maze: Heterogeneous Ligand Unbinding along Transient Protein Tunnels
Jakub Rydzewski

TL;DR
The paper introduces 'maze', a new PLUMED 2 module that efficiently samples multiple ligand unbinding pathways from proteins, providing detailed atomistic insights into dissociation processes without extensive parameter tuning.
Contribution
It presents a novel implementation of a method for sampling heterogeneous ligand unbinding pathways, integrated into an open-source enhanced sampling framework.
Findings
Successfully reconstructed ligand unbinding pathways in model systems.
Demonstrated the method's ability to identify multiple pathways.
Showed the approach's flexibility and minimal parameter dependence.
Abstract
Recent developments in enhanced sampling methods showed that it is possible to reconstruct ligand unbinding pathways with spatial and temporal resolution inaccessible to experiments. Ideally, such techniques should provide an atomistic definition of possibly many reaction pathways, because crude estimates may lead either to overestimating energy barriers, or inability to sample hidden energy barriers that are not captured by reaction pathway estimates. Here we provide an implementation of a new method [J. Rydzewski \& O. Valsson, J. Chem. Phys. {\bf 150}, 221101 (2019)] dedicated entirely to sampling the reaction pathways of the ligand-protein dissociation process. The program, called \texttt{maze}, is implemented as an official module for PLUMED 2, an open source library for enhanced sampling in molecular systems, and comprises algorithms to find multiple heterogeneous reaction…
Click any figure to enlarge with its caption.
 Figure 1
Figure 1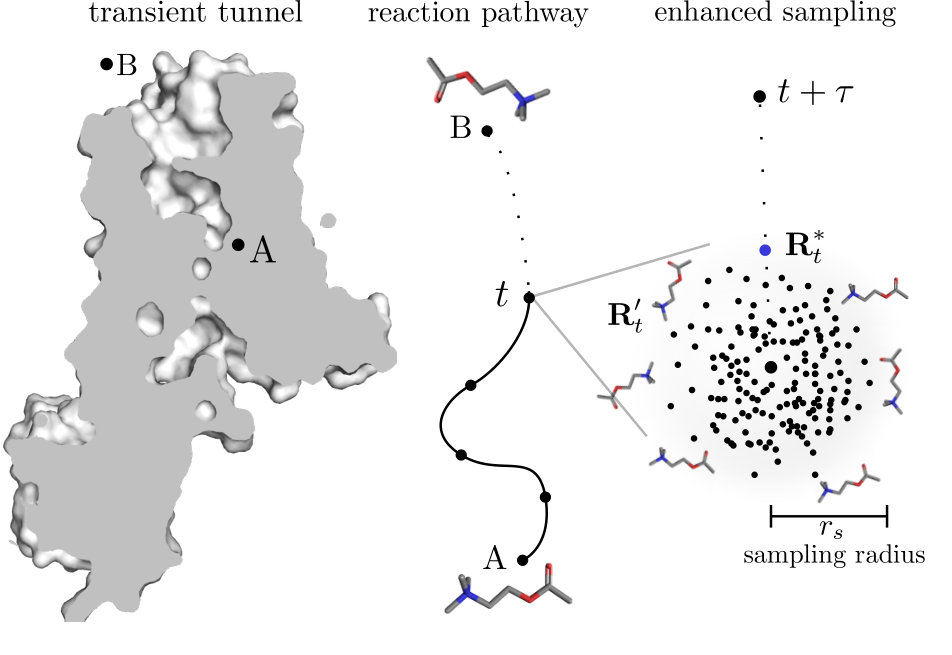 Figure 1
Figure 1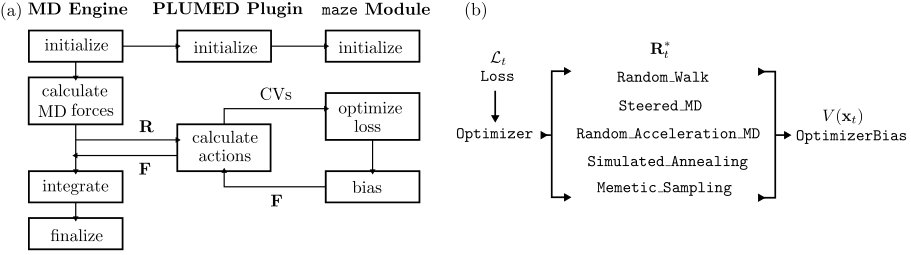 Figure 2
Figure 2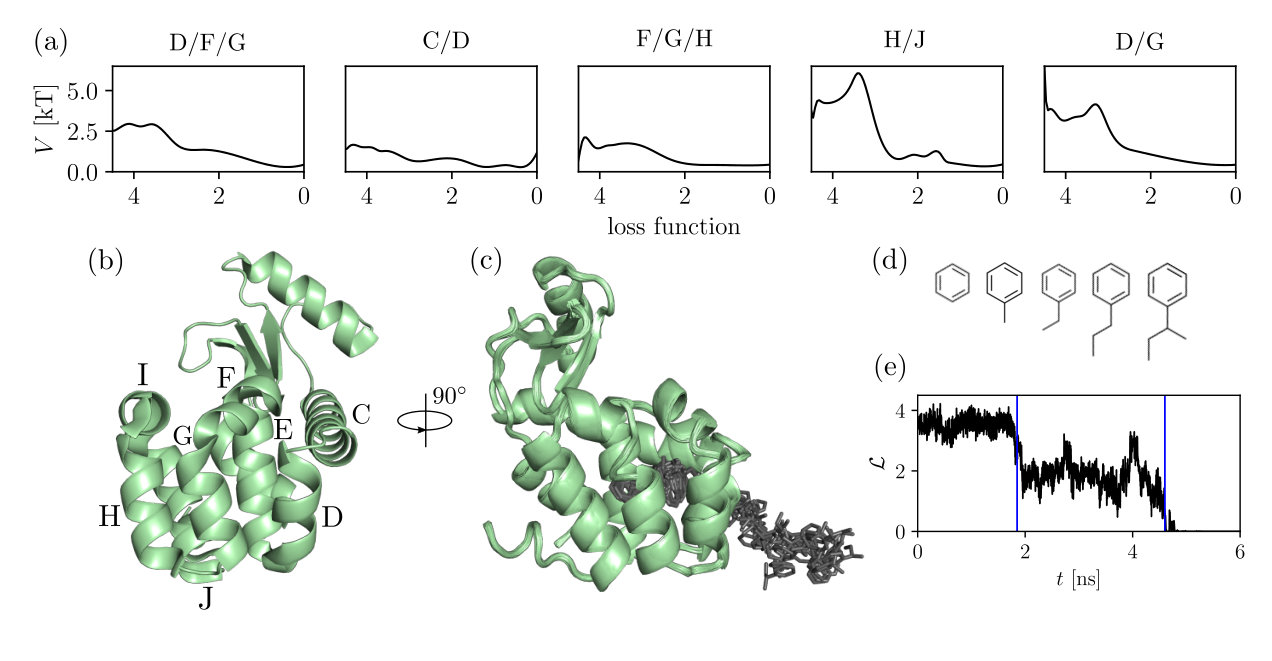 Figure 3
Figure 3| Program title: | maze | |
| Program version: | 1.0 | |
| Program files DOI: | http://dx.doi.org/10.17632/x5zsgzxcnx.1 | |
| Licensing provisions: | L-GPL-3.0 | |
| Programming language: | C++11 | |
| Nature of problem: | Finding heterogeneous unbinding reaction pathways of ligand transport processes such as dissociation and diffusion in ligand-protein systems during molecular dynamics simulations. | |
| Solution method: | Enhanced sampling molecular dynamics method with non-convex optimization techniques performed on-the-fly during biased simulations. | |
| Unusual features: | maze is a module for the PLUMED 2 software, it must be used in conjunction with this software, embedded in a molecular dynamics code such as Gromacs, NAMD, LAMMPS, OpenMM, and Amber. | |
| Additional comments: | Program Github repository: https://maze-code.github.io |
| PDB IDa | ligand | biasing rate [Å/ps] | no. simulations | no. pathways | pathways |
| 4w52 | benzene | 0.02 | 300 | 5b | D/F/G, C/D, F/G/H, H/J, D/G |
| 4w53 | toluene | 0.02 | 50 | 4 | C/D, F/G/H, H/J, D/G |
| 4w54 | ethylbenzene | 0.03 | 50 | 4 | D/F/G, C/D, F/G/H, H/J |
| 4w55 | -propylbenzene | 0.03 | 50 | 5 | D/F/G, C/D, F/G/H, H/J, D/G |
| 4w56 | -butylbenzene | 0.03 | 50 | 5 | D/F/G, C/D, F/G/H, H/J, D/G |
| a All the protein-ligand system are reported in Ref. Merski et al., 2015. | |||||
| b Reported in Ref. Rydzewski and Valsson, 2019. | |||||
| method | simulation time | requirements | reference |
|---|---|---|---|
| MTD | s | putative reaction coordinate for path-CVs | Laio and Parrinello Laio and Parrinello (2002) |
| infrequent MTD | s | putative reaction coordinate for path-CVs | Wang et al. Wang et al. (2018) |
| TRAM | s | combining biased and long unbiased simulations | Wu et al. Wu et al. (2016) |
| SGOOP | — | initial guess of CVs monitored during test runs | Tiwary and Berne Tiwary and Berne (2016) |
| RAVE | ns | putative reaction coordinate | Ribeiro and Tiwary Lamim Ribeiro and Tiwary (2018) |
| volume-based MTD | ns | — | Capelli et al. Capelli et al. (2019) |
| maze | ns | — | Rydzewski and Valsson Rydzewski and Valsson (2019) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
maze: Heterogeneous Ligand Unbinding along Transient Protein Tunnels
Jakub Rydzewski
Institute of Physics, Faculty of Physics, Astronomy and Informatics, Nicolaus Copernicus University, Grudziadzka 5, 87–100 Torun, Poland
Abstract
Recent developments in enhanced sampling methods showed that it is possible to reconstruct ligand unbinding pathways with spatial and temporal resolution inaccessible to experiments. Ideally, such techniques should provide an atomistic definition of possibly many reaction pathways, because crude estimates may lead either to overestimating energy barriers, or inability to sample hidden energy barriers that are not captured by reaction pathway estimates. Here we provide an implementation of a new method [J. Rydzewski & O. Valsson, J. Chem. Phys. 150, 221101 (2019)] dedicated entirely to sampling the reaction pathways of the ligand-protein dissociation process. The program, called maze, is implemented as an official module for PLUMED 2, an open source library for enhanced sampling in molecular systems, and comprises algorithms to find multiple heterogeneous reaction pathways of ligand unbinding from proteins during atomistic simulations. The maze module requires only a crystallographic structure to start a simulation, and does not depend on many ad hoc parameters. The program is based on enhanced sampling and non-convex optimization methods. To present its applicability and flexibility, we provide several examples of ligand unbinding pathways along transient protein tunnels reconstructed by maze in a model ligand-protein system, and discuss the details of the implementation.
Manuscript version 1.0 release. See https://arxiv.org/abs/1904.03929 for all versions.
Date. .
ligand unbinding; enhanced sampling; reaction pathways; collective variables; non-convex optimization
Program Summary
I Introduction
Atomistic simulations such as molecular dynamics (MD) provide sufficient temporal and spatial resolution to study complex physical processes. However, despite substantial progress in method development, conventional MD simulations are unable to access the longtime scales on which infrequent events occur Rydzewski and Valsson (2019); Valsson et al. (2016); Rydzewski and Nowak (2017a, b); Noé (2018). This so-called ergodic breakdown is associated with high energy barriers separating important energy minima. If these barriers are much higher than the thermal energy , the system will be kinetically trapped in a metastable energy minimum during the course of simulations. To access longer time scales and reach the regime of high energy barriers of rare events enhanced sampling methods are needed. Finding collective variables (CVs) and estimating reaction pathways for rare events in a crude manner often leads to the overestimation of energy barriers, and thus, underestimation of exponentially dependent kinetic rates, which arise from inability to capture intrinsic degrees of freedom. This is related to the degeneracy of microscopic configurations originating from sampling wrong CVs and reaction pathways, which is likely to mask hidden energy barriers.
A typical example of an event that occurs on the long time scales is ligand-protein dissociation. The main computational hurdle, which makes reaction pathways for ligand-protein unbinding difficult to sample during the conventional MD simulation, stems from accounting for transient internal features of proteins, such as tunnels. However, these features are important for mechanisms, thermodynamics, and kinetics of ligand unbinding, as the structural flexibility of tunnels and channels allows proteins to facilitate binding by adapting to ligands along possibly multiple pathways to the binding site. This transient dynamics poses a severe challenge even for enhanced sampling methods that have been used to sample reaction pathways in ligand unbinding so far. For instance, such methods either do not account for protein dynamics, interpolate reaction pathways linearly between bound and unbound states Heymann and Grubmüller (2000), or sample protein tunnels by employing simple heuristics as random walks Lüdemann et al. (2000).
New enhanced sampling methods, however, are often not used in the wider community because easy to use implementations are not available. A few of open source platforms serving as plugins to MD engines, e.g., PLUMED 2 Tribello et al. (2014); Bussi and Tribello (2018), MIST Bethune et al. (2018), SSAGES Sidky et al. (2018), and i-PI Kapil et al. (2018) are available. These open source platforms have significantly lowered the technical barrier for the application of advanced sampling methods in MD simulations. Considering the potential impact that enhanced sampling-based methods have on atomistic simulations, it is of considerable interest to develop new methods for already existing open source platforms that serve as the interface between enhanced sampling and atomistic simulations packages, e.g., LAMMPS Plimpton (1995), Gromacs Abraham et al. (2015), or NAMD Phillips et al. (2005). The contribution of this work is to provide the implementation of the maze code which can be interfaced with many MD engines via PLUMED 2. We provide a specific-use package dedicated entirely to simulating ligand-protein unbinding. Given the utilities provided by maze, running MD simulations of ligand unbinding is much easier through a simple input file for PLUMED 2, and enables enormous flexibility in creating new components for the method such as biasing potentials and optimizers.
The manuscript is organized as follows. In Sec. II, the theoretical framework of the maze software is provided. We show in detail how ligand-protein interactions are calculated, and further optimized using a coarse-grained CV for the adaptive bias potential. Next, we provide a protocol showing how the bias potential is used in maze. We focus on the software and technical detail of the implementation in Sec. III, including data preparation, numerical examples, and availability. Finally, we conclude our work in Sec. VI.
II Method
The method, which is the base component of maze, is able to find multiple diverse reaction pathways of ligand unbinding along transient protein tunnels Rydzewski and Valsson (2019). In this section, we describe how this can be achieved. The method does not require an initial guess of intermediates nor a unbound state, which is very important, as many existing methods for calculating reaction pathways rely on it. Its only prerequisite is the knowledge of the X-ray binding site the ligand resides within. Since the searching procedure is performed iteratively during MD simulations, the method takes into account protein dynamics which is important to observe the openings and closings of transient tunnels and exits while probing the time scale on which these conformational changes occur.
The method relies on the following concepts:
- –
Carefully selected CV describes interactions in a ligand-protein system. In our protocol, this CV is optimized on-the-fly during the MD simulation of ligand unbinding. Interestingly, as this part of the method resembles many similarities with machine learning Goodfellow et al. (2016); Mehta et al. (2019), the CV can be seen as a specific loss function tailored to the optimization problem of ligand unbinding, in the sense that a loss function is a function that maps an event onto a real number intuitively representing some “cost” associated with that event. Thus, we will use these two names interchangeably throughout this article (Sec. II.1).
- –
Optimization method seeks to minimize this loss function. A non-convex optimization method is used to minimize this loss function with specific constraints that are based on the surrounding protein tunnel or channel. These constraints define a local CV space to sample ligand conformations within the protein tunnel. In this manner maze learns the protein tunnels accessible for ligand transport (Sec. II.2).
- –
Adaptive biasing potential enforces ligand dissociation. An adaptive bias potential is employed to enforce the transition between the ligand current conformation and a localized minimum of the CV in the following MD simulation steps. Once the bias reaches the computed minimum of the loss function, and if the ligand is still within the protein matrix, maze repeats the cycle (Sec. II.3).
In the following subsections we describe the aforementioned concepts in detail, and show how each component of the method is connected to each other.
II.1 Loss Function for Ligand Unbinding
Let consider a ligand-protein system consisting of a -set of ligand coordinates and a -set of protein coordinates . A CV that successfully models ligand unbinding needs to fulfill several important characteristics that are based on the notion that the problem of finding reaction pathways of ligand unbinding can be solved using optimization techniques Rydzewski and Valsson (2019). The loss function needs to:
- –
Tend to infinity as the ligand moves too close to the protein. This prohibits the method from sampling ligand configurations that would clash with the protein.
- –
Decrease as the ligand unbinds from the protein. This provides a coarse estimate of how deeply ligand conformations are buried within the protein tunnels.
Such loss functions can be used to obtain ligand unbinding pathways through iterative minimization within MD simulations, however, we note that selecting a particular CV for this may depend on the decision if one wants to study effective interaction energy in a ligand-protein system, or use an approximate formula fulfilling the listed criteria. We note that in any case the reference ligand unbinding pathways should be used as an initial guess for methods like metadynamics Laio and Parrinello (2002); Barducci et al. (2008) or variationally enhanced sampling Valsson and Parrinello (2014) if thermodynamic and kinetic properties of the system are to be reconstructed.
In the current version maze implements an exponential loss function that can be used to describe ligand unbinding reaction pathways:
[TABLE]
where is the rescaled distance between atoms of the ligand and the protein of the th pair, is the number of ligand-protein atom pairs, and are positive scaling parameters (or hyperparameters). Several combinations of these parameters have been tested in our previous studies Rydzewski and Valsson (2019); Rydzewski et al. (2018a). We underscore that the number of ligand-protein atom pairs is based on recalculating the neighbor list between the ligand and the protein, and it varies during the unbinding process as the ligand neighborhood changes.
The loss function is calculated in the ligand neighborhood, and thus it needs a particular type of bounds for sampling solutions only from this neighborhood. Clearly, the procedure should not sample possible ligand conformations in the full accessible conformational space, because that may render spurious trajectories, e.g., the next ligand conformation may be behind some steric barrier, and overcoming it would result in an nonphysical dissociation process. To this aim, we automatically estimate the ligand neighborhood during the optimization procedure as a sphere centered on the ligand conformation from MD, of radius calculated as the minimal distance between ligand-protein atom pairs,
[TABLE]
For a schematic figure showing the ligand neighborhood, see the right panel of Fig. 1. Using such adaptive constraints limits the search for a global minimum of the loss function to an easily found local minimum in the proximity of the ligand conformation, and allows to sample non-linear reaction pathways in any ligand-protein system during enhanced sampling simulations. Thus, the local neighborhood of the ligand depends only on the current ligand position and the minimal distance between ligand-protein atoms, which can be roughly described as the width of the sampled protein tunnel Rydzewski and Valsson (2019).
The loss function resembles many similarities to coordination number, and can be seen as a coarse-grained CV which describes how deep the ligand is buried within the protein matrix, or as an effective interaction energy of a ligand-protein complex. For an example of how the loss function behaves during an MD simulation, see Fig. 3e. The loss function identifies intermediate states along unbinding pathways, and although may fluctuate during simulations, should decrease as the ligand unbinds from the protein. We note also that this CV can be biased during simulations by any CV-based enhanced sampling method, e.g., metadynamics Laio and Parrinello (2002). Similar CVs were used by enhanced sampling method in ligand binding studies Do et al. (2013); Spitaleri et al. (2018).
II.2 Finding Minima of the Loss Function
The minimization of the loss function can be performed using any robust non-convex optimization method. There are examples of many approaches of using such techniques in atomistic simulations Hansmann and Wille (2002); Rydzewski and Nowak (2015); Rydzewski et al. (2018b). The maze code implements several such methods to optimize the CV chosen for ligand unbinding in the local space near the ligand. The full list of the implemented optimizers with their corresponding keywords in PLUMED 2 is given below:
- –
MAZE_SIMULATED_ANNEALING: simulated annealing Kirkpatrick et al. (1983); optimizes the biasing direction based on the dynamically-adjusted Metropolis-Hastings method. For detailed description, see Ref. Rydzewski and Valsson, 2019.
- –
MAZE_MEMETIC_SAMPLING: memetic algorithm Rydzewski and Nowak (2015, 2016, 2017c); Rydzewski et al. (2018a) with learning heuristics such as stochastic hill climbing and an adaptive Solis-Wets search; performs exhaustive search using evolutionary algorithms.
In these methods, the ligand is encoded as its center-of-mass position. The sampling procedure performed during the optimization phase draws a random translation vector for the ligand conformation from the current step in the MD simulation. If the translated ligand position is preferable in terms of the loss function, it is taken as the next conformation to which the ligand needs to be bias toward, as this direction of biasing results in the decreases of the loss function. This sampling procedure is repeated during the optimization phase to unveil the position of the lowest loss function value. At each step of MD, the center-of-mass difference between the MD ligand conformation and the optimal one serves as the direction of ligand unbinding.
Apart from the above optimizers, the maze implements also standard techniques for sampling ligand-protein dissociation that are currently used by the community, and can be used instead of the optimizers:
- –
MAZE_RANDOM_WALK: random walk; steers the ligand unbinding in a random direction.
- –
MAZE_STEERED_MD: steered MD Heymann and Grubmüller (2000); steers the ligand unbinding in a predefined direction.
- –
MAZE_RANDOM_ACCELERATION_MD: random acceleration MD Kokh et al. (2018); changes the direction of the ligand unbinding randomly if the ligand does not overcome a predefined threshold distance.
The above methods are not optimizers per se, but they can be used as samplers for the biasing direction. In contrast to the optimizers, which provide an optimal direction of biasing, the standard methods do not provide optimal solutions for the adaptive bias, but return some direction of biasing (given by the above listing).
II.3 Adaptive Biasing of Reaction Pathways
Let denote sampled ligand-protein conformations. Once the optimal ligand conformation is calculated, the ligand is biased in the direction of the optimal solution at time along a selected transient protein tunnel. This stage is performed by biasing the positions of the ligand atoms with an adaptive harmonic potential defined as:
[TABLE]
where is the biasing rate, is the interval at which the loss function is minimized, and is a force constant. The bias potential given by Eq. 3 is a generalization of a simple harmonic biasing potential introduced in Ref. Heymann and Grubmüller, 2000, modified to be able to bias curvilinear unbinding pathways.
The biasing procedure is schematically depicted in Fig. 1, and summarized as follows:
Initialize the MD simulation and set time . 2. 2.
Sample ligand conformations within the protein tunnel using constraints defined by Eq. 2. 3. 3.
Solve using a non-convex optimization technique. 4. 4.
Calculate a bias potential for the ligand driving toward the optimal conformation using Eq. 3. 5. 5.
Run steps of the enhanced sampling simulation with the defined bias potential . 6. 6.
Set . 7. 7.
Repeat the steps 2–5 during the MD simulation until reaches numerical zero when the ligand unbinds fully from the protein tunnel. 8. 8.
Stop the MD simulation. 9. 9.
Return putative reaction pathway coordinates learned during many optimization swaps.
To find multiple unbinding pathways many simulations must be performed.
III Software
III.1 Installation
The latest release of maze can be downloaded from the maze web page maz (a). It is also possible to clone the maze Github repository by appending the maze repository address to the command git clone. The maze code is an optional module of maze and thus it needs to be enabled when configuring the compilation with the --enable-modules=maze flag (or simply --enable-modules=all) when running the configure script. Further information on compiling and installing PLUMED 2 can be found on in the PLUMED documentation plu . The maze module is currently available in the official development version of PLUMED 2, and will be released in PLUMED 2.6 soon.
III.2 Implementation
Fig. 2a summarizes the workflow used in maze to perform a ligand unbinding simulation. As maze is a module for PLUMED 2, it can be used with a wide range of MD codes, i.e., Gromacs and NAMD. Input files are defined using the PLUMED input syntax. Within an input file every line is an instruction for PLUMED to perform some action: calculating CVs, performing analysis or running an enhanced sampling simulation. The PLUMED syntax as well as all keywords are available in the PLUMED documentation plu .
The maze module comprises three base classes located within src/maze of the main directory of the installation (with the PLUMED 2 base classes in parentheses):
- –
Loss (Colvar): the loss function which act as the CV for ligand unbinding,
- –
Optimizer (Colvar): the optimizer which provides a method to minimize the loss function,
- –
OptimizerBias (Bias): the adaptive bias potential to enforce ligand-protein dissociation along transient tunnels.
The implementation is provided in the object-oriented paradigm of programming, and it is very easy to extend by adding new loss functions, optimizers (or standard techniques), and biases. For a dependency scheme of the maze module, see Fig. 2b. Classes in maze can be extended by providing new subclasses. The Loss and Optimizer classes are base classes from which any derived class can inherit functions.
This setup allows for new ligand unbinding methods to be developed using a single unified interface, fully decoupled from MD codes. Every subclass that derives from Optimizer must provide a registerKeywords function that parses method-specific variables, and an optimize function which is responsible for finding the optimal direction to bias ligand unbinding. For instance, the Random_Walk’s optimize function returns a random unit vector and calculates the loss function to score a particular ligand-protein configuration. The optimize functions of Steered_MD and Simulated_Annealing return a predefined unit vector, and the optimal biasing direction calculated by simulated annealing, respectively. In that sense, the maze module provides an unified implementation of methods capable of calculating ligand unbinding reaction pathways. For a schematic workflow of the maze module, see Fig. 2a, which depicts how an MD engine is connected through the PLUMED plugin to the maze module.
Regarding performance, the current maze version support inter-process communication via OpenMP to parallelize the computation of the critical slower parts of the implementation such as calculating the loss function for each ligand and updating the neighbor list of ligand-protein atoms, which altogether render the sampling fast, as opposed to other implementations used currently for finding ligand exit pathways from proteins, for instance random acceleration MD implemented as a TCL script in NAMD.
III.3 Data Preparation
From the user perspective, before running the unbinding simulations using maze, a chosen ligand-protein system of interest must be modeled. Although PLUMED is interfaced with many MD codes, here we describe the preparation of a ligand-protein complex only using Gromacs. For a very useful tutorial for beginners regarding ligand-protein complexes, we redirect the readers to Ref. Lemkul, 2018. If the force-field parameters are not accessible within a standard force field, we refer to a server for parametrizing small organic molecules Dodda et al. (2017). Once the ligand-protein complex is modeled and ready for the production runs, the user needs to define the loss function (List. 1) that is to be biased during the simulations. Next, the optimizer must be defined (List. 2), and then the biasing potential (List. 3). All method-specific keywords and examples of usage are described and available in the PLUMED documentation maz (b).
The maze package requires very few ad hoc parameters, with the main information that used needs to provide being the X-ray structure of a ligand-protein complex. Ideally, the ligand conformation should be inferred from the crystallographic structure, but if the crystallographic binding site with the bound ligand is not know, then a docking procedure should suffice. Apart from the structure, the user needs to provide a simple configurational file which consists of the loss function (List. 1), the optimizer for the loss function (List. 2), and the adaptive bias potential for the optimizer (List. 3).
In List. 1 we show how to define a loss function. As shown in Eq. 1, the loss function suitable for ligand unbinding simulations is parametrized by the positive arguments, and they can be provided using the PARAMS argument as a tuple. The defined loss function can be then passed to the definition of an optimizer.
Aside from optimizer-specific parameters and the loss function, every optimizer should define the ligand and protein atoms using the LIGAND and PROTEIN keywords, the number of minimization steps (N_ITER), and the stride (in MD steps) at which the optimization is launched during the simulation. Here, we used simulated annealing as an example optimizer to minimize the loss function. The initial value of the temperature-like parameter (PROBABILITY_DECREASER) was modified according to the geometric cooling scheme: , where is the iteration number, and (COOLING). The defined optimizer should be then passed to the bias definition. We used the input files shown in Lists. 1–3 to run MD simulations of ligand unbinding.
All questions regarding maze module for PLUMED can be asked using Gitter; for details see the Supporting Materials.
IV Example Ligand Unbinding Pathways
IV.1 Model Ligand-Protein System
To show how the calculations done by the maze module are performed, example ligand unbinding pathways from the T4 lysozyme L99A mutant (T4L) were calculated for the following series of congeneric ligands bound to T4L: benzene, toluene, ethylbenzene, -propylbenzene, and -butylbenzene (Tab. 1). T4L is frequently used as a model system to study ligand unbinding from proteins (Fig. 3b). The ligands were parametrized using the LigParGen server Dodda et al. (2017) from the group of W. L. Jorgensen. The OPLS/AA force field Kaminski et al. (2001) was used in the simulations; the SPC water model Berendsen et al. (1981) was used to solvate the system. The system was neutralized by adding 6 CL ions. The modeling was performed using Gromacs 5.1.3 Abraham et al. (2015).
The MD simulations were launched using Gromacs 5.1.3 Abraham et al. (2015) with a 2-fs time step. The system was simulated using periodic boundary conditions and the particle mesh Ewald method for long range electrostatics Darden et al. (1993). The system was first minimized, and then equilibrated in the NVT ensemble (10 ns) and in the NPT ensemble (10 ns) with the Parrinello-Rahman barostat Martoňák et al. (2003) set at 1 bar. The stochastic velocity rescaling thermostat Bussi et al. (2007) was used to keep temperature at 300 K with T4L and benzene coupled. Bonds were constrained using LINCS Hess et al. (1997). The unbinding simulations, in which the maze module was employed, were simulated in the NVT ensemble.
To efficiently calculate distances between the ligand and the protein we used a parallelized neighbor list search which was recomputed every 0.5 ps with a cut-off of 0.7 nm using OpenMP. The optimization procedure was run every 200 ps using simulated annealing and the loss function given by Eq. 1. The bias rate was set to or depending on the ligand size, and the biasing constant was . To sample different ligand unbinding pathways for the ligands (Fig. 3d), we run 200 10 ns trajectories.
Our results show that the resulting ligand unbinding trajectories from the T4L system can be classified into several different reaction pathways. Although thermodynamic and kinetic quantities are important for studying ligand unbinding, the structural representation of reaction pathways is as important. Clearly, the pathways show different structural mechanisms of the benzene unbinding, which is underlined additionally by the different values of the bias potential deposited along the optimized loss function (Fig. 3a). Since, the detailed analysis of the benzene unbinding pathways was published recently Rydzewski and Valsson (2019), here we only focus on a qualitative comparison of the reaction pathways. The sampled reaction pathways are in very well agreement with recent computational studies. Namely, we found two unbinding pathways reported by Mondal et al. Mondal et al. (2018), a dominant unbinding pathway found by Wang et al. Wang et al. (2016, 2017), one pathway that resulted from using Gaussian-accelerated MD Miao et al. (2015), and four benzene escape pathways found using temperature-accelerated MD Nunes-Alves et al. (2018). For a detailed analysis of benzene unbinding pathways from T4L, see Ref. Rydzewski and Valsson (2019).
Additionally, the ligand selectivity of choosing a particular T4L pathway can be observed also for more complex then benzene ligands: toluene, ethylbenzene, -propylbenzene, and -butylbenzene. Our results show that even for more complex ligands, the reaction pathways are very similar to the benzene dissociation pathways (Tab. 1), which can be explained intuitively as ligands similar in size to -butylbenzene are not enough to change the state of the apolar T4L cavity from the closed to open conformation Merski et al. (2015).
V Discussion
V.1 Related Methods
While the residence time and binding free energy are a very important for ligand unbinding, an equally if not more important are the unbinding pathway adopted by the ligand as the kinetic and thermodynamic quantities depend on the structural definition of the ligand unbinding reaction pathways Pramanik et al. (2019). Therefore, here we compare the method with other computational techniques that are able to sample ligand unbinding reaction pathways.
Tab. 2 shows frequently used CV-based enhanced sampling methods as well as newly introduced methods. The methods are compared by the MD simulation time required to get a ligand unbinding reaction pathway and requirement for obtaining such pathways. Methods like metadynamics (MTD), and its variant which allows for calculating kinetic quantities for biased MD simulations, infrequent MTD Wang et al. (2018), need an initial reaction pathway on which path-collective variables Branduardi et al. (2007) can be defined. Recently introduced SGOOP Tiwary and Berne (2016) and RAVE Lamim Ribeiro and Tiwary (2018) require a rough estimate of several pre-selected CVs that define a multidimensional initial pathway that is improved during simulations. A different approach which combines biased MD simulations with long unbiased trajectories is the transition-based reweighting analysis method (TRAM) Wu et al. (2016).
Currently, a volume-based variant of MTD Capelli et al. (2019) and the method implemented in the maze module for PLUMED are capable of calculating ligand unbinding reaction pathways with no requirement for an initial guess of the pathway. One important distinction should be underlined here: the variant of MTD requires an extensive post-processing procedure to recover unbinding pathways Capelli et al. (2019), unlike the method presented here which gives a ligand unbinding pathway as a result of a biased MD simulation. At this stage, however, maze is unable to calculate free energy barriers along the pathways, which is not a problem for any MTD based method, but only a rough estimate of energy barriers by calculating averaged biased potential along the reaction pathways Rydzewski and Valsson (2019). This averaged bias potential, in the limit of slow pulling should give a correct energy barrier. Therefore, we think that maze can serve as an ideal starting point for MTD, SGOOP or RAVE.
V.2 Parameters Transferability
The parameters required by the optimization procedure in the maze module are not sensitive, and therefore can be applied to other ligand-protein complexes. This is because of the local conformational space of the diffused ligand, which adapts as the ligand dissociates through the protein by estimating the tunnel width. The local space of the ligand is calculated automatically by Eq. 2, which makes it adaptable to any ligand-protein system. The only parameters that need to be selected before the production runs are the biasing rate introduced in Eq. 3 which defines how fast the biasing potential moves along the optimized reaction pathway, and the interval at which the optimization is re-launched during an MD simulation. Since the reaction pathways found by the maze module are expressed as piecewise functions that are linear over all intervals, the interval should be chosen as the minimal number of unbinding direction re-calculations needed to approximate a curved reaction pathway for a given ligand-protein complex.
V.3 Convergence
The convergence of the biased simulations can be tested ensuring the times of ligand dissociation from a long-lived metastable bound state obey Poisson statistics Salvalaglio et al. (2014). Analysis of the unbinding time distribution obtained from the biased simulations can be performed by comparing the Poisson cumulative distribution function (CDF; ) with the empirical cumulative distribution function (ECDF) obtained from the unbinding probability distribution (i.e., the histogram of the number of unbinding events observed in the biased simulations over time) from trajectories for each unbinding pathway Salvalaglio et al. (2014). The distance between CDF and ECDF which quantify their similarity can be calculated using a Kolmogorov-Smirnov test. This procedure should be used to calculate the distance for every reaction pathway found by the method, and, if the distances between ECDF and CDF are small then no additional production runs are necessary. For an example of using this analysis, we refer to Ref. Rydzewski and Valsson, 2019.
VI Conclusion, Limitations, and Future Works
We introduced the maze module for the PLUMED 2 software, which implements a method to find multiple heterogeneous reaction pathways of ligand unbinding from proteins. To the best of our knowledge, it is the first contribution of enhanced sampling methods fully dedicated to ligand-protein unbinding. The module is implemented in C++ and interfaced to PLUMED 2 as a new module, similarly to, e.g., the VES code ves . The method performs enhanced sampling of a coarse-grained variable that is optimized during MD simulations to enforce ligand unbinding. The maze code can be used with MD codes that can be interfaced with PLUMED 2, e.g., Gromacs, or NAMD.
In addition, we provided the detailed description of the sampling in maze that requires using adaptive biasing and non-convex optimization. We showed the performance of the maze module on a model ligand-protein system, and analyzed reaction pathways for ligand unbinding from the T4 lysozyme L99A mutant, including input files needed to run the simulations.
As for the limitations of the method—we note that the bias potential can be used to estimate the energy barriers along the pathways only in a limited manner, and for a full thermodynamic description methods like metadynamics Laio and Parrinello (2002); Barducci et al. (2008), or variationally enhanced sampling are needed Valsson and Parrinello (2014). At the current stage of development the maze package enables finding multiple heterogeneous ligand unbinding reaction pathways which can be used as an initial reaction pathway for more advanced enhanced sampling methods.
The maze module for PLUMED 2 provides a robust and efficient method to enforce ligand unbinding, and by providing a good guess of ligand unbinding pathways it may be used to limit the computational time spend to sample thermodynamic and kinetic properties of complex physical systems with other methods, also provided by the PLUMED 2 software. In future work, we plan to focus on using different bias potentials that would allow to compute free energy estimates for each reaction pathway.
All the data and PLUMED input files required to reproduce the results reported in this paper are available on PLUMED-NEST (www.plumed-nest.org), the public repository of the PLUMED consortium The PLUMED Consortium (2019), as plumID:19.056.
Acknowledgements.
During its development maze was partially supported by the National Science Center grants (2015/19/N/ST3/02171, 2016/20/T/ST3/00488, and 2016/23/B/ST4/01770). The example MD simulations were computed at the Interdisciplinary Center for Modern Technologies, Nicolaus Copernicus University, Poland. We would like to thank all the people who provided valued suggestions, and made time to discuss the code privately with the author. In particular, we thank O. Valsson, M. Zieliński, and P. Różański for critically reading the article, G. Bussi for help in including the maze module in the official PLUMED 2 release, and H. Grubmüller, W. Nowak, and M. Parrinello for guidance.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rydzewski and Valsson (2019) J. Rydzewski and O. Valsson, J. Chem. Phys. 150 , 221101 (2019).
- 2Valsson et al. (2016) O. Valsson, P. Tiwary, and M. Parrinello, Annu. Rev. Phys. Chem. 67 , 159 (2016).
- 3Rydzewski and Nowak (2017 a) J. Rydzewski and W. Nowak, Phys. Life Rev. 22–23 , 58 (2017 a).
- 4Rydzewski and Nowak (2017 b) J. Rydzewski and W. Nowak, Phys. Life Rev. 22–23 , 85 (2017 b).
- 5Noé (2018) F. Noé, ar Xiv preprint ar Xiv:1812.07669 (2018).
- 6Heymann and Grubmüller (2000) B. Heymann and H. Grubmüller, Phys. Rev. Lett. 84 , 6126 (2000).
- 7Lüdemann et al. (2000) S. K. Lüdemann, V. Lounnas, and R. C. Wade, J. Mol. Biol. 303 , 797 (2000).
- 8Tribello et al. (2014) G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni, and G. Bussi, Comput. Phys. Commun. 185 , 604 (2014).