlated systems, we address open questions about the Fermi surface topology and its s, p, d, f character in the recently synthesized RPt4Ge12 filled skutterudites with R = La, Ce or Pr. Our study is driven by the large body of experimental work on the RPt4Ge12 , that demonstrate an array of novel properties Morelli and Meisner (1995); Takeda and Ishikawa (2000); Sekine et al. (1997); Maple et al. (2007); Maisuradze et al. (2010); B. Maple et al. (2008). Existing experimental results (summarized in Section II) point to the need for comparative study of electronic properties in RPt4Ge12 compounds. Such a study would be useful to understand similarities and differences between these materials and the consequence of these for macroscopic properties. Despite some existing density of states (DOS) calculations Gumeniuk et al. (2008, 2011); Chandra et al. (2016), a comprehensive study across the range of these compounds including spin-orbit coupling (SOC) and the Hubbard parameter is still lacking. As pointed out in Ref. Zhang et al., 2013, such calculations may shed light on possible multi-band nature of superconductivity and the origin of multiple and/or complicated pairing mechanisms. Thus our paper reports a systematic and comparative study of the RPt4Ge12 electronic structure.
The complex crystal structure of the filled skutterudites (shown in Fig. 1a) is stabilized by adding an electropositive ion to the empty voids of the skutterudites (see Fig. 1b). Besides stabilizing the crystal structure it turns out that the electropositive ion (which in our work is La, Ce or Pr) also gives rise to novel electronic states Gumeniuk et al. (2010a) and improved thermoelectric properties Sales et al. (1996). Considering the complex crystal structure and the fact that usually -electron systems pose challenges for DFT methods Peters et al. (2014); Petit et al. (2016) we confirm the robustness of our main results, by carrying out an extensive study under various theoretical scenarios such as DFT+SOC and DFT+SOC+U with the -electrons treated as core or valence electrons. Among our goals is to understand the changes in the electronic structure and Fermi surface (FS) topology and its character, as we progress from La to Ce and Pr, possessing, respectively, 0, 1, and 2 -electrons in their pure elemental states.
We find that independently of the theoretical approximation used in our calculations: (I) multiple large Fermi surfaces exists in RPt4Ge12 , a feature similar to that in MgB2 where multi-band superconductivity is found Xi (2008); Kortus et al. (2001); (II) The large FS have anistropic orbital character on the FS, a feature similar to the FS in Pb where multi-band superconductivity is also found Floris et al. (2007); Blackford and March (1969); (III) in all RPt4Ge12 compounds the states at are dominated by Ge- states; (IV) states are always present at for CePt4Ge12 and PrPt4Ge12 independently of the values of the Hubbard parameter U; (V) SOC lifts band degeneracy, shifts bands and affects the topology of the Fermi surfaces; (VI) DOS show deep pseudogaps above which may have implications for thermoelectric properties. In addition, our DFT+SOC+U calculations qualitatively explain features observed in photoemission experiments, while revealing a need for improved treatment of the -electrons systems. We also find that the deduced values of the mass enhancement are not insignificant signaling the existence of modest correlations in these materials.
The paper is organized as follows. In Section II, we give a brief overview of the experimental behavior of filled skutterudites. Section III is devoted to a discussion of the methods used for our calculations and the different theoretical scenarios considered. This is followed by Section IV where we present the results for the rare-earth-filled skutterudites, RPt4Ge12 (R=La, Ce, Pr) under different scenarios, namely PBE, PBE + SOC, PBE+SOC +U. Results for band structure, energy bands and FS without the the inclusion of Hubbard like correlation are presented in Sections IV.1, IV.2, and IV.3 respectively, and the effects of including on DOS and FS are presented in Section IV.4. In Section IV.5, based on measurements of low-temperature specific heat, we provide simple estimates of the effective mass enhancements over the band mass, for the various theoretical scenarios considered. We discuss possible thermoelectric application of the RPt4Ge12 in Section IV.6. We end the main text with discussions in Section V and acknowledgments in Section VI. We note that we provide additional details in appendices in Section VII.
II Experimental Background
Experiments on BaPt4Ge12 , SrPt4Ge12 , and RPt4Ge12 skutterudites reveal a diverse array of interesting properties: while Ba-, Sr-, La-, Pr-Pt4Ge12 exhibit superconductivity at 5.1, 5.35, 8.3, 7.9 K respectively Bauer et al. (2007); Rosner et al. (2009); Gumeniuk et al. (2008), CePt4Ge12 does not show superconductivity, and instead it is believed to demonstrate mixed valence or Kondo lattice behavior, or possibly a behavior in between. Masatoshi et al. (2008); Gumeniuk et al. (2011) NdPt4Ge12 and EuPt4Ge12 are characterized by anitferromagnetic transitions at 0.67 and 1.7 K respectively Gumeniuk et al. (2008). SmPt4Ge12 exhibitsGumeniuk et al. (2010b) heavy fermion behavior with a large value of the electronic specific heat coefficient ( 450/mol/K2), indicating the possibility of strong correlations in this material.
In the Ba, Sr and La compounds, experiments point to conventional nodeless s-wave BCS pairing; however, sub-linear behavior of the field-dependent electronic part of specific heat and thermal conductivity may indicate two equal-sized s-wave superconducting gaps in La-Pt4Ge12 . Pfau et al. (2016); Chandra et al. (2012, 2016) The nature of pairing in PrPt4Ge12 is less settled. Low-T specific heat and muon spin resonance measurements claim evidence for unconventional pairing with point nodes in the gap Maisuradze et al. (2009). But, several other experiments point to multi-band superconductivity with two superconducting gaps. Lower and upper critical field measurements and photoemission spectroscopy (PES) Nakamura et al. (2010a); Nakamura et al. (2012), and upturn in temperature dependence of upper critical field Chandra et al. (2012, 2016); Gurevich (2007) indicate multi-band paring. Strong indication of two-gap superconductivity (as in MgB2 Tsuda et al. (2004, 2007); Xi (2008); Kortus et al. (2001) and Pb Floris et al. (2007); Blackford and March (1969); Lykken et al. (1971)) has been found in the analysis of superfluid density in single crystal PrPt4Ge12 Zhang et al. (2013), the only single crystal measurement in this material that we are aware of. Multi-band pairing with one nodeless and one nodal gap has been inferred Singh et al. (2016) from fitting low-T specific heat data in PrPt4Ge12 and Pr1-xCexPt4Ge12 , while other work based on transport and thermodynamic measurements on Pr1-xCexPt4Ge12 Huang et al. (2014), and PrPt4Ge12-xSbx Jeon et al. (2016) leave open the possibility of nodal versus multi-band superconductivity. 73Ge nuclear quadrupole resonance Kanetake et al. (2010) shows a coherence peak below pairing transition temperature, typical of BCS pairing.
A detailed study of the electronic structure of RPt4Ge12 may be able to shed some light on the extent to which the Pr 4 electrons play a role in these couplings, and thereby indirectly playing a role in multi-band pairing and gaps. The strong experimental indication of multi-band pairing in PrPt4Ge12 , and possibly in LaPt4Ge12 , is suggestive of a complex band structure with several bands crossing the Fermi surface. Therefore a detailed study of the electronic properties in RPt4Ge12 is necessary.
The RPt4Ge12 materials studied here do not exhibit long range magnetic order. LaPt4Ge12 is diamagnetic at all temperatures, while the -electrons in CePt4Ge12 and PrPt4Ge12 obey Curie-Weiss behavior at sufficiently high temperatures. For PrPt4Ge12 , the magnetic susceptibility starts to exhibit a Curie-Weiss behavior for T 100 K Masatoshi et al. (2008) with experimental average effective magnetic moment of 3.59/f.u. Masatoshi et al. (2008) or 3.69/f.u. Huang et al. (2014); Gumeniuk et al. (2008) ( being Bohr magneton) that are close to that of the free Pr3+ ion (3.58 ) with the electronic configuration 4f2 and the 3H4 Hund rule ground state multiplet, indicating the presence of local moments on the Pr ions. However, the magnetic susceptibility saturates at low temperatures, indicating a non-magnetic ground state, scenario that is consistent with the crystal field splitting of the degenerate 3H4 Hund rule ground state multiplet Masatoshi et al. (2008); Gumeniuk et al. (2008).
In the case of CePt4Ge12 , the magnetic susceptibility has a also Curie-Weiss behavior down to 200 K Masatoshi et al. (2008) with experimental average effective magnetic moment of 2.58/f.u. or 2.51/f.u. Gumeniuk et al. (2011) that are close to that of the free Ce3+ ion (2.54 ) with the electronic configuration 4f1 and the 2F5/2 Hund rule ground state multiplet, indicating the presence of local moments on the Ce ions. Below 200 K, the susceptibility deviates from Curie-Weiss and exhibits a broad maximum at 80 K, followed by an upturn below 20K Masatoshi et al. (2008); Gumeniuk et al. (2011). The broad maximum can be interpreted as a characteristic features of intermediate valence Masatoshi et al. (2008) or as indicative of local moment screening with a large characteristic energy Gumeniuk et al. (2011).
From all RPt4Ge12 compounds studied here, CePt4Ge12 is the only one where a long range order, of antiferromagnetic type, can be stabilized at low temperatures upon Sb substitution of Ge Nicklas et al. (2012). Due to the absence of local moments in LaPt4Ge12 or the non-magnetic ground state of the Pr ions in PrPt4Ge12 , no long range magnetic order is found in LaPt4Ge12 or PrPt4Ge12 compounds.
In addition to superconducting, magnetic, and transport properties, filled skutterudites appear promising with respect to their thermoelectric capabilities Humer et al. (2013). Since the capabilities of a thermoelectric material is a consequence of the interplay between the transport quantities such as the Seebeck coefficient S, the electrical resistivity , and the thermal conductivity , optimizing these quantities in a material could give rise to a large thermoelectric figure of merit, ZT1, that is required for reasonable performance of a thermoelectric material. Recently it was shown that ZT could be further improved in filled skutterudites by tuning the thermal conductivity based on the so-called Phonon Glass and Electron Crystal (PGEC) concept which was developed for cage-forming structures in general Slack (1995); Beekman et al. (2015); Slack and Tsoukala (1994). In addition, related to the materials studied in this work, it has been shown that by substituting Ge by Sb in LaPt4Ge12 compound, the Seebeck coefficient and resistivity could be enhanced by an order of magnitude at room temperature Humer et al. (2013), thus increasing the playground to optimize the figure of merit ZT.
III Methods
All our calculations are based on the electronic density functional theory method Hohenberg and Kohn (1964); Kohn and Sham (1965) with the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation Perdew et al. (1996) to the exchange-correlation potential. Calculations are performed on the experimental crystal structures having the Im-3 cubic space group Gumeniuk et al. (2010a).
For each compound, we studied in detail the electronic band structure, DOS and FS under various theoretical scenarios such as PBE, PBE+SOC and PBE+SOC+U. For each of these scenarios the rare-earth -electrons were treated either as valence electrons or core electrons. We include, SOC and U in our calculations due to the fact that they are important energy scales for rare earth elements.
Owing to the experimentally reported absence of magnetism in the RPt4Ge12 compounds studied in this work, we performed all our calculations in non-spin polarized states by constraining the total spin moment per unit cell to zero. At the same time, we made sure that all our solutions also have the orbital moments equal to zero for each theoretical scenario considered in this work. Since we set the initial atomic moments to zero, every band should obey time reversal symmetry, . In addition, our structure has inversion symmetry, so we expect which we checked to be the case here. As a consequence, each FS presented in this paper is doubly degenerate.
In this work we combine results from VASP and WIEN2k codes, to take advantage of their strengths. We note that WIEN2k is an all electron code (core + valence electrons), while VASP works only with the valence electrons (while the potential of the atom nuclei and the core electrons is replaced by an effective potential known as a pseudopotential). Even though the basis sets in the two codes differ, as long as the basis set in each code is almost complete basis, the results obtained by the two codes should be very similar. We compared the electronic band structure of the RPt4Ge12 obtained by the two codes for each theoretical scenario used in this work and in each case we found very similar electronic structure, as expected.
Below, we give specific parameters used for the calculations in each code.
VASP: Electronic band structure and DOS calculations utilize the plane-wave based DFT code VASP Kresse and Furthmuller (1996) with the all-electron projector augmented wave (PAW) method Kresse and Joubert (1999). We take the standard VASP PAW potentials, keeping the -orbitals as valence states (except where we specify in core). Increasing plane wave energy cutoffs had no discernible impact on the band structure so we apply the VASP defaults. However, we use the full FFT grids consistent with the energy cutoff to represent charge densities so as to avoid wrap-around errors. Electronic -point meshes are increased to in order to converge the electronic DOS, which we evaluate using tetrahedron integration followed by 0.01eV Gaussian smearing.
WIEN2k: FS and the associated electronic band structure were performed using the full-potential linearized augmented plane wave (FP-LAPW) method as implemented in the WIEN2k code Blaha et al. (2018). The calculations were performed on a 31 31 31 -point mesh. The muffin-tin radii were chosen as 2.50, 2.45 and 2.22 Bohr units for the rare earth ions, Pt and Ge, respectively. In order to have accurate calculations we used RmtKmax = 8. The energy which separates the core and the valence states was chosen to be 10 Ry. Self-consistent criteria for the energy and charge were 10*-4* Ry and 10*-4* electrons, respectively. All other input parameters were used with their default values.
IV Results
IV.1 Overview of Band Structure
Fig. 2 presents electronic band structure and atom-projected DOS for PBE and PBE+SOC approximations, where -electrons are treated as valence electrons. For each band structure plot, the -point path that connects special points in the first Brillouin zone (BZ) is defined as in Ref. Setyawan and Curtarolo, 2010: , , , and .
Several things may be noted in the set of plots in Fig. 2:
(I) From the electronic band structure plots for the RPt4Ge12 compounds for a given approximation (rows in Fig. 2), we see that by going from La to Pr (which is equivalent with increasing the number of -electrons, nf, from 0 to 2) new bands appear above the Fermi level (EF) in the energy range (0.2 - 0.6) eV. The new bands are related to the Ce and Pr -electrons, and although they hybridize with the other bands in the system, we see that the bands in the vicinity of the EF don’t change their shape drastically in CePt4Ge12 and PrPt4Ge12 when compared to LaPt4Ge12 , but they are shifted down in energy with respect to the EF. Since the intersections of the bands with the EF defines points on the 3D FS, by shifting the bands around the EF will change the topology of the FS. Thus, we expect that the topology and the number of the FS in CePt4Ge12 and PrPt4Ge12 to be different from the FS in LaPt4Ge12 .
(II) The atom-projected DOS gives information about the contribution/weight of each atom to the electronic band structure at a given energy. Thus our calculations show that in the La compound, the states at the EF come mostly from the Pt and Ge atomic states and there are few or no La states. By contrast, in the Ce and Pr compounds, besides the Pt and Ge atomic states, we also have states from the rare-earth elements. As we will discuss later in paper this can have implications for the single- versus multi-band superconductivity in RPt4Ge12 compounds.
(III) Comparing plots with and without SOC for a given compound, see Fig. 2, panels (a)-(c) versus (d)-(f), we see that SOC lifts the band degeneracy at special points and along various k-paths in the first BZ and shifts some of the bands by up to a few hundred meV. For example, these effects are especially evident at the and H points and along the -P path. Since SOC has an impact on the electronic band structure, it also has a direct effect on the topology and degeneracy of the FS.
It is well-known that for rare earth elements, the occupied -states can behave as inert localized states in some compounds (and in those cases the -electrons must be treated as core in the theoretical approximations) or the -states can hybridization with the other states in the system (and in those cases the -electrons must be treated as valence electrons). In Appendix A, we show the electronic band structure and atom projected DOS, for the case where the -electrons are treated as core, thereby eliminating hybridization with the other states in the system. From the calculations with -electrons in core, we learn that for a given approximation, these compounds have almost identical electronic band structure and thus almost the same FS as expected (see rows in Fig. 6). Thus, the theoretical approximations where the -electrons are treated as core electrons can not explain the differences between the LaPt4Ge12 and CePt4Ge12 or PrPt4Ge12 compounds. The effects of SOC on the band structure are similar, independently of the way we treat the -electrons (in valence or in core).
IV.2 Projected DOS
In Fig. 3(a)-(c), we show the total DOS per formula unit of RPt4Ge12 compounds together with the atom-projected DOS. In addition, in Fig. 3(d)-(l), we also show the orbital-projected DOS for each atom within the RPt4Ge12 compounds. These calculations were done using the PBE+SOC approximation within VASP with the -electrons treated as valence electrons. In this figure we also show the comparison of our calculations with the available experimental soft X-ray photoemission spectra Nakamura et al. (2010b). We digitized the experimental data and we plot it on top of our calculated DOS in Fig. 3(a) and (c). Since the experimental data is in arbitrary units, we scaled it such that we get the best agreement between the experiment and theory in the energy range around -4 eV. We see that when comparing the experimental data with the total DOS per formula unit, by construction we get a good agreement for the energy range around -4 eV, but the agreement is poor for energy closer to the EF. The reason for this discrepancy is the fact that the photoemission spectra were measured at an incident energy of 1.2 KeV, energy where mostly Pt d states were probed, the other states practically being invisible to this probe due to the very small scattering cross-section at this energy Nakamura et al. (2010b). These discrepancies are expected since the total DOS per formula unit is a sum of all states with equal probability without considering the experimental scattering cross-sections. But if we consider the scattering cross-sections, and we compare the photoemission spectra only with the Pt d DOS as shown in Fig. 3(g) and (i), we see that the agreement between experiment and theory is much better over the full energy range.
Looking at all orbital projected DOS, see Fig. 3(d)-(l), we see that the orbital contributions of the s, p and d states at the EF is similar for all three compounds. The difference is that in the case of Ce and Pr compounds, -states are also present at the EF. This suggests that the electrons forming the Cooper pairs in PrPt4Ge12 might have some additional character besides the , and , while in LaPt4Ge12 their character is mostly , and . Based on our results, and making an analogy with the case of Pb Floris et al. (2007), where the anisotropic contributions of the s, p and d orbital character to the wavefunctions forming the states at the EF leads to anisotropic electron-phonon coupling and multi-band superconductivity, we suggest the possibility of multi-band superconductivity for PrPt4Ge12 and single- or multi-band superconductivity for LaPt4Ge12 . The pseudogaps found in the total DOS of the RPt4Ge12 compounds, might be relevant to possible improvement of their thermoelectric effects, as we will discuss later on in the paper.
IV.3 3D Fermi Surfaces
In Fig. 4 we show the 3D FS (labeled by symbols Sn, n=1 to 6) for the RPt4Ge12 . These FS are calculated in WIEN2k, using the PBE+SOC approximation and treating the -electrons as valence electrons. In addition to the FS, we also show the electronic band structure for each compound in a narrow energy range around the Fermi energy.
As can be seen in Fig. 4(a), LaPt4Ge12 has six FS. The hole or electron pocket character can be ascribed to these FS by considering the corresponding band structure in Fig. 4(b). S1, S2 and S3 are hole pockets, and S5 and S6 are electron pockets. The character of S4 is not as clear from the band structure, see Fig. 4(b). Thus it is interesting to examine the changes of the FS with increasing number of - electrons. With 1 additional electron (compare Ce 4 with La 4), the Fermi energy increases, and the S1 and S2 hole pockets get filled while the S3 pocket is nearly filled, see Fig. 4(c). With the addition of another electron (the case of Pr 4) the S3 Fermi hole pocket is completely filled, see Fig. 4(e). The upshot is that CePt4Ge12 has four FS and PrPt4Ge12 has only three, see Fig. 4(c) and (e). Within the PBE+SOC approximation, the three compounds have in common three large FS, namely S4, S5 and S6. As the number of -electrons increases, S4 shrinks in size and hence has dominant hole character, while S5 grows and S6 is hardly affected. The S1-S3 hole pockets present in LaPt4Ge12 and absent in CePt4Ge12 and PrPt4Ge12 within the PBE+SOC approximation, show up within the PBE+SOC+U approximation for both CePt4Ge12 and PrPt4Ge12 compounds.
The orbital character of the FS may influence the nature of superconductivity. It is claimed that Pb has two Fermi surfaces Floris et al. (2007); Blackford and March (1969), one spherical and one more complex. Owing to the anisotropic orbital character of the wave functions (, and across the two FS), the electron-phonon coupling varies across the FS and between the two FS, thereby giving rise to multi-band superconductivity. It may also be recalled that in MgB2, multi-band superconductivity is given by two FS, both with character (one FS is in character and the other is in character) Tsuda et al. (2004, 2007); Xi (2008); Kortus et al. (2001); Gurevich (2007). If the mechanism for superconductivity in the La and Pr compound is also phonon-mediated, since there are multiple sizable FS as in MgB2, this implies the possibility of multi-band superconductivity in RPt4Ge12 , as pointed out by experiments. In addition, since the Pr compound is shown to have character on the FS (as shown by DOS in Fig. 3), this could lead to different electron-phonon couplings across the FS, similar to the anisotropic electron-phonon coupling found in Pb. Thus, the electron-phonon couplings may be different in PrPt4Ge12 compared to that in the LaPt4Ge12 case, thereby resulting in differences in the type of pairing. In order to ascertain the anisotropic orbital character across the FS, besides looking at the orbital projected DOS which gives an average of the orbital contribution over the all k-points in the BZ, we also looked at the ”fat-band” representation Schwarz et al. (0002) around the EF which gives a detailed description of the orbital contribution at specific k-points in the BZ. The ”fat-band” plots are shown in Appendix B. From the ”fat-band” representation, we find anisotropic orbital character to the 3D FS for all the orbital states coming from R, Pt and Ge atoms.
IV.4 Effects of correlations on DOS and FS
Photoemission measurements on PrPt4Ge12 where the incident energy is scanned through the -resonance of the Pr atoms, reveal the presence states at EF and below. For example, see Fig. 5b where we show the digitized data from Ref. Nakamura et al., 2010b for two incident energies, 1.2 and 0.9 KeV. For incident energy of 1.2 KeV we probe mostly the Pt states (magenta curve) and for incident energy of 0.9 KeV, in addition to Pt states, we also probe the Pr states (black curve). Thus, the difference between the magenta and black curves in Fig. 5b represents the contribution of the Pr states to DOS. Besides small contributions at the EF, the Pr states appear to manifest as two peaks centered around -1 and -4.5 eV which disagrees with our DOS calculations for the PBE+SO approximation shown in Fig. 3f. In order to explain the experimentally observed contribution to DOS, shown in Fig. 5b we need to go beyond the DFT approximations. Repulsive interactions among the tightly bound -orbitals of the Lanthanide atoms can create electronic correlations that are not properly modeled by our PBE density functional. A first step in correcting this effect is to supplement the DFT Hamiltonian with onsite Coulomb energies Dudarev et al. (1998); Anisimov et al. (1993), which we take into account using a single effective Hubbard parameter . We take eV, a typical value for Lanthanides, to illustrate the qualitative effect. For comparison, we also explore other values of .
DOS computed within the PBE+SOC+U with =0 eV shown in Fig. 3 reveal that the -band is partly occupied, with the net occupation of =0.85 for Ce and =2.07 for Pr. For , occupied and empty orbitals split proportionately to for Pr, but the splitting is far weaker for Ce, consistent with the respective values of . For example, in Fig. 5 we show the Pt(d)+Pr(f) DOS for CePt4Ge12 and PrPt4Ge12 compounds computed using 2.5 and 5.0 eV. As expected, the effect of U is to increase the splitting between the occupied and unoccupied states, thus moving the theoretical peak of occupied states in the Pt(d)+Pr(f) DOS, to lower energies. Comparing our theoretical DOS with the experimental data, we find that the position of the theoretical DOS could explain the experimental peak shape at -1eV for U = 2.5 eV or the peak at -4.5 eV for U = 5.0 eV, but our theory can not explain both experimental peaks at the same time. This suggests that PBE+SOC+U method captures part of the electronic correlation, but in order to explain the experimental findings a theory beyond DFT+ is required. One such theory, where correlation effects are treated more rigorously is the density functional theory + embedded dynamical mean field theory (DFT+eDMFT) Haule et al. (2010); Haule (2018); web .
Since DOS at the EF are affected by U, the FS of CePt4Ge12 and PrPt4Ge12 will also be affected. Since the positions of bands with respect to directly relate to the topology of the FS, we also expect changes in the topology of the FS. While additional details are given in Appendix B (see Fig. 10 for CePt4Ge12 and Fig. 11 for PrPt4Ge12 ), we note a few salient points here. The band corresponding to the S6 FS does not change too much with increasing indicating that it is mostly , and in character. The bands corresponding to S4 and S5 change with increasing , S4 being more affected than S5, suggesting that these FS having stronger character than S6. To confirm the contribution of states to the S4 and S5 FS for finite values, we have also computed the FS for the case where the -electrons are treated as core electrons (in this case the Fermi surface has no contribution). The fact that the topology of the FS for finite differs from the case where the -electrons are treated as core electrons (labeled in Appendix B Fig. 10 and Fig. 11), confirms once again the presence of character in the Fermi surface of CePt4Ge12 and PrPt4Ge12 , independently of the strength of correlations. In addition, we also learn that the presence or absence of the S1, S2 and S3 FS in CePt4Ge12 and PrPt4Ge12 depends on the strength of the correlations.
IV.5 Effective mass
While our calculations show that the rare-earth filled skutterudites have complex band structures and DOS, simple estimates of effective mass can nevertheless be obtained by considering the linear coefficient of the specific heat obtained experimentally in these materials. The electronic specific heat in metals, for ( being the Fermi temperature), is given by: Kittel (2005)
[TABLE]
For a spherical FS with parabolic bands, the linear coefficient of specific heat, , is related to the electronic DOS at the EF as , with being the single isotropic effective mass , and the Fermi momentum. Assuming that the theoretical density of states at the EF, , can likewise be related to a single band mass , we obtain estimates of the mass enhancement over the band mass, , by comparing the measured (obtained from ) and the computed density of states at EF, . While in reality, the density of states and effective masses are expected to be anisotropic, thermodynamic measurements, such as specific heat, are averages over FS; hence simple analyses as this may provide a reasonable estimate of .
Table 1 summarizes results drawn from the various theoretical scenarios we have considered in this work. In particular we show the calculated total DOS at the EF, D and mass enhancement over the band mass, . For a given theoretical scenario, different values of estimated for each skutterudite correspond to ’s obtained from specific heat measurements by different groups. The estimated mass enhancements, , in the range of 2-4, are giving an indication of the correlation strength.
IV.6 Thermoelectric Applications
There has been sustained interest in the skutterudites as possible next generation thermoelectric materials. Viability for such application is measured in terms of the figure of merit, (preferably greater than 1) given by
[TABLE]
where contains electronic and lattice contributions to thermal conductivity, is the electrical conductivity, and the Seebeck coefficient. To achieve requires an optimal interplay of various quantities. Our theoretical calculations suggest that these materials are metallic, with a sizable DOS at and thus a small Seebeck coefficient is expected at room temperature. One of the ways to increase , is to increase the Seebeck coefficient which, in simple terms, is given by: Ziman (1972); Ketterson (2016)
[TABLE]
where , , , , and are the charge, carrier density, density of states, mobility, and the Fermi energy, respectively. Thus S depends on the density of states and inversely on the carrier density, as well as the logarithmic derivative of the carrier mobility with energy. In the above derivation of , conductivity , and mobility have been taken to be: and , where is the carrier lifetime which can be energy-dependent, and the carrier effective mass. Thus in effect, the second term in can depend on the energy variation of the lifetime. The derivative of accounts for the way in which electron current is distributed in energy. Thus, if is an increasing function of energy, a relatively higher proportion of current will be carried by the more energetic carriers, and these will transport a larger amount of heat. The above simple result provides a sense of the expected behavior of thermopower for metals at low temperatures . In a more realistic scenario, one would need to express conductivity in terms of Fermi surface area, and treat density of states and lifetime in more details.
As was discussed in Ref. Humer et al., 2013, large S can be achieved by driving the system close to a metal to insulator transition where the charge carrier density, , diminishes. is not expected to be appreciable in the metallic state of these materials due to large charge carrier density, n and large density of states at the EF, . The predicted small values of S in RPt4Ge12 compounds has been confirmed experimentally Humer et al. (2013); White et al. (2013); Gumeniuk et al. (2011); Huang et al. (2014). Although S is small in the stoichiometric compounds, doping LaPt4Ge12 with electrons, by replacing Ge with Sb, namely LaPt4Ge12-xSbx, drives the system close to a metal-insulator transition, and beyond = 5 the system is insulating. Close to the metal-insulator transition, the thermoelectric properties of the LaPt4Ge12-xSbx compound are greatly enhanced vs. the undoped LaPt4Ge12 compound Humer et al. (2013). Experiments showed that increasing the electron doping enhances the overall temperature dependence of the resistivity and the charge carrier density , both consistent with an overall enhanced temperature dependence of S Humer et al. (2013). To understand these experimental findings, in Ref. Humer et al., 2013, the temperature dependence of the Seebeck coefficient S(T) was computed for various values and it was shown that S(T) increases drastically in the vicinity of the metal-insulator transition, e.g. by an order of magnitude at room temperature. For the LaPt4Ge12 , the possibility of the metal-insulator transition was connected with the existence of a pseudogap above for =0.
Our total DOS calculations for RPt4Ge12 (see Fig. 3) show the existence of notable pseudogaps around 1-1.5 eV above , independently of the theoretical scenario we used in our calculations (PBE, PBE+SOC or PBE+SOC+U). Based on Eq. (3) for , the presence of such a pseudogap suggests that large values of S could be produced by tuning these metallic systems towards an insulating state or to a ”bad metal” state by electron doping. In addition, the measurements of the temperature dependent resistivity in PrPt4Ge12-xSbx Jeon et al. (2016) which show the same trend as in LaPt4Ge12-xSbx Humer et al. (2013) further suggests the possibility of increasing ZT in RPt4Ge12 .
Based on our total DOS calculations showing pseudogap features above the EF and using the similarity of previous resistivity measurements in RPt4Ge12 , we suggest that electron doped CePt4Ge12 and PrPt4Ge12 compounds could also exhibit larger figures of merit than the stoichiometric compounds, as it was already proved for doped LaPt4Ge12 .
V Discussion
We have presented a comprehensive study of band structure, DOS and FS for f-electron systems of type RPt4Ge12 (where R = La, Ce, Pr), using various theoretical scenarios such as PBE, PBE+SOC and PBE+SOC+U within the two choices of treating the -electrons, as valence or core electrons. Our calculations show several bands crossing the EF giving rise to multiple FS.
As expected, when the -electrons are treated as part of core, the three compound show almost identical band structure, and thus similar FS, for a given theoretical scenario. Scenarios where SOC is present or absent, show different FS due to the fact that SOC lifts band degeneracy and shift bands, thus increasing the number of FS (especially at point) and changing slightly the FS topology.
When the -electrons are treated in valence the three compounds show some slight differences. Although SOC has the same trends on the band structure as mentioned before, the increasing number of -electrons from 0 to 2 and the increase of correlations by adding the Hubbard parameter U on top of the PBE+SOC approximation, makes things more interesting. Our calculations within PBE+SOC approximation reveal the presence of six FS for the LaPt4Ge12 , four of them having a hole like character and the other two electron like character. While increasing the number of -electrons, the hole pockets get filled and three of them disappear going from LaPt4Ge12 to PrPt4Ge12 , while the electron pockets increase their surface. The effects of filling the hole pockets at point are reversed with increasing correlations effects between the -electrons within the PBE+SOC+U approximation. Although these compounds have two electron-like and one hole-like FS in common, independently of the theoretical scenario used, the hole pockets at point are dependent on the theoretical scenario we used in our calculations. Thus, de Hass van Alphen Rourke and Julian (2012); Blake et al. (2015); Brasse et al. (2013) experiments are desirable to validate the topology and the number of FS in RPt4Ge12 compounds.
While our theoretical calculations cannot directly explain superconductivity, they may have significance with respect to the multi-band superconductivity or anisotropic nature of the pairing, as indicated by experiments on PrPt4Ge12 and LaPt4Ge12 . Our calculations find a small amount of Pr-4 states admixed with Ge- states in PrPt4Ge12 , a feature obviously different from that in LaPt4Ge12 . This is confirmed by soft X-ray photoemission experiments on PrPt4Ge12 Nakamura et al. (2010a); Nakamura et al. (2012) that show a finite contribution of Pr 4 to the states near . Additionally, superconducting gaps are effected Singh et al. (2016); Jeon et al. (2016) by Ce substitution of Pr, i.e. Pr1-xCexPt4Ge12 , suggesting that the filler atoms may affect superconductivity. Based on these, it is conceivable that superconductivity is a consequence of Cooper pairs formed by the s, p and d electrons of the Pt and Ge atoms in the LaPt4Ge12 , while superconductivity in the PrPt4Ge12 is more unusual since in addition to s, p, d electrons we also have -electrons at the EF. As we have remarked earlier, since the PrPt4Ge12 has anisotropic character on the FS, while the LaPt4Ge12 does not, this may lead to different electron-phonon couplings, and hence different forms of pairing. Thus it would be interesting to calculate electron-phonon couplings in these systems.
In addition, we have shown that the total DOS has a pseudogap which could imply the tunability of these materials through a metal to insulator transition and consequently the possibility of improving the thermoelectric properties of these materials. While DFT calculations are not able to treat the energy variation of mobility (and hence lifetime) that can modify results for , a method such as DMFT, which is able to calculate finite-T lifetime, may provide further understanding of .
Finally we mention that our results are consistent with previous DOS calculations Gumeniuk et al. (2008, 2011) that comprise a limited set of results and a small subset of our more extensive work. However, our band structure and FS results differ substantially from those presented in a recent paper Chandra et al. (2016), which used the PBEsol approximation. We also performed calculations using the PBEsol approximation and found the results to be very similar to those obtained using the PBE approximation. Our results also reveal challenges confronted by DFT methods and its extension for correlated materials. We showed that the DFT +U method can only qualitatively explain the soft X-ray photoemission experiments on PrPt4Ge12 . Thus, it would be desirable to use more advance methods for correlated materials, such as DFT + eDMFT, in order to explain quantitatively the experimental results.
VI Acknowledgements
MW acknowledges support from the Department of Energy under grant DE-SC0014506. KQ acknowledges a QuantEmX grant from ICAM and the Gordon and Betty Moore Foundation, Grant GBMF5305, which partly funded this work. G.L.P. and K.H. were supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, as a part of the Computational Materials Science Program, funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. We thank Di Xiao, G. Malcolm Stocks, Gabriel Kotliar, Greg Boebinger, and Carmen Almasan for useful discussions.
VII Appendix
VII.1 Electronic band structure: -electrons in ’Core’
In some systems, the states of -electrons behave as inert localized states, hybridizing very little or at all with the other states in the system. To understand the effects of -electron hybridization, we present the results of calculations when the -electrons are treated as core electrons. In Fig. 6 we show these calculations within the PBE and PBE+SOC approximations. We find that the electronic band structure within a given approximation for the RPt4Ge12 compounds is almost identical when the -electrons are treated as core. This implies that all the interesting low temperature properties of CePt4Ge12 and PrPt4Ge12 are mostly due to the electrons states and their hybridization with the other states in the system.
VII.2 Fat-band Representation
By projecting the band wave-function onto the atomic orbitals of a particular atom, we obtain the band structures shown by solid lines + open circles, where the size of the circles at an energy and momentum point are proportional to the absolute value of the wave-function amplitude corresponding to that given atom Schwarz et al. (0002). It may be noted that in these plots the size of the circle corresponds to the atomic character and is shown for one atom only, even thought the band wave-functions forming the bands have contributions from all the atoms (of the same type) within the unit cell.
In Fig. 7, we plot the electronic band structure with the atomic character for LaPt4Ge12 . Looking at the (a), (b) and (c) panels corresponding to the La, Pt and Ge atomic character, we see that around the Fermi energy the electronic character comes mostly from the Ge and Pt atoms, consistent with the previously presented atom projected DOS in Fig. 2. At the same time, looking at the the first column in Figure 3, we see that for Pt atoms the largest DOS at the EF comes and states and for the case of Ge ion the maximum DOS comes from states. Thus, it may tempting to say that for the LaPt4Ge12 compound the electrons involved in superconductivity would mostly be Ge states plus some and Pt states. With reference to the notation in Fig. 4 for FS, here we can see that the S1, S2 and S6 FS are mostly Ge in character, while S3, S4 and S5 have contributions from both Ge and Pt ions. Notably, La is largely absent from the FS.
Figure 8 shows fat-band plots for CePt4Ge12 . The discussion here follows that of Fig. 7 for LaPt4Ge12 , with the additional fact that for CePt4Ge12 the FS have moderate character in addition.
Likewise, in Fig. 9, we show fat-band plots for PrPt4Ge12 . A similar discussion can be made as before for the case of LaPt4Ge12 and CePt4Ge12 with the additional fact that for PrPt4Ge12 the FS have strong character. In addition, for the PrPt4Ge12 compound if we look at Fig. 9(a), where we show the Pr contribution to the FS, we see that the character of the FS is anisotropic. For example, compare the strength (size of the circles) of Pr character at the two intersections of the S4 band with the EF between points P and N. In analogy with Pb, one could suggest that anisotropic band character could lead to anisotropic electron-phonon couplings for these FS. This makes for a strong case for calculating electron-phonon couplings in the LaPt4Ge12 and PrPt4Ge12 skutterudites, and exploring if -electron coupling leads to multi-gap pairing in the case of PrPt4Ge12 .
VII.3 Effects of on Bands and Fermi Surfaces
From band structure plots, Fig. 10 (a) - (b), and Fig. 11 (a) - (b), it is obvious that the band corresponding to the S6 FS doesn’t change much as we increase the correlation, thus this FS is quite robust which indicates that it is probably mostly , and in character. On the other hand, plotting the S4 and S5 FS versus U (see Fig. 10 (c) - (d), and Fig. 11 (c) - (d), we find that S4 is the most affected, followed by S5. This suggests that the S4 and S5 FS have stronger character than S6. To confirm the contribution of states to the S4 and S5 FS for , we have also computed the FS for the case where the -electrons are treated as core electrons (in this case the FS has no contribution). These results are shown in Fig. 10 (c) - (d), and Fig. 11 (c) - (d), labeled as . The fact that the topology of the FS for is different than that of , confirms once again the presence of character in the FS of CePt4Ge12 and PrPt4Ge12 . In addition, if we compare the bands in Fig. 4d (=0) for CePt4Ge12 with Fig. 10 () we see for example, that with increasing , band S3 falls below along the path P-H, but rises above at .
Comparing Fig. 4(f) (=0) for PrPt4Ge12 with Fig. 11 (), we see that S3 similarly rises above at , creating a new FS sheet. Thus, in both CePt4Ge12 and PrPt4Ge12 compounds, we find that depending on the strength of correlation parameter U, the S1, S2 and S3 FS can be present or absent. As mentioned in the discussion section, de Haas-van Alphen measurements could sort out the puzzle of FS number and indirectly give information of the correlation strength in the CePt4Ge12 and PrPt4Ge12 compounds.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jeitschko and Braun (1977) W. Jeitschko and D. Braun, Acta Crystallographica Section B 33 , 3401 (1977), URL https://doi.org/10.1107/S 056774087701108 X . · doi ↗
- 2Gumeniuk et al. (2010 a) R. Gumeniuk, H. Borrmann, A. Ormeci, H. Rosner, W. Schnelle, M. Nicklas, Y. Grin, and A. Leithe-Jasper, Zeitschrift f u ¨ ¨ 𝑢 \ddot{u} r Kristallographie Crystalline Materials 225 , 531 (2010 a).
- 3B. Maple et al. (2008) M. B. Maple, Z. Henkie, R. E. Baumbach, T. A. Sayles, N. P. Butch, P.-C. Ho, T. Yanagisawa, W. M. Yuhasz, R. Wawryk, T. Cichorek, et al., Journal of the Physical Society of Japan 77 , 7 (2008), eprint https://doi.org/10.1143/JPSJS.77SA.7, URL https://doi.org/10.1143/JPSJS.77SA.7 . · doi ↗
- 4Aoki et al. (2007) Y. Aoki, T. Tayama, T. Sakakibara, K. Kuwahara, K. Iwasa, M. Kohgi, W. Higemoto, D. E. Mac Laughlin, H. Sugawara, and H. Sato, Journal of the Physical Society of Japan 76 , 051006 (2007), eprint https://doi.org/10.1143/JPSJ.76.051006, URL https://doi.org/10.1143/JPSJ.76.051006 . · doi ↗
- 5Maisuradze et al. (2009) A. Maisuradze, M. Nicklas, R. Gumeniuk, C. Baines, W. Schnelle, H. Rosner, A. Leithe-Jasper, Y. Grin, and R. Khasanov, Phys. Rev. Lett. 103 , 147002 (2009), URL https://link.aps.org/doi/10.1103/Phys Rev Lett.103.147002 .
- 6Maisuradze et al. (2010) A. Maisuradze, W. Schnelle, R. Khasanov, R. Gumeniuk, M. Nicklas, H. Rosner, A. Leithe-Jasper, Y. Grin, A. Amato, and P. Thalmeier, Phys. Rev. B 82 , 024524 (2010), URL https://link.aps.org/doi/10.1103/Phys Rev B.82.024524 .
- 7Takeda and Ishikawa (2000) N. Takeda and M. Ishikawa, Journal of the Physical Society of Japan 69 , 868 (2000), eprint https://doi.org/10.1143/JPSJ.69.868, URL https://doi.org/10.1143/JPSJ.69.868 . · doi ↗
- 8Yamamoto et al. (2006) A. Yamamoto, S. Wada, I. Shirotani, and C. Sekine, Journal of the Physical Society of Japan 75 , 063703 (2006), eprint https://doi.org/10.1143/JPSJ.75.063703, URL https://doi.org/10.1143/JPSJ.75.063703 . · doi ↗