On structure-stabilizing electronic interferences in bcc-related phases. A research report
Heinrich Solbrig

TL;DR
This paper investigates how electronic interferences stabilize structures in bcc-related phases, using first-principles calculations and a unified interference concept to explain stability, hybridization, and electronic properties.
Contribution
It introduces a VEC-based Hume-Rothery framework with joined planar-radial interferences to analyze electronic stabilization in cubic phases near bcc sublattices.
Findings
Electronic interference enhances charge redistribution and lowers band energy.
Stabilizing networks and hardness relate to specific interference patterns.
Comparison with FLAPW results confirms the interference-based interpretation.
Abstract
This study deals with cubic crystals where the contents of the simple cubic unit cells are close to nnn-bcc sublattices ( = 2: diamond- and zinc-blende type, = 3: -brasses). First-principle results on the electronic structure are obtained from augmented LMTO-ASA calculations and interpreted within a VEC-based Hume-Rothery concept which employs joined planar-radial interferences to treat interference and hybridization on the same footing. We show that the charge redistribution supports enhanced electronic interference which causes the band energy to decrease. Several topics are included such as stabilizing networks, hardness and -to- transfer, co-operation of interferences, interplay between local radial order and global planar order, electron-per-atom ratio, and the comparison with recent FLAPW-based results.
Click any figure to enlarge with its caption.
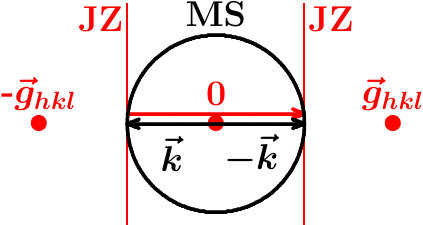 Figure 1
Figure 1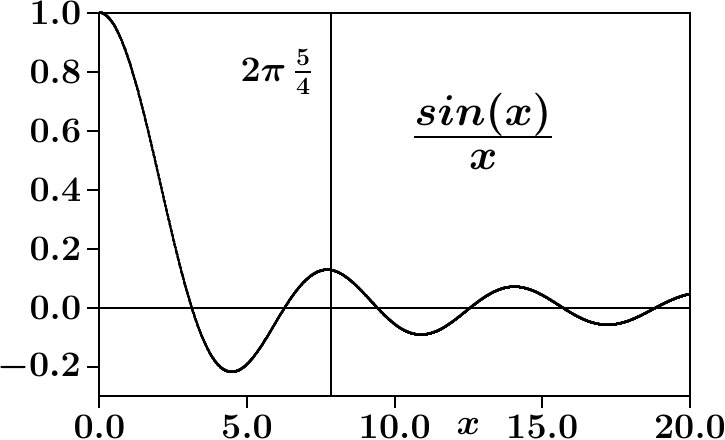 Figure 2
Figure 2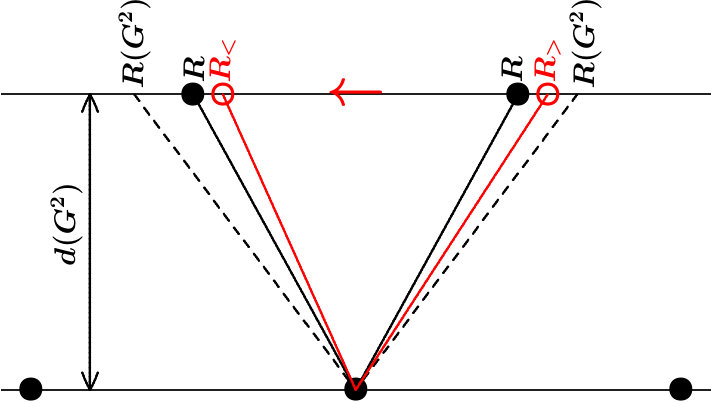 Figure 3
Figure 3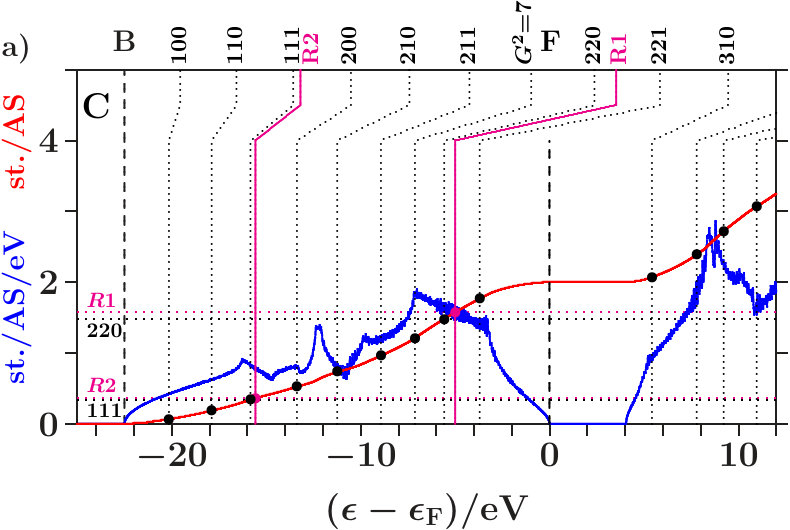 Figure 4
Figure 4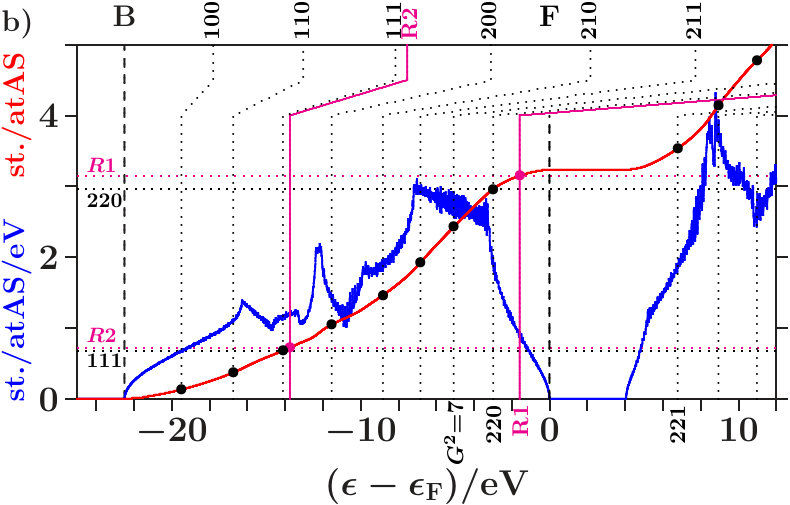 Figure 4
Figure 4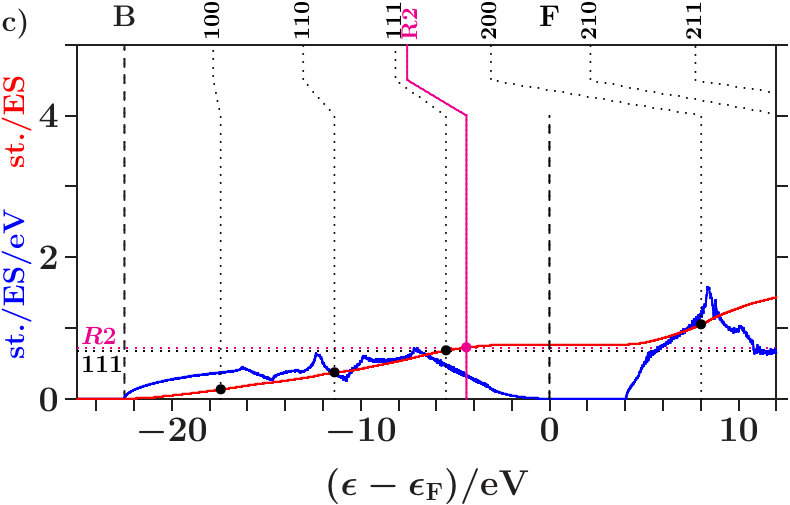 Figure 4
Figure 4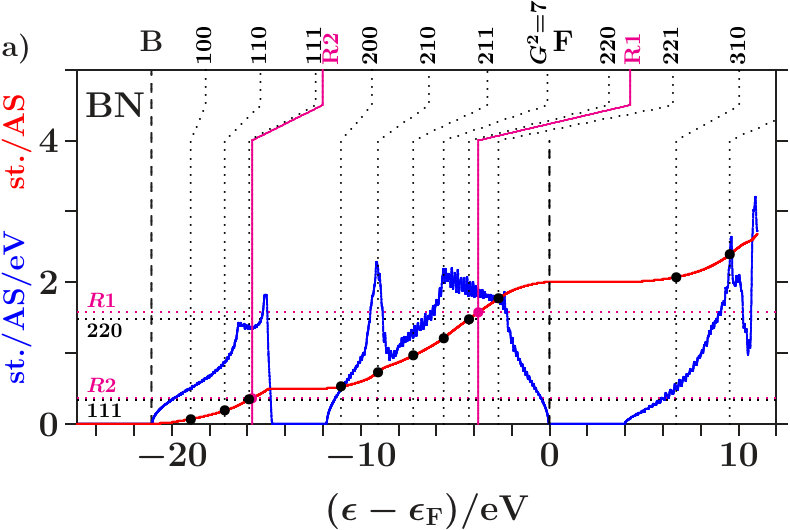 Figure 5
Figure 5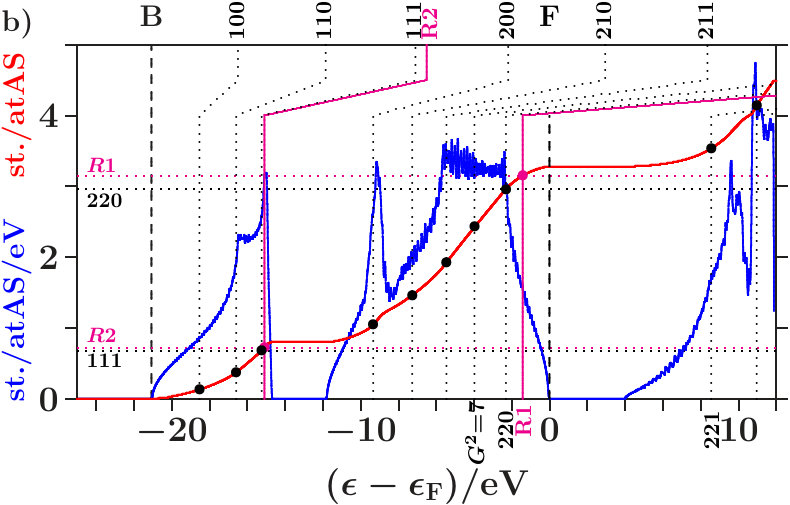 Figure 5
Figure 5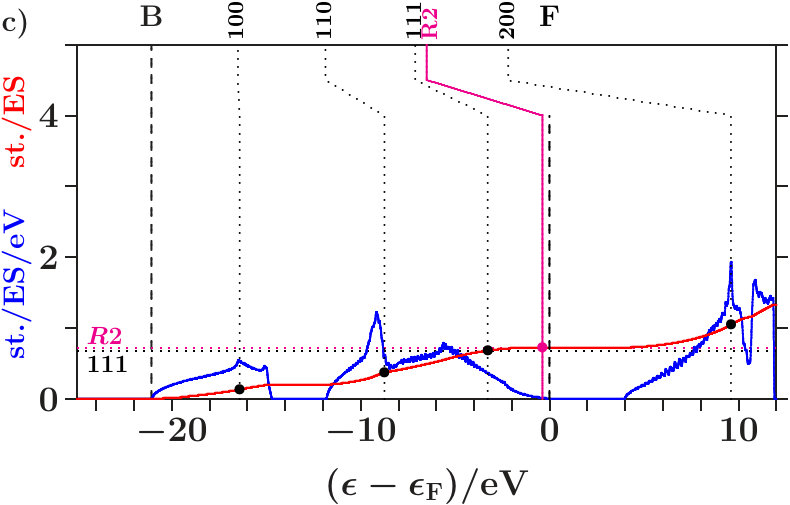 Figure 5
Figure 5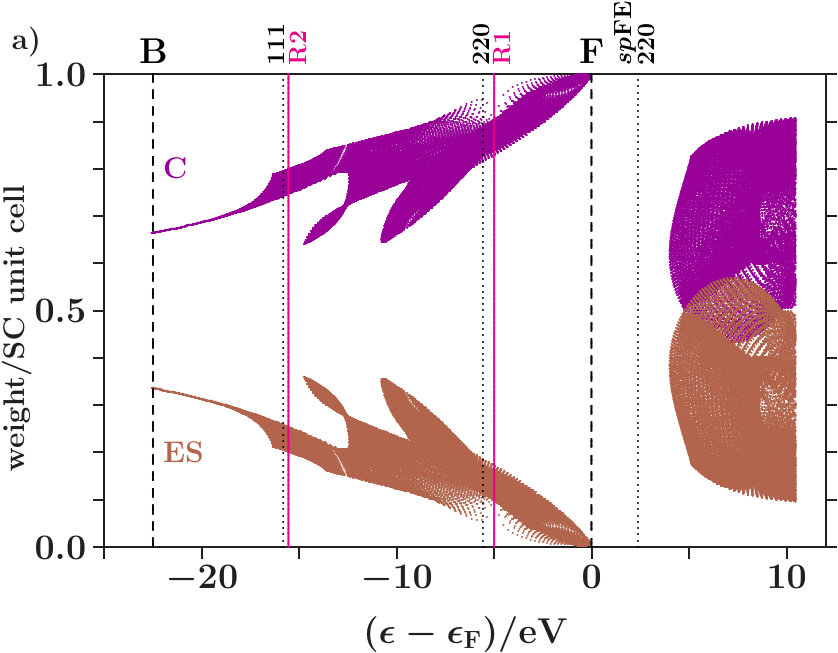 Figure 6
Figure 6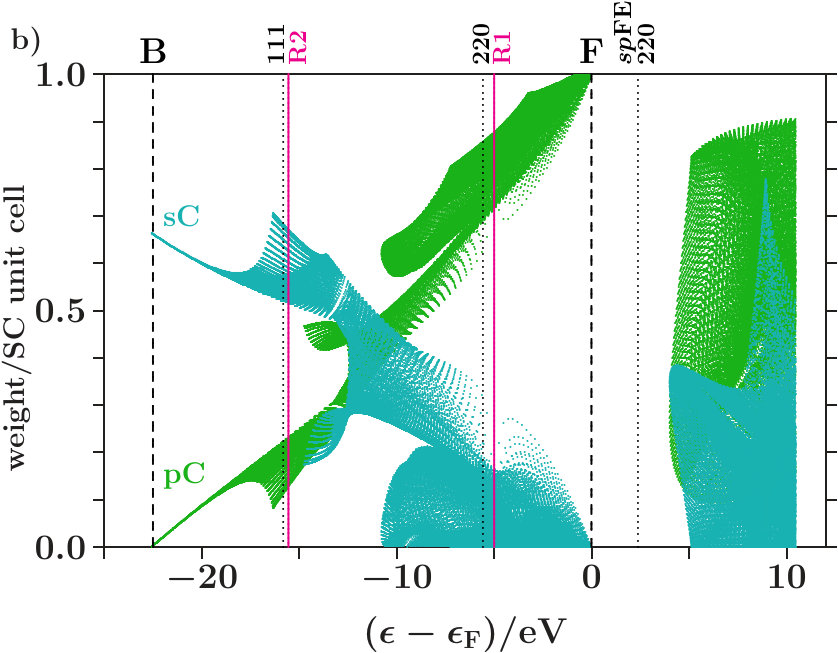 Figure 6
Figure 6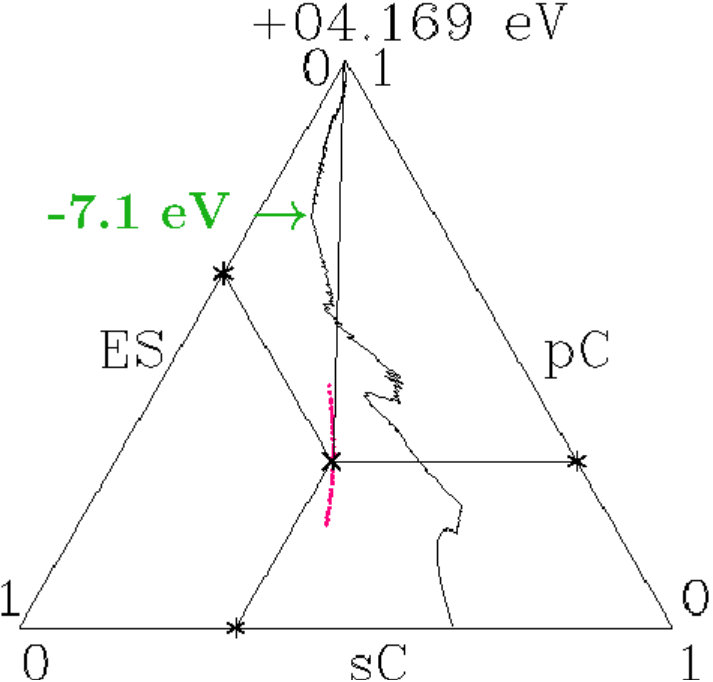 Figure 7
Figure 7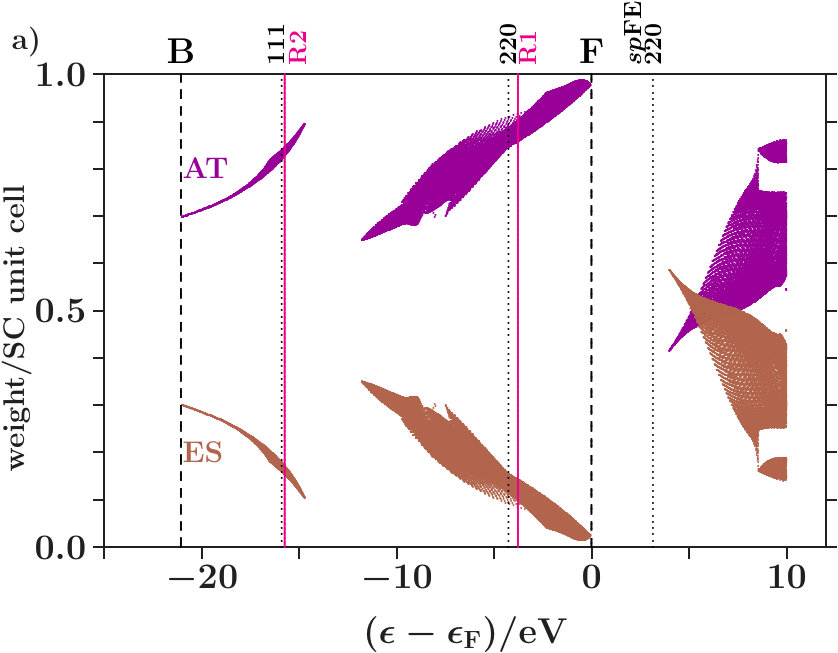 Figure 8
Figure 8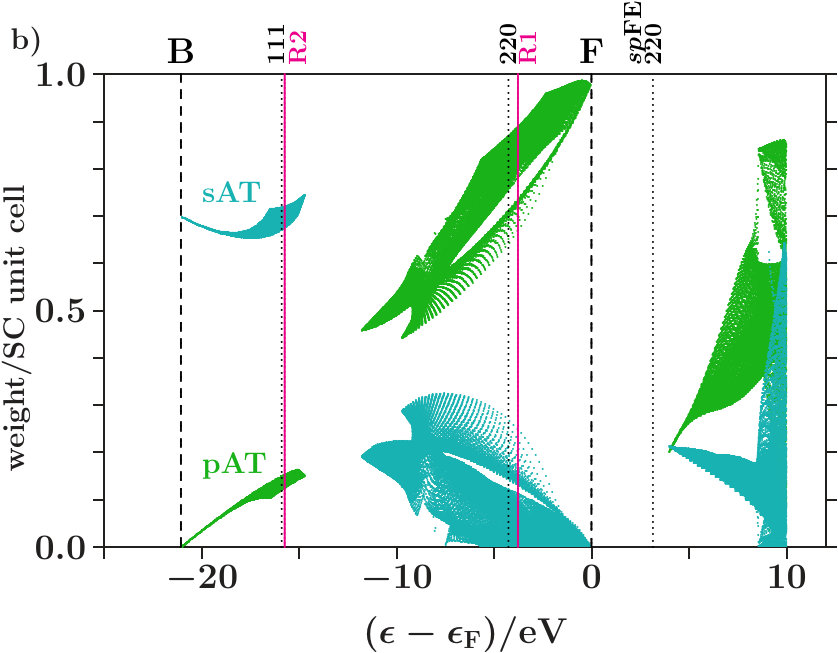 Figure 8
Figure 8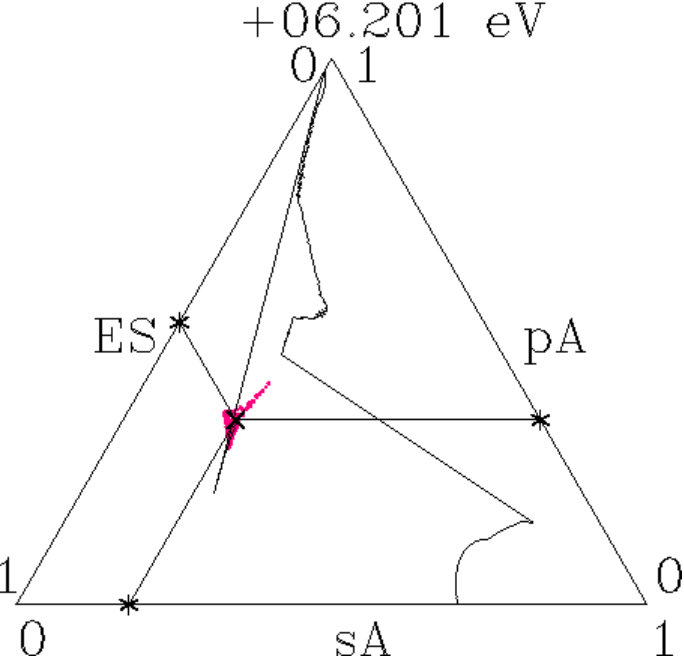 Figure 9
Figure 9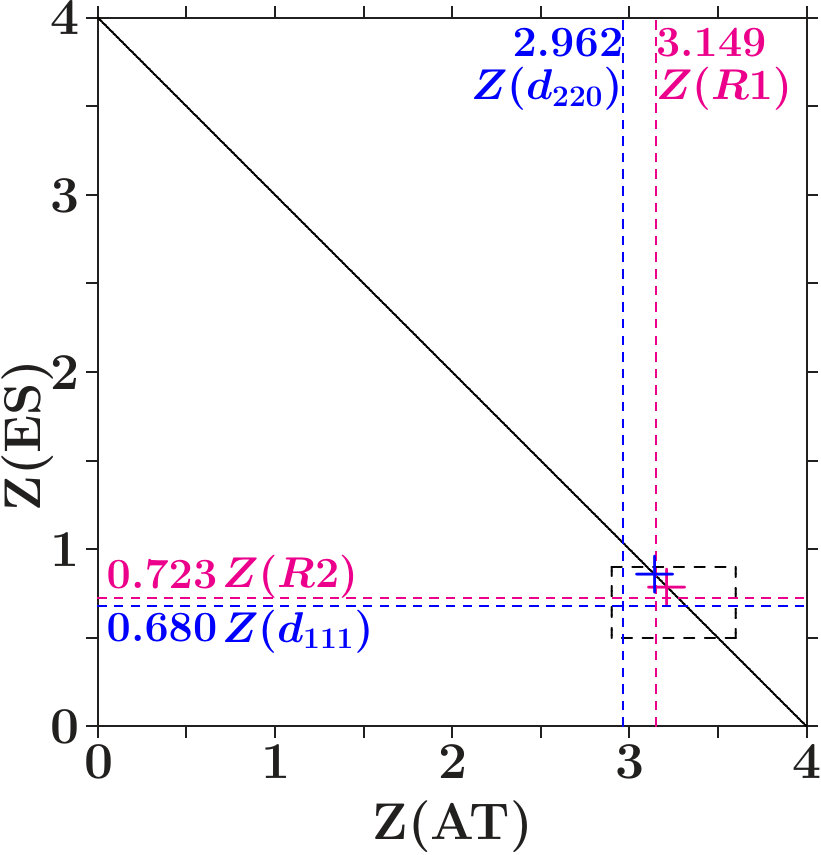 Figure 10
Figure 10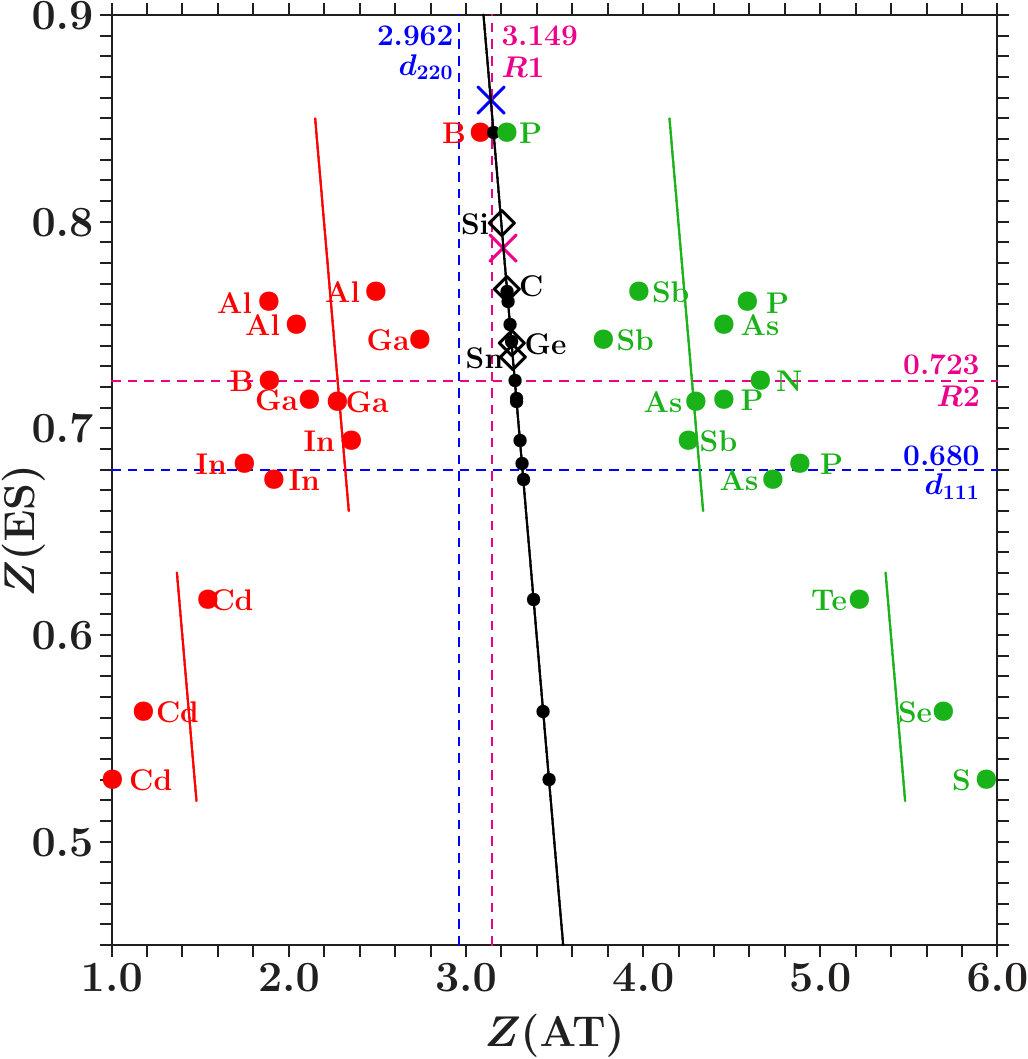 Figure 11
Figure 11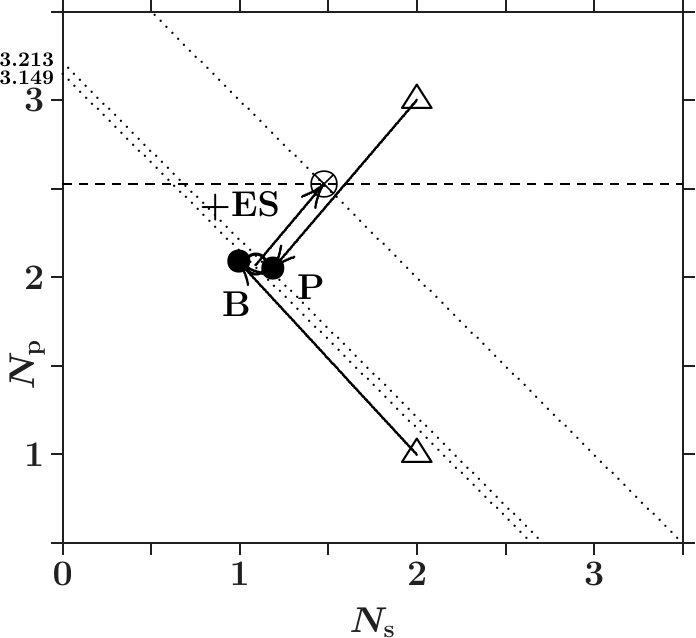 Figure 12
Figure 12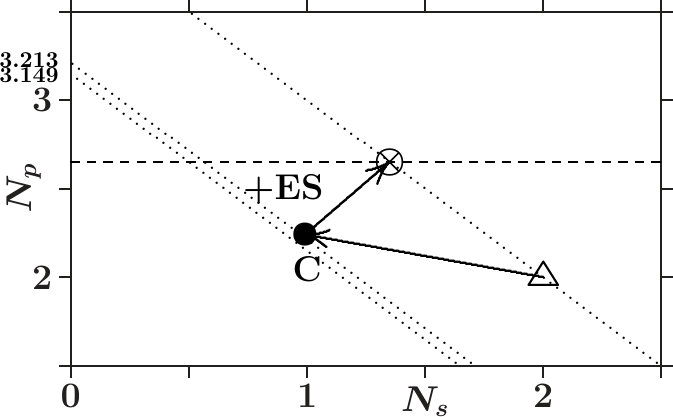 Figure 13
Figure 13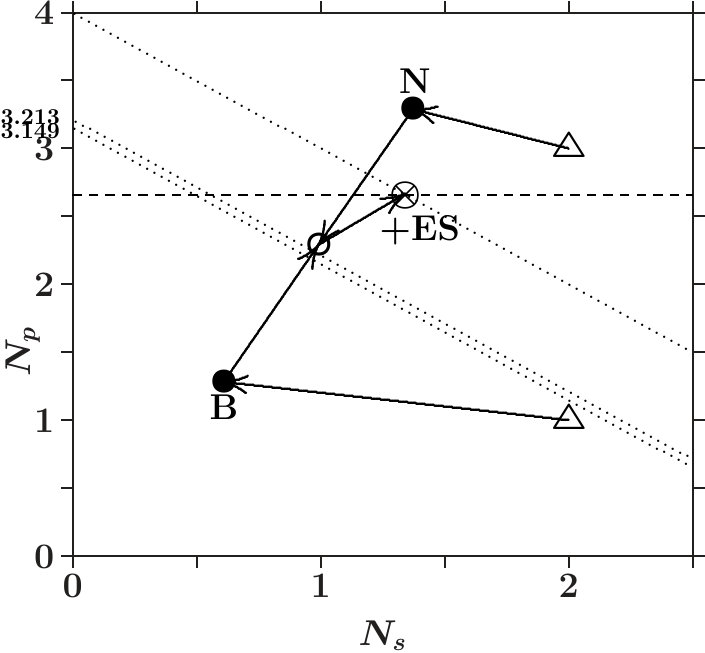 Figure 14
Figure 14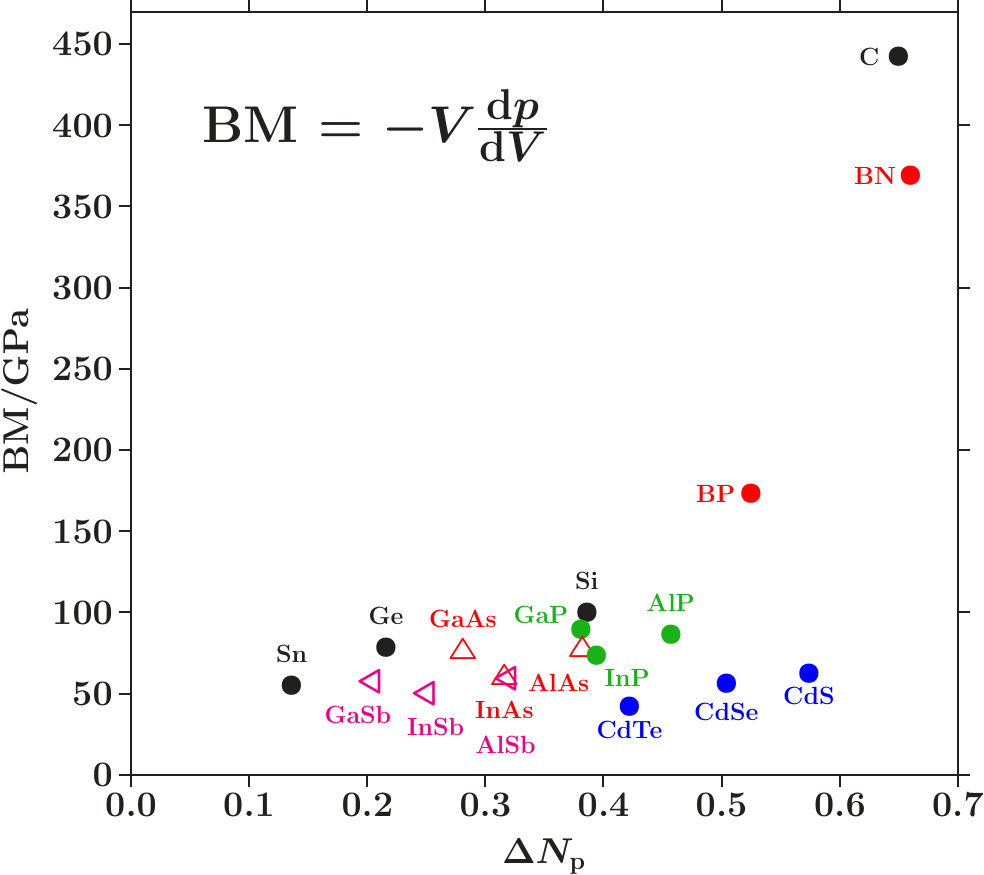 Figure 15
Figure 15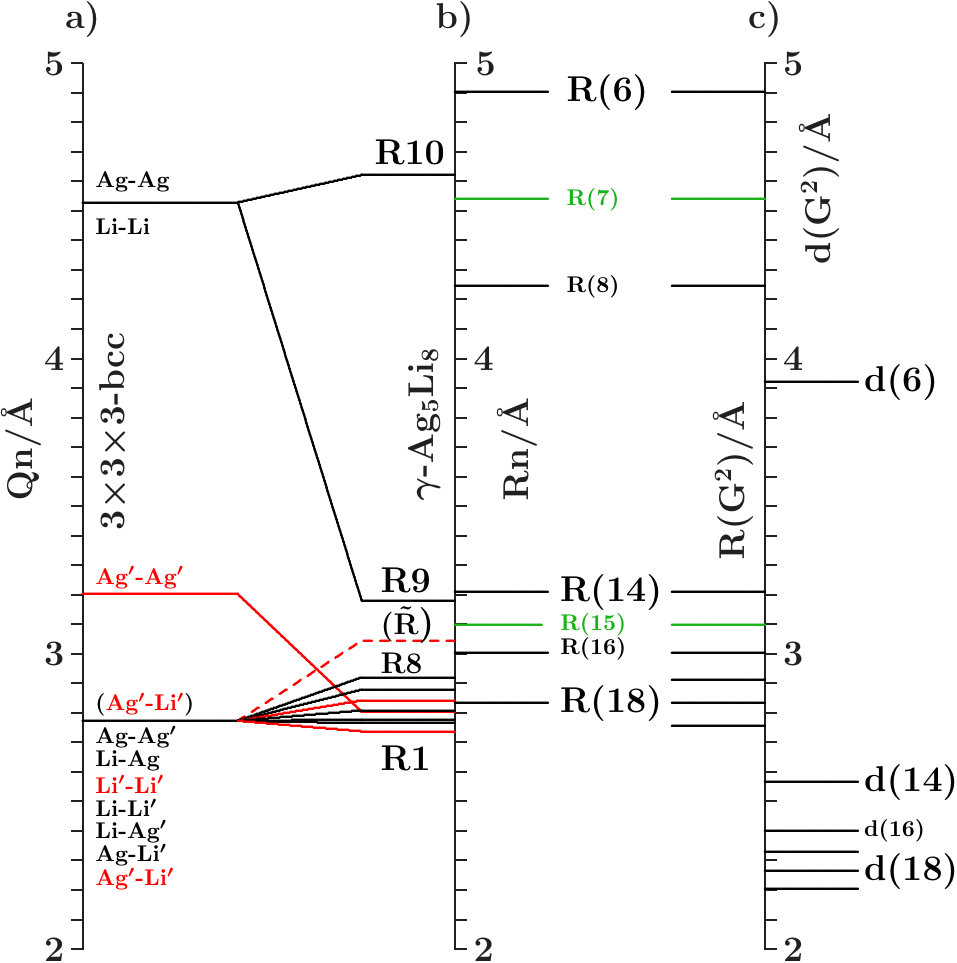 Figure 16
Figure 16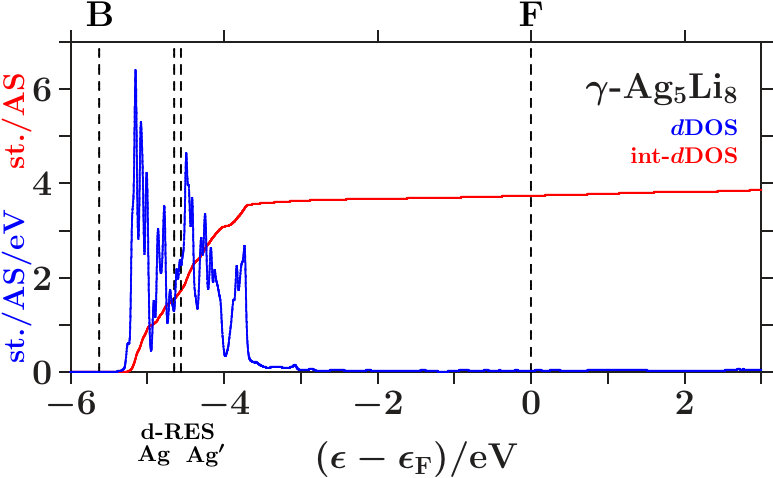 Figure 17
Figure 17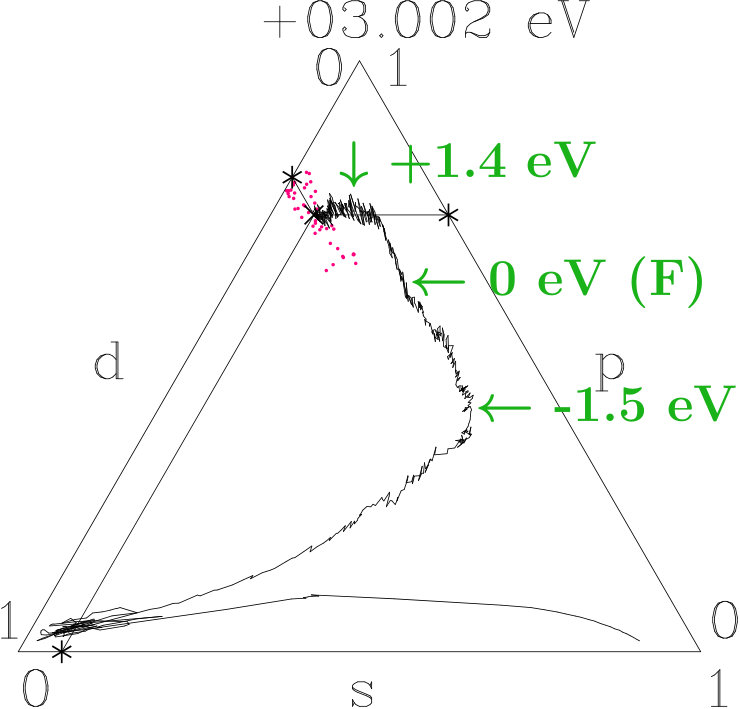 Figure 18
Figure 18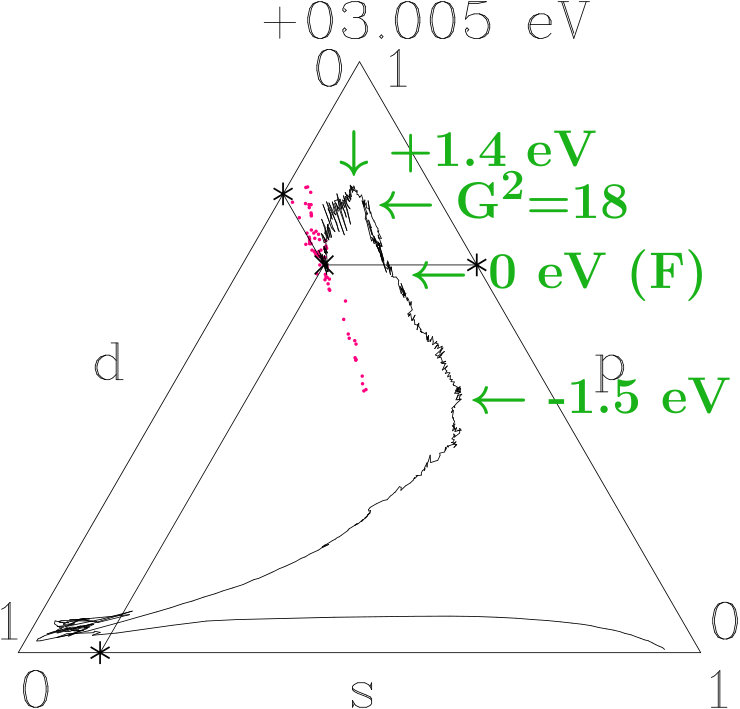 Figure 19
Figure 19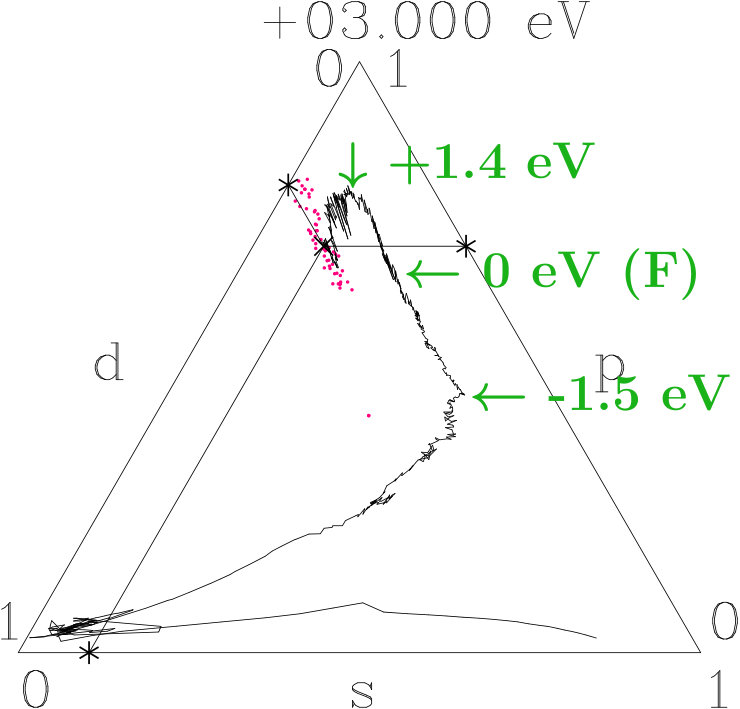 Figure 20
Figure 20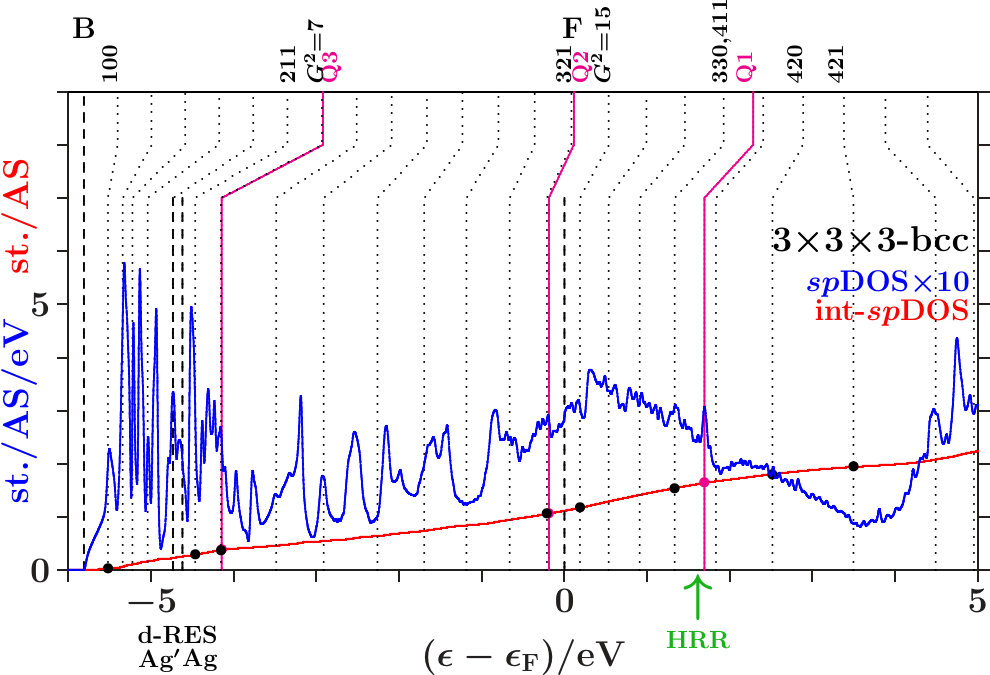 Figure 21
Figure 21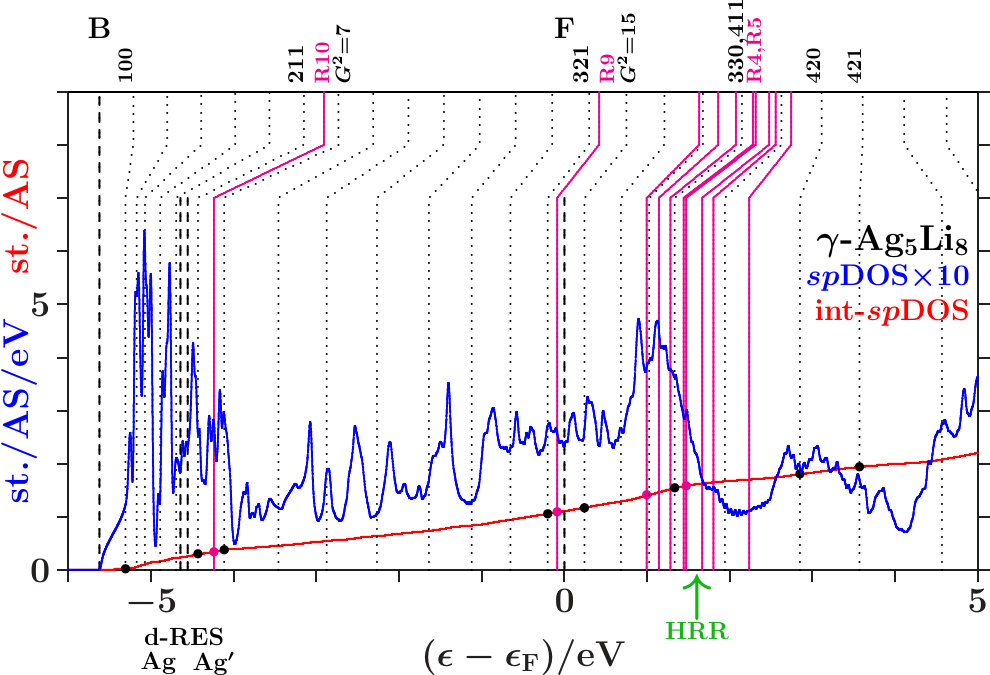 Figure 22
Figure 22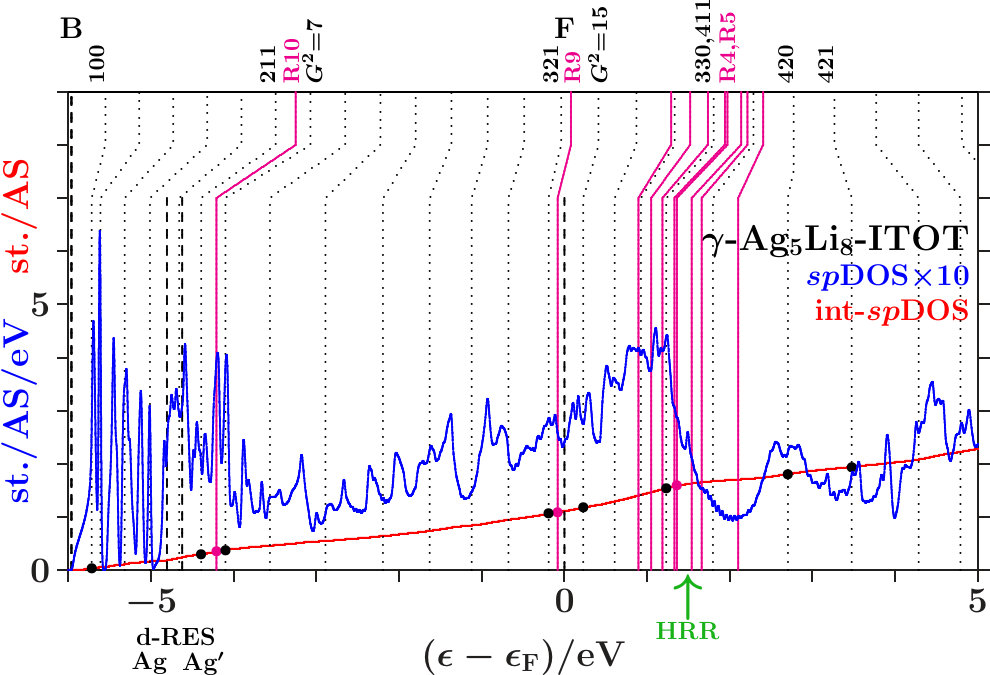 Figure 23
Figure 23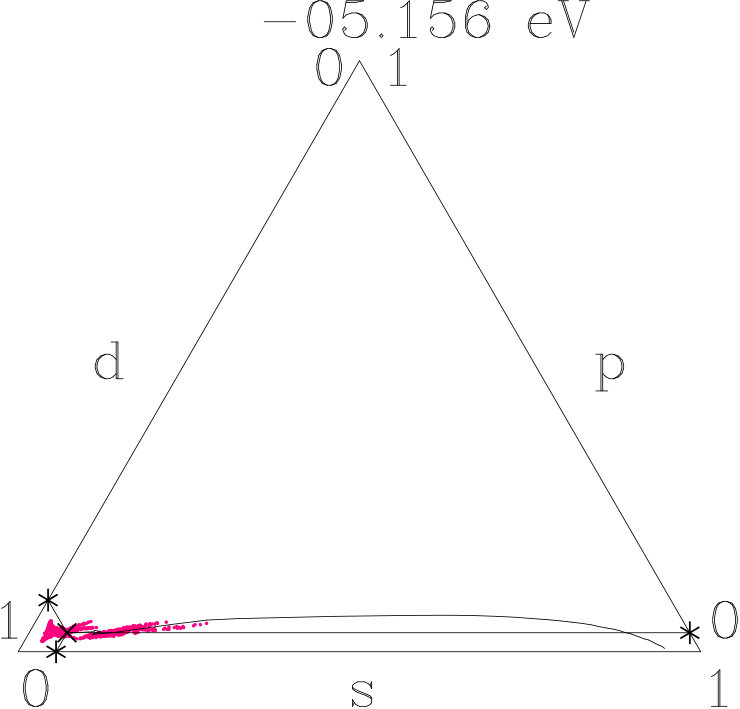 Figure 24
Figure 24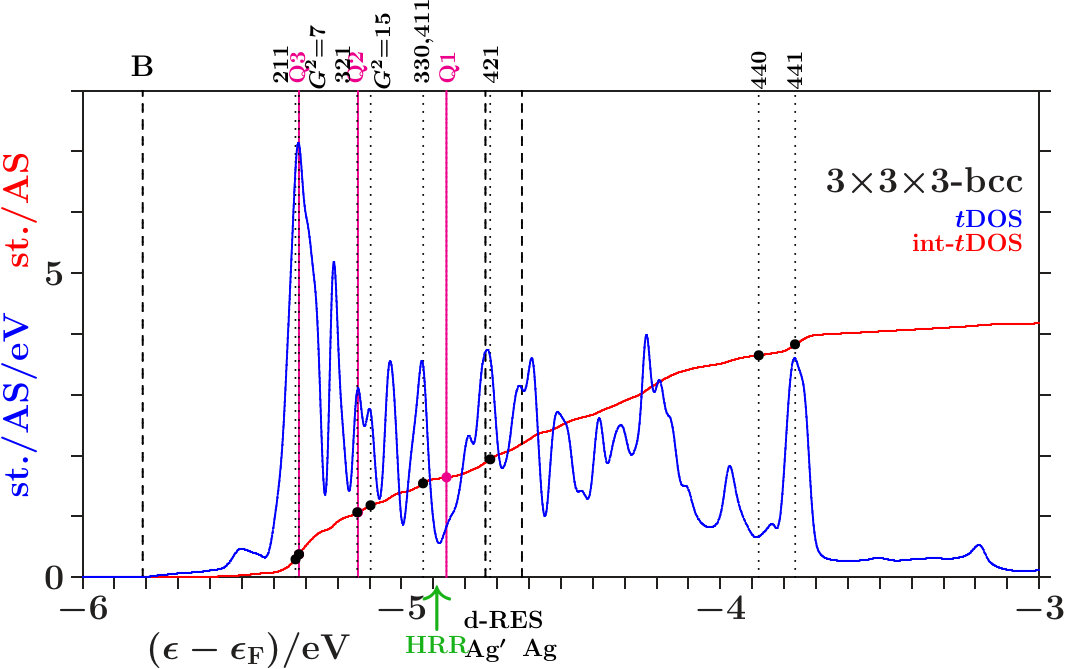 Figure 25
Figure 25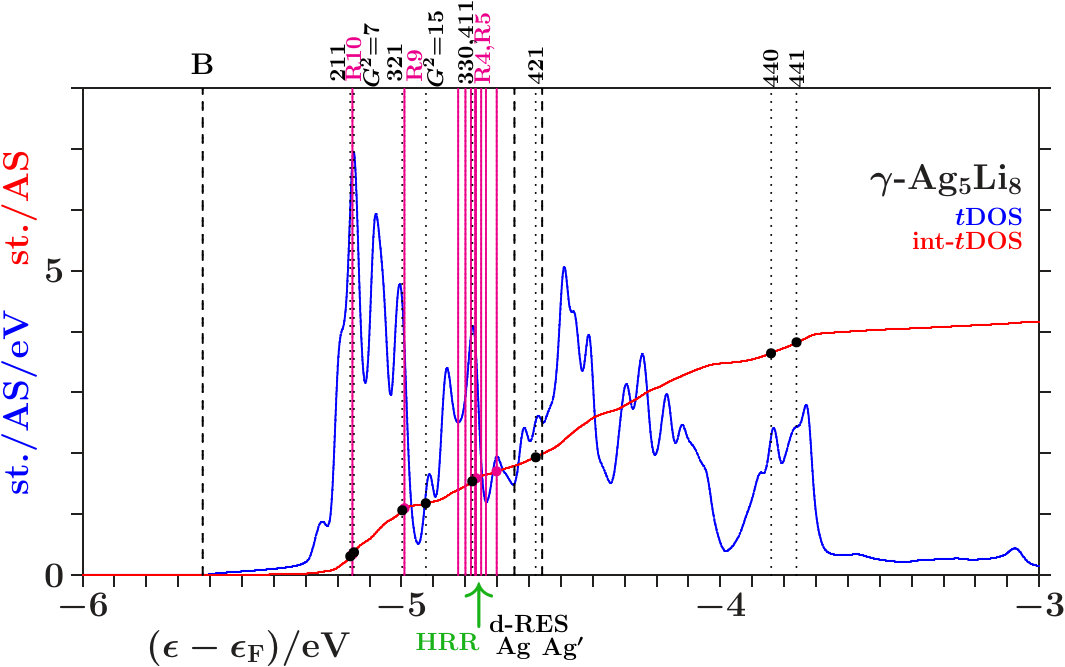 Figure 26
Figure 26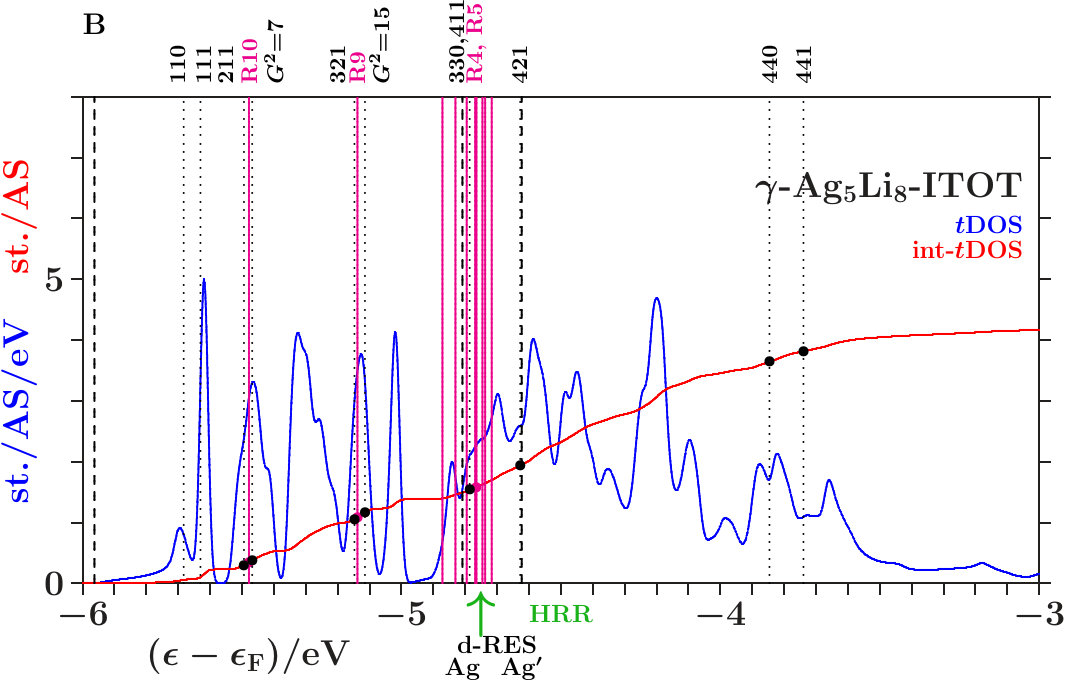 Figure 27
Figure 27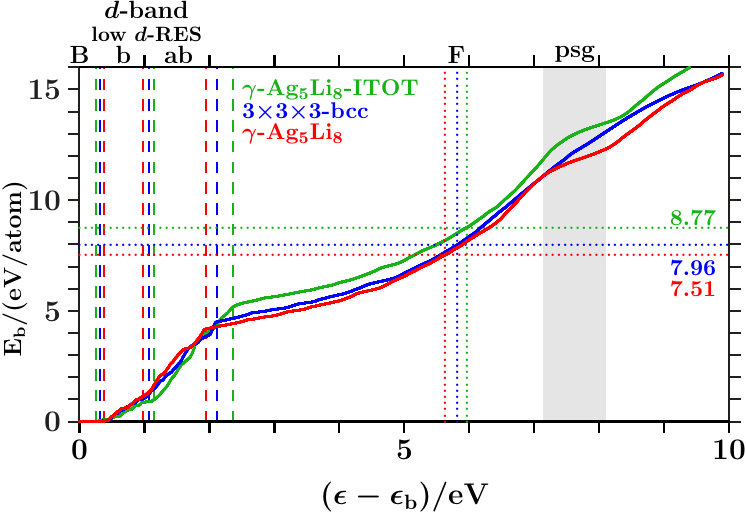 Figure 28
Figure 28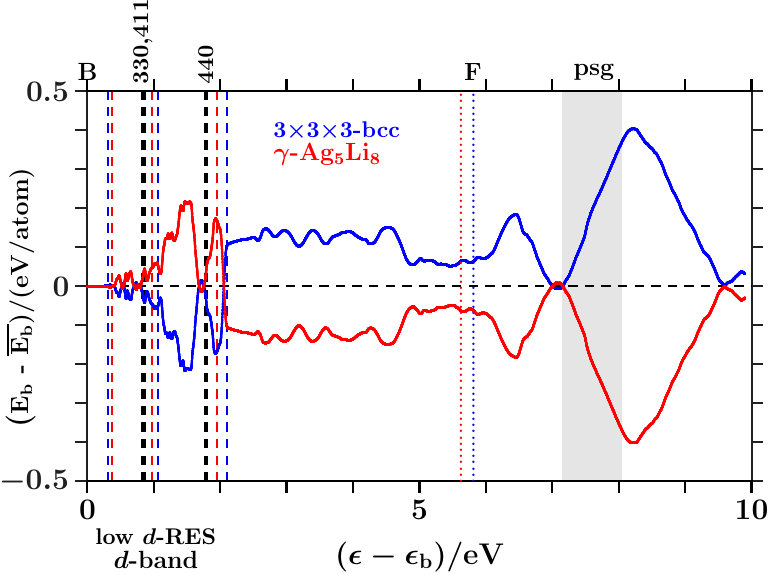 Figure 29
Figure 29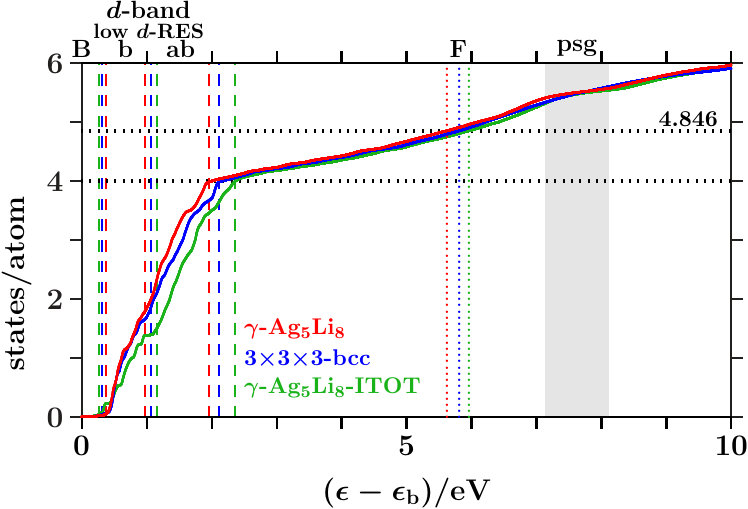 Figure 30
Figure 30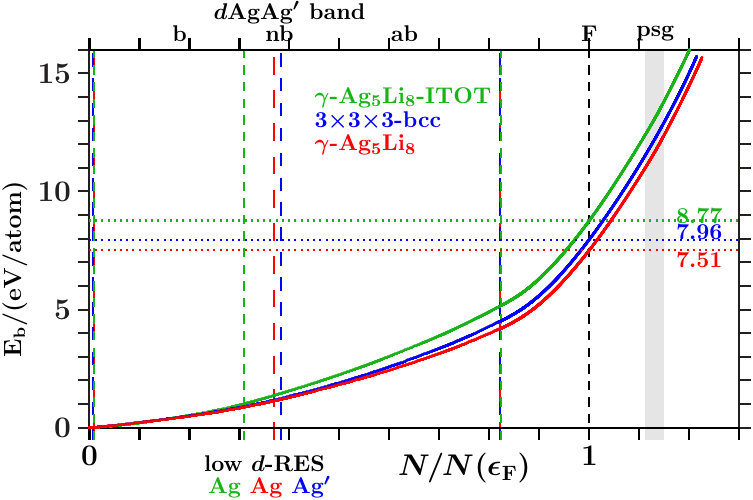 Figure 31
Figure 31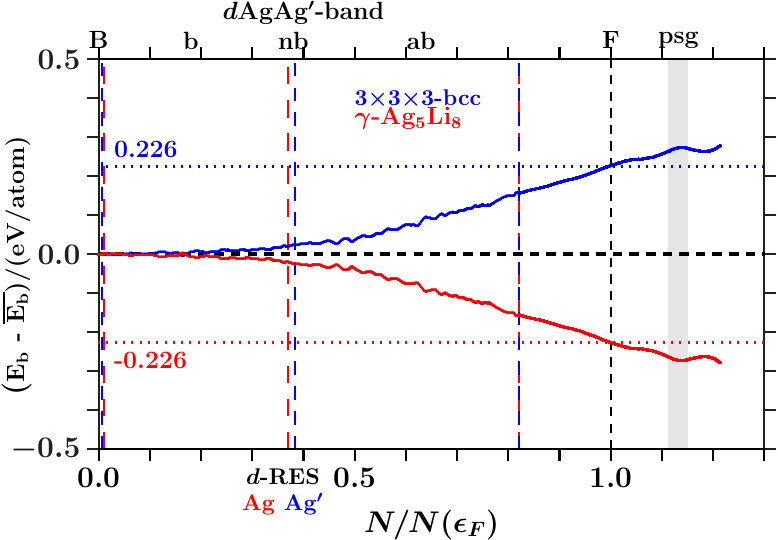 Figure 32
Figure 32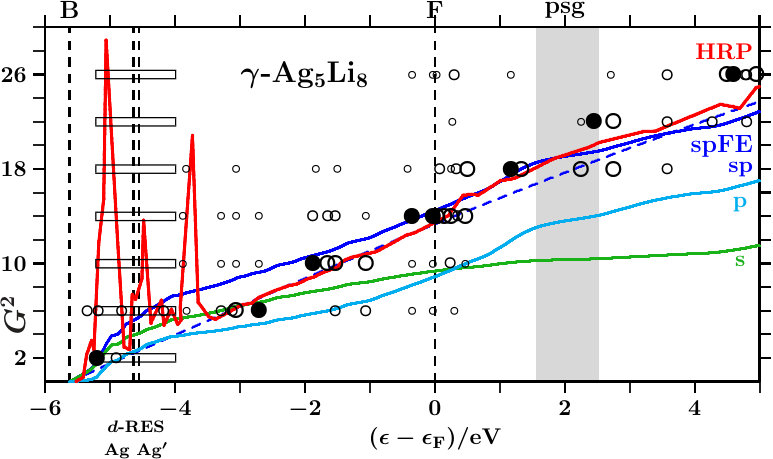 Figure 33
Figure 33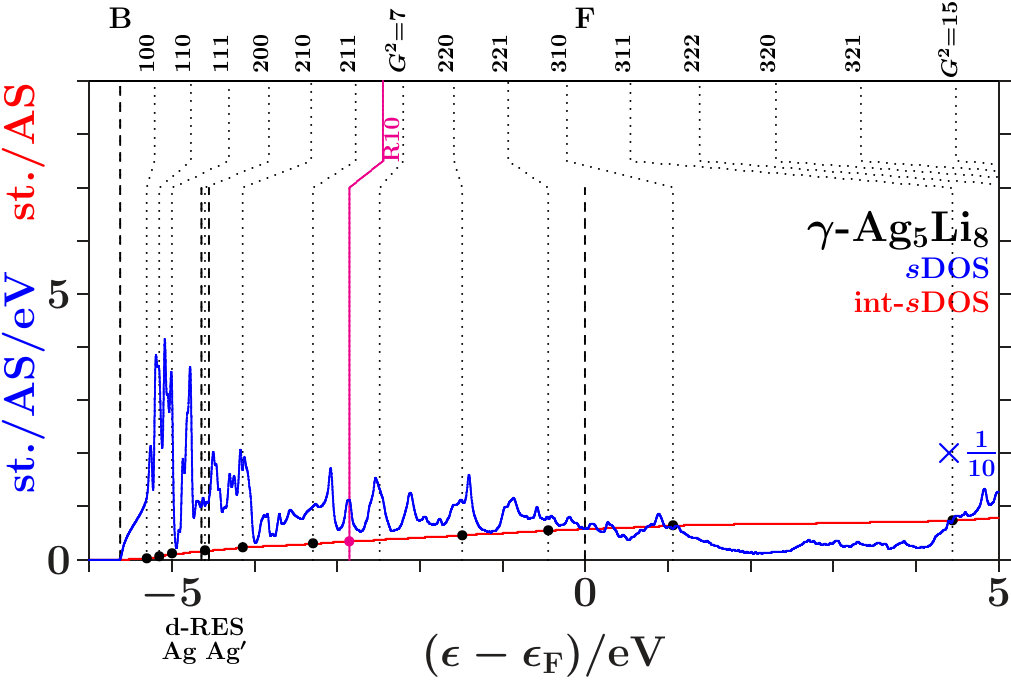 Figure 34
Figure 34| bcc | fcc | dia | ||
| (hkl) | , 2 | , 4 | , 8 | |
| 1.574 | ||||
| 1.446 | 0.723 | |||
| 3.149 | ||||
| (110) | 1.481 | |||
| (111) | 1.360 | 0.680 | ||
| (200) | 4.189 | 2.094 | ||
| (211) | 7.695 | |||
| (220) | 5.924 | 2.962 |
| sub- | sites | planar | radial | ||
|---|---|---|---|---|---|
| cells | (nn0) | (nn1) | |||
| 2 | ((2+1)) | ||||
| 1 | 1 | 2 | 1.481 | 2.721 | 1.574 |
| 2 | 8 | 16 | 1.481 | 1.767 | 1.574 |
| 3 | 27 | 54 | 1.481 | 1.606 | 1.574 |
| 4 | 64 | 128 | 1.481 | 1.551 | 1.574 |
| 5 | 125 | 250 | 1.481 | 1.526 | 1.574 |
| AS-types | |||
|---|---|---|---|
| Li(IT) | -0.555 | +0.616 | +0.132 |
| Ag(OT) | -0.227 | +0.415 | -0.492 |
| Ag′(OH) | -0.229 | +0.445 | -0.460 |
| Li′(CO) | -0.546 | +0.586 | +0.118 |
| to 333- | -0.414 | +0.527 | -0.113 |
| to - | -0.011 | +0.005 | +0.006 |
| AS-types | |||
|---|---|---|---|
| Al(IT) | -0.941 | +0.491 | +0.349 |
| V(OT) | -1.494 | +0.685 | +0.707 |
| V′(OH) | -1.441 | +0.777 | +0.717 |
| Al′(CO) | -0.914 | +0.570 | +0.385 |
| to 333-bcc | -1.136 | +0.610 | +0.526 |
| to - | +0.007 | +0.014 | -0.021 |
| AS-types | |||
|---|---|---|---|
| Zn(IT) | -1.107 | +1.010 | -0.034 |
| Cu(OT) | -0.303 | +0.881 | -0.507 |
| Cu′(OH) | -0.270 | +0.901 | -0.498 |
| Zn′(CO) | -1.077 | +1.048 | -0.021 |
| to 333-bcc | -0.772 | +0.985 | -0.213 |
| to - | -0.003 | -0.002 | +0.005 |
| F1 | F2 | F3 | F4 | |
|---|---|---|---|---|
| F1 | 2 | 1 | 0,3 | 1 |
| F2 | 2 | 1 | 0,3 | |
| F3 | 2 | 1 | ||
| F4 | 2 |
| phase | F1 | F2 | F3 | F4 |
|---|---|---|---|---|
| diamond (#227) | C | C | empty | empty |
| cubic BN (#227) | B | N | empty | empty |
| Heusler (#225) | Al | Co | Cr | Co |
| phase AB | BM/GPa | EN(A) | EN(B) | avEN |
|---|---|---|---|---|
| C | 442 | 2.55 | 2.55 | |
| BN | 369 | 2.04 | 3.04 | 2.54 |
| BP | 173 | 2.04 | 2.19 | 2.12 |
| Si | 100 | 1.9 | 1.9 | |
| AlP | 86 | 1.61 | 2.19 | 1.90 |
| GaP | 89 | 1.81 | 2.19 | 2.00 |
| Ge | 78 | 2.01 | 2.01 | |
| GaAs | 76 | 1.81 | 2.18 | 2.0 |
| AlAs | 77 | 1.61 | 2.18 | 1.9 |
| CdS | 62 | 1.69 | 2.58 | 2.14 |
| InP | 73 | 1.78 | 2.19 | 1.99 |
| CdSe | 56 | 1.69 | 2.55 | 2.12 |
| InAs | 60 | 1.78 | 2.18 | 1.98 |
| GaSb | 57 | 1.81 | 2.05 | 1.93 |
| AlSb | 59 | 1.61 | 2.05 | 1.83 |
| InSb | 47 | 1.78 | 2.05 | 1.92 |
| CdTe | 42 | 1.69 | 2.1 | 1.9 |
| Sn | 55 | 1.96 | 1.96 |
| subcluster | atoms | - | 333-bcc |
|---|---|---|---|
| links | Å | Å | |
| OH-CO | Ag′-Li′ | R1 = 2.737 | Q1 = 2.773 |
| OT-CO | Ag-Li′ | R2 = 2.766 | Q1 = 2.773 |
| IT-OH | Li-Ag′ | R3 = 2.778 | Q1 = 2.773 |
| OH-OH | Ag′-Ag′ | R4 = 2.803 | Q2 = 3.202 |
| IT-CO | Li-Li′ | R5 = 2.808 | Q1 = 2.773 |
| CO-CO | Li′-Li′ | R6 = 2.842 | Q1 = 2.773 |
| IT-OT | Li-Ag | R7 = 2.879 | Q1 = 2.773 |
| OT-OH | Ag-Ag′ | R8 = 2.921 | Q1 = 2.773 |
| IT-IT | Li-Li | R9 = 3.179 | Q3 = 4.529 |
| OT-OT | Ag-Ag | R10 = 4.682 | Q3 = 4.529 |
| OH-CO | Ag′-Li′ | = 3.045 |
| subcluster | atoms | - | 333-bcc |
|---|---|---|---|
| links | Å | Å | |
| OH-CO | V′-Al′ | R4 = 2.687 | Q1 = 2.662 |
| OT-CO | V -Al′ | R1 = 2.647 | Q1 = 2.662 |
| IT-OH | Al-V′ | R2 = 2.659 | Q1 = 2.662 |
| OH-OH | V′-V′ | R6 = 2.726 | Q2 = 3.074 |
| IT-CO | Al-Al′ | R5 = 2.712 | Q1 = 2.662 |
| CO-CO | Al′-Al′ | R7 = 2.768 | Q1 = 2.662 |
| IT-OT | Al-V | R3 = 2.680 | Q1 = 2.662 |
| OT-OH | V-V′ | R9 = 2.782 | Q1 = 2.662 |
| IT-IT | Al-Al | R8 = 2.771 | Q3 = 4.348 |
| OT-OT | V-V | R10 = 4.434 | Q3 = 4.348 |
| OH-CO | V′-Al′ | = 3.034 |
| subcluster | atoms | - | 333-bcc |
|---|---|---|---|
| links | Å | Å | |
| OH-CO | Cu′-Zn′ | R2 = 2.548 | Q1 = 2.559 |
| OT-CO | Cu -Zn′ | R1 = 2.547 | Q1 = 2.559 |
| IT-OH | Zn-Cu′ | R4 = 2.581 | Q1 = 2.559 |
| OH-OH | Cu′-Cu′ | R3 = 2.557 | Q2 = 2.955 |
| IT-CO | Zn-Zn′ | R6 = 2.631 | Q1 = 2.559 |
| CO-CO | Zn′-Zn′ | R7 = 2.643 | Q1 = 2.559 |
| IT-OT | Zn-Cu | R5 = 2.611 | Q1 = 2.559 |
| OT-OH | Cu-Cu′ | R9 = 2.7044 | Q1 = 2.559 |
| IT-IT | Zn-Zn | R8 = 2.7038 | Q3 = 4.179 |
| OT-OT | Cu-Cu | R10 = 4.320 | Q3 = 4.179 |
| OH-CO | Cu′-Zn′ | = 2.856 |
| 1 | 0.0201 | 11 | 0.7347 | 21 | 1.9380 |
| 2 | 0.0570 | 12 | 0.8371 | 22 | 2.0781 |
| 3 | 0.1046 | 13 | 0.9439 | (23) | 2.2214 |
| 4 | 0.1611 | 14 | 1.0549 | 24 | 2.3678 |
| 5 | 0.2252 | (15) | 1.1699 | 25 | 2.5173 |
| 6 | 0.2960 | 16 | 1.2889 | 26 | 2.6698 |
| (7) | 0.3730 | 17 | 1.4116 | 27 | 2.8253 |
| 8 | 0.4557 | 18 | 1.5379 | (28) | 2.9838 |
| 9 | 0.5437 | 19 | 1.6678 | 29 | 3.1450 |
| 10 | 0.6368 | 20 | 1.8012 | 30 | 3.3091 |
| model | from F | from F | from MT0 |
| to MT0 | to B | to B | |
| 333-bcc | -6.028 | -5.811 | +0.217 |
| - | -6.044 | -5.624 | +0.420 |
| --ITOT | -6.202 | -5.962 | +0.240 |
| model | from MT0 | from F | from B |
| 333-bcc | |||
| Ag | +1.406 | -4.622 | +1.189 |
| Ag′ | +1.291 | -4.737 | +1.074 |
| - | |||
| Ag(OT) | +1.399 | -4.645 | +0.978 |
| Ag′(OH) | +1.485 | -4.559 | +1.065 |
| --ITOT | |||
| Ag(IT) | +1.393 | -4.809 | +1.153 |
| Ag′(OH) | +1.579 | -4.623 | +1.339 |
| model | from F | from F | from MT0 |
| to MT0 | to B | to B | |
| 333-bcc | -9.642 | -10.288 | -0.646 |
| - | -9.534 | -10.010 | -0.476 |
| model | from MT0 | from F | from B |
| 333-bcc | |||
| V | +10.127 | +0.485 | +10.773 |
| V′ | +10.009 | +0.367 | +10.655 |
| - | |||
| V(OT) | +9.852 | +0.318 | +10.328 |
| V′(OH) | +9.905 | +0.370 | +10.381 |
| model | from F | from F | from MT0 |
| to MT0 | to B | to B | |
| 333-bcc | -9.310 | -11.001 | -1.691 |
| - | -9.183 | -10.742 | -1.559 |
| model | from MT0 | from F | from B |
| 333-bcc | |||
| Cu | +6.073 | -3.237 | +7.764 |
| Cu′ | +5.821 | -3.489 | +7.512 |
| Zn | +2.432 | -6.878 | +4.123 |
| Zn′ | +1.815 | -7.495 | +3.506 |
| - | |||
| Cu(OT) | +5.701 | -3.482 | +7.261 |
| Cu′(OH) | +5.840 | -3.343 | +7.400 |
| Zn(IT) | +2.040 | -7.143 | +3.600 |
| Zn′(CO) | +2.211 | -6.973 | +3.771 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHigh-pressure geophysics and materials · Boron and Carbon Nanomaterials Research · Diamond and Carbon-based Materials Research
On structure-stabilizing electronic interferences in bcc-related phases.
A research report
H. Solbrig0
Chemnitz University of Technology, Institute of Physics, D-09107 Chemnitz, Germany
**Abstract
** This study deals with cubic crystals where the contents of the simple cubic unit cells are close to nnn-bcc sublattices ( = 2: diamond- and zinc-blende type, = 3: -brasses). First-principle results on the electronic structure are obtained from augmented LMTO-ASA calculations and interpreted within a VEC-based Hume-Rothery concept which employs joined planar-radial interferences to treat interference and hybridization on the same footing. We show that the charge redistribution supports enhanced electronic interference which causes the band energy to decrease. Several topics are included such as stabilizing networks, hardness and -to- transfer, cooperation of interferences, interplay between local radial order and global planar order, electron-per-atom ratio, and the comparison with recent FLAPW-based results.
PACS numbers: 81.30.Bx, 83.10.Tv, 61.66.Dk, 62.20.de, 64.60.aq, 61.50.Lt, 61.72.jd, 71.20.Be.
00footnotetext: [email protected]
Contents
Introduction 1
Structure stability and electronic 3
interference
2.1 Momentum sphere 3
2.2 The general interference condition 4
2.3 Planar interference 4
2.4 Joined planar-radial interference 5
and hybridization
2.5 Virtual valence 7
Diamond and zinc blende phases 8
3.1 Stability of essential length scales 10
3.2 Stabilizing -networks 12
3.3 Summarizing 15
Electronic interference in -brasses 15
4.1 - 15
4.2 - 24
4.3 - 29
Conclusions 35
Appendices 37
1 Introduction
Since the 1920th materials science has developed from purely empirical roots to an applied branch of natural sciences. It was William Hume-Rothery [1] who did a decisive step in 1926. He announced a tight connection between the crystal structures of certain alloys and the estimated numbers of valence electrons per atom (the -ratio). This finding defines the origin of the Hume-Rothery rules which predict the crystal structures depending on the -ratios. Even today the rules are useful for two reasons: (i) Simple estimates of the -ratios can be obtained from the -electron configurations of the component free atoms. (ii) The Hume-Rothery rules provide a valuable classification tool for the wealth of crystalline alloys [2].
However, going into detail, the -ratio thus estimated checks the involved free atoms regarding the itinerant quantum weight they could possibly acquire up to the Fermi energy. Hence, the bonding effect is supposed around the Fermi energy and nearly-free electron like. This must cause difficulties if atomic -states are involved or if the bonding contributions arise deep in the valence band. Following Raynor [3] amended empirical rules are available which employ even negative valences for transition metals [4].
In 1936, Mott and Jones [5] have contributed the basic ideas for more realistic treatments of the electron-structure interrelations. They attributed structure stability to so-called ”Fermi-surface Brillouin-zone interactions“. This means that structures are considered stable if they allow for electron transitions on the surface of a momentum sphere (MS) in the extended -space which are supported by Bragg-reflections. The MS encloses the itinerant weight of the valence electrons up to the Fermi energy, i.e. the contributions of all atoms in the unit cell. However, applications have clarified that the proper MS differs from the real Fermi body [6, 7, 8]. Since the 1930th the community is busy with the determination of appropriate -values. Recently, the activities of the group around Mizutani have been reviewed [9].
Among the great amount of experimental and theoretical contributions to the Hume-Rothery stabilization, a paper by Watanabe and Ishii [10] reveals an interesting aspect of the cooperation between interference and hybridization. The paper deals with simple Al-Mn alloys where - hybridization and -band splitting dominate around the Fermi energy, clearly not a standard case of Hume-Rothery stabilization. The authors emphasize: The bonding effect at the Fermi-energy is hybridization-based. However, unhybridized band states allow for interference around the lower bound of the Mn-band which configures the partial weights for hybridization close to the Fermi energy. Hence, interference clearly below the Fermi energy supports the hybridization close to the Fermi energy. We will show that this kind of cooperations happens in -brasses, too.
The present study examines cubic crystals where the contents of the cubic unit cells are close to n$$\times$$n$$\times$$n-bcc sublattices. There belong with = 1 the CsCl-type phases (number 221), with = 2 the diamond- (number 227), zinc-blende- (number 216), and Heusler-phases (number 225), with = 3 the -brasses (number 217), and with = 4 certain quasicrystalline approximants such as of Al-Cu-Fe with 128 atoms in the simple cubic (SC) unit cell (number 198). This study deals only with cases = 2 and = 3.
Using published structure models in augmented LMTO-ASA calculations we obtain various projected densities of states together with the compositions of the band states in the atomic-site angular-momentum representation. The main purpose of the study is the subsequent interpretation of the first-principle results within a Hume-Rothery concept based on the calculated valence-electron concentrations (VEC).
For diamond and related phases with pronounced covalent bonding character optimized quantum-chemical approaches are available. The present attempt within the atomic-sphere approximation with local XC-potential may thus appear quite strange. Admittedly, the resulting gaps are not wide enough. However, we put emphasis on treating different phases on equal footing regarding the stabilizing aspects of electronic interference. It will turn out that the mentioned drawback does not devalue the subsequent analysis of the first-principle results.
Three -phases will be examined which were formerly proved quite special, namely the exceptions to the Hume-Rothery rule - [11] and - [12] and the prototype alloy - of the group-one -brasses [9] which matches the Hume-Rothery rule perfectly. In each case, we put emphasis on the consequences of the improved hybridization after the transition from 333-bcc to the -phase. Both systems differ only slightly in view of the planar order and the assigned planar interferences.
We focus on those properties of the valence electrons which promote the electronic influence and ensure this way structure stability. Such properties are low band energy, enhanced planar interference, and efficient hybridization on the atomic scale. However, despite of many successful applications of each separate principle in the literature, more insight into the mutual cooperation is required. The following two issues will be tackled in this study:
(i) For planar interferences a quantitative description is available in terms of interference conditions in the extended -space. For hybridization, on the contrary, this level is not yet achieved. Generally, only evidence for the influence of hybridization is announced [4]. We show that efficient hybridization at an atom goes along with efficient radial interference of inward backscattered waves by the sequence of neighbor shells. Radial interference conditions can be found which reveal possible cooperation with nearby planar interferences via the common momentum pool. We use such joined planar-radial interferences at specific energies to analyze the results of the first-principle calculations.
(ii) Low band energy and enhanced joined planar-radial interference should be seen as equivalent orientations on the way to structure stability. They are both indicators of electronic reinforcement. We show, in particular for -brasses, that structure relaxation which improves the interference status does also reduce the electronic band energy. Improved interference concentrates spectral weight at fulfilled interference conditions which provides a practicable tool to evaluate the achieved interference status.
We deal with interferences in transitions on the surface of a MS in the extended -space. The size of the MS is derived from the integrated DOS projected to the respective active part of the electron quantum space (EQS), e.g. the -subspace or the -subspace of certain atomic components. To identify active subspaces we inspect the fluctuating decompositions of the norm One of band states and search for preferences of the fluctuation patterns. This procedure is a generalization of the reduction to unhybridized band states in the approach by Watanabe and Ishii [10].
Joined planar-radial interferences link the local radial order with the extended planar order. The crystalline translation symmetry thus acts on the content of the unit cell, because certain interatomic distances get access to strong planar reflections of the crystal. The shape and the size of the equilibrium unit cell must depend on the inside-outside tuning by guiding joined interferences.
This concept opens a wide field of possible applications. There belong also cases, such as quasicrystals, where this equilibrium is not achieved with lattice periodicity in three dimensions. The long-range planar partners of the local radial processes reveal rather Fibonacci-like arrangements. One may conjecture that the formation of icosahedral clusters drives the system to develop this kind of long-range order.
In amorphous phases the long-range co-ordination is suppressed despite of well developed local radial interference and hybridization. In wide composition fields of thin-film amorphous phases, the experiments by P. Häussler and coworkers [13, 14, 15] have verified the concept of structure stability due to resonance between the electrons and the average neighbor-shell order.
Even gaps in the sequence of planar interferences at certain values of prove important. In that case, the planar interference can join a radial interference somewhat above which operates inside a larger spectral range for optimized hybridization without being hampered by a second planar interference. In particular the absent simple-cubic interferences = 7 and 15 ensure freedom for local radial interferences and hybridizations in the examined phases.
Section 2 gives a detailed introduction to the concept of joined planar-radial interferences. A few examples of the planar-radial interplay support the confidence into the reality of this unification. Interference conditions for the planar and radial components of the joined interferences are supplied for the subsequent applications in the Sections 3 and 4. Section 3 shows that diamond- and zinc-blende-type phases distribute the charges according to interference aspects. In particular the role of voids (empty spheres) will be examined. Moreover, -dominated networks are shown to be highly important for phase stability. Section 4 goes the step to three -brasses which give rise to remember the intrinsic physical content of the Hume-Rothery rules.
2 Structure stability and electronic interference
2.1 Momentum sphere
Suppose a cubic crystal with effective atoms (atomic spheres, AS) per SC unit cell (side length ) and spin-orbitals per AS. This defines a space-filling atomic-site angular-momentum basis of the total electron quantum space, dimension =. Each electron band state is specified by a normalized set of orbital amplitudes which are subjected to fermionic restrictions with respect to other band states at lower energies. Various integrated subspace-projected densities of states, , describe the resulting utilization of the EQS up to given energies, .
The present study obtains the by means of the LMTO-ASA [16] with the special feature [17] that the atomic spheres contain muffin-tin potentials (non-overlapping). This LMTO-ASMT ensures some proximity to both the muffin-tin scattered-wave (MT-SW) concept and the FLAPW method (full-potential linearized augmented plane wave [18, 19]). Above all, we intend to describe various systems on equal footing, knowing that the AS-approximation has drawbacks (e.g. reduced gap widths). However, charges can definitely be assigned to atomic sites. On this basis we will examine the role of electronic interference in the stabilization of structures.
Following Hume-Rothery, Mott, and Jones [1, 5] (HMJ-concept) we adopt that structure stability on a given length scale, , can arise from electronic interference in an active space (total EQS or subspace) around a specific energy, e.g. an interplanar distance, the -subspace, and the Fermi energy, . An active space is spanned by all AS-orbitals which are clearly involved in the interference process.
For estimating electronic interference in valence band states we skip to the extended momentum space (-space). Band states at the energy must be subjected to equivalent conditions in the momentum representation as in the atomic-site angular-momentum representation. Since the very beginning, the adequate characterization of these conditions turned out to be the crucial point. Generally, the HMJ-concept has proved successful if interference is estimated at a -space sphere instead of the real Fermi surface [6, 7, 8]. At the energy , we adopt a -space sphere of the volume , hereafter referred to as the active momentum sphere (MS). This ensures that the band states in both representations are subjected to fermionic restrictions with respect to the really active quantum volume.
Checking interference conditions with the MS refers to the self-consistently determined valence-electron concentration (VEC) in the active space. At specific VEC values, the size of the MS allows for constructive interference, and the band states are notably affected by this interference. Consequently, spectral weight is accumulated in the active space. Pronounced spectral features of the DOS projected to the active space can thus be labelled by the generating interferences.
Note that the influence of electronic interference is two-fold. On the one hand, depending on the size of the MS, fulfilled interference conditions determine the composition of each band state. On the other hand, the number of band states per unit energy (total DOS, DOS) is affected by fulfilled interference conditions, too. The DOS projected to the active space reveals the average common effect.
Besides the MS we will apply the -space sphere of the corresponding free-electron space, counted from the bottom of the valence band. The deposition of spectral weight in the active space acts bonding if the MS indicates fulfilled interference conditions at lower energies than this free-electron -space sphere.
2.2 The general interference condition
Interference in the backscattering along the diameter of the MS (true backscattering, Figure 1) is most important because there are many related cases nearby. True backscattering by a stack of -spaced obstacles (lattice planes, shells of neighbor atoms) is supported by constructive interference once the phase gain, , on going forth and back between successive obstacles is 2. The diameter of the MS should thus equal 2, and the -space volume enclosed is . We define which is twice the number of cells inside the MS or, in other terms, the quantum volume in the active space to be enclosed by the MS at interference on the length scale . This provides the general interference condition,
[TABLE]
Suppose that =N_{\rm act}$$\nu_{\rm act} is the dimension of the active space where sites contribute each spin-orbitals. Hence, the required quantum volume per SC unit cell can be decomposed into contributions per participating site,
[TABLE]
will be referred to as the virtual valence for interference on the length scale . This is just the classical result of Mott and Jones [5].
We ask moreover for the energy where interference on the scale affects the band states which implies . After replacing by the integrated DOS projected to the active space per participating site we obtain
[TABLE]
We insert (2) with (3) into (1) and arrive at the implicit condition for the energy ,
[TABLE]
This shows that the present study follows a rather classical concept: The required momentum transfer in the extended -space results from an empty-lattice consideration whereas the size of the MS accounts for both the actual deposition of quantum weight and the fermionic restrictions.
In the following, we will ever start with the search for the a possible active space. Thereafter we calculate the DOS projected to the active space by the LMTO-ASMT and apply (4) with the right-hand side specified to the sequence of planar respectively radial length scales of the employed structure model.
2.3 Planar interference
In the real space, the SC crystal is composed of equidistant lattice planes with interplanar distances,
[TABLE]
The Miller indices are integers without a common measure. Lattice planes with the same interplanar distances form a group labelled by =++.
The sequence of of the SC lattice reveals gaps for two reasons: (i) There are values such as 7 which cannot arise from three squared integers. (ii) There are other values such as 8 which cannot arise from three squared integers without a common measure. Below we will deal with cases where sublattices are inscribed into the SC unit cell, and the SC lattice itself serves as a reference. Some triples with common measures may thus represent occupied lattice planes, too, such as (220) with half the planar spacing of the (110) planes in the case of an inscribed 222-bcc sublattice.
The SC reciprocal lattice vectors including a factor of 2, =, point along the normal directions of the lattice planes , and the interplanar distance is specified by =(2=2. Jones zones (JZ) are assigned to the vectors as the perpendicular bisecting planes (Figure 1). Once the MS touches a pair of JZ the plane waves and - at the contact points are related by the fulfilled Bragg condition in the true backscattering,
[TABLE]
For such planar interferences the general interference condition (1) with (2) reads as
[TABLE]
Electron-structure interrelations via Bragg effects emerge in the true backscattering with high statistical weights and continue acting at rising energy as specular reflections with lower statistical weights. This causes some broadening of spectral features.
Note that only the utilized quantum volume per SC unit cell is determined by (7) at the planar interference . The assigned virtual valence, , depends on the number of contributing sites, .
Note moreover that structure-specific selection rules of the diffraction theory, such as =3,8,11,16,19,…for diamond structures, are due to single scattering and distant fields. The band electrons contribute close fields and are subjected to multiple scattering. One may rather expect the whole sequence of the Bragg effects in the SC reference crystal with some preference for allowed diffraction events.
Even gaps in the sequence of mentioned above may be significant due to the absence of planar interference which allows for undistorted local processes.
2.4 Joined planar-radial interference and hybridization
One may suppose that the electronic stabilization of structures is finally due to the charge redistribution on the shortest interatomic length scales, i.e. due to hybridization on these scales. Inside the band states, hybridization adjusts the orbital amplitudes at single sites (such as ) or at sites in contact (molecule-like bonding/anti-bonding superpositions). Regarding the HMJ-concept, however, the term hybridization is often restricted to the integration of resonant -orbitals into band states besides -orbitals. There is confident evidence for close interrelation between this kind of hybridization and the HMJ-stabilization of structures. For various systems, it has been demonstrated that the stabilizing pseudogaps are weakened or even disappear after removing hybridizing links in LMTO calculations [20, 4, 12, 21].
The HMJ-concept rests on interference conditions for plane waves. Interference aspects are thus exclusively attributed to the extended planar order whereas hybridization is treated as a competing local effect. However, the discovery of amorphous Hume-Rothery phases [22] has proved that interference-based stabilization occurs due to the medium-range radial order around reference atoms even without long-range planar order. The stability of many amorphous phases has been experimentally investigated by Häussler [14, 15] within the concept of the resonance between the electrons and the spherically periodic sequence of neighbor-shells.
Following Watanabe and Ishii [10], the ranking of the elementary processes in the stabilization of structures should be reconsidered with some preference for hybridization. However, on-site hybridization at a given AS requires backscattering by a non-spherical environment, and this includes the radial interference of the reflected waves from the neighbor shells. A planar interference at a related momentum transfer can link the local radial interferences throughout the crystal to become an extended phenomenon. Hence, at given translation symmetry of the crystal (size and shape of the unit cell) certain hybridizations inside the unit cells (interatomic distances) must be favored.
In the following we focus on two issues, (i) the momentum transfers where radial interferences and the assigned hybridizations occur and (ii) the formation of joined planar-radial interferences.
. Radial order appears in the average radial density of neighbor atoms around reference atoms, 4, where is the average atom number density of the system. The pair correlation function, , reveals the sequence of neighbor shells around reference atoms by ( - 1), i.e. by the deviation from the homogeneous distribution of neighbors.
Suppose a strong spectral component, , of a band state close to a fulfilled planar interference condition in the true backscattering by a stack of lattice planes orthogonal to . How does it notice the existing radial order? The situation resembles a diffraction experiment. For estimation purposes we are only interested in general structure-related properties and
confine thus to identical -scatterers at all lattice sites. Calculating the intensity of the true backscattering of this spectral component, , now accounting for the average radial order, one arrives at the structure factor with the momentum transfer ,
[TABLE]
The structure factor (8) gets large at the main diffraction peak (MDP), , where the sequence of the neighbor-shell radii, , and are tuned as to encounter nearly the maxima of the interference function, (Figure 2),
[TABLE]
Scattered waves of all neighbor shells interfere constructively with each other. We conclude that the average neighbor-shell sequence at maximum coupling to an assigned set of lattice planes is restricted to spherical periodicity,
[TABLE]
Similar to the planar case (6), the momentum transfer in (9) is related to the neighbor shell spacing, , by =2/. A second relation can be drawn from (9), =(5/4), which has no analog in the planar case. It indicates that the spherically periodic neighbor-shell sequence (10) is pinned at the central atom. Hence, the radial interference condition is twofold,
[TABLE]
and the general interference condition (1) with (2) can be specified to radial interferences by
[TABLE]
The strongest planar-radial coupling via mutual true backscatterings requires the momentum transfer at the MDP (11) close to the momentum transfer at the assigned planar interference (6). Hence, we define the equivalent interatomic distances, , of an interplanar distances, , by
[TABLE]
In the following, the term ”joined planar-radial interferences and hybridizations“ (short ”joined interferences“) will be applied to interferences which fulfil approximately the conditions (13).
, , . The periodic sequence of neighbor-shells (10) around central atoms must cause interferences of the inward backscattered spherical waves which are due to a singular outgoing spherical wave emerging from the central atom. This kind of radial interference is part of the one-particle Green function, and hence it must affect both the AS-DOS and the hybridization at the central atom. We will show that just the interference conditions (11) specify such situations.
Searching for a diffraction-related representation of the AS-DOS in a finite atom cluster [23, 24, 25] we have used the Green function in terms of a scattering-path matrix,
[TABLE]
which is a special adaption of the scattering-path operator (Gyorffy and Stott [26]). The denote the real scattering phase shifts of the AS number (AS), and the matrices and contain the interatomic vacuum-wave propagators respectively the scattering amplitudes of the AS (cf. Appendix 6.2). is the amplitude of the regular orbital in the AS, angular momentum =, which appears in response to a singular outgoing wave with unit amplitude in the AS, angular momentum .
Let be the partial DOS of the bare AS (without environment, a function of ). The diagonal elements, , specify the relative environment contribution in the partial DOS, , of the embedded AS (with environment) [23, 24, 25],
[TABLE]
For estimation purposes we confine (i) to single scattering in the environment, , suppose (ii) identical -scatterers at all atomic sites ( = (0,0) 0, phase shift , scattering amplitude ), and take (iii) the average, , of all individual in the large cluster.
[TABLE]
Each element implies the sum of all neighbors at around the reference atom at , but only matters, not the orientation of . Hence, we replace the explicite summations in (17) by an integral with the pair-correlation function, .
[TABLE]
The term between the exponential factors in (18) describes the average inward reflection of vacuum waves whereas the factors are due to the transitions from inside the AS to the vacuum and vice versa.
As a result of neutron optics, the prefactor is the inward-reflection coefficient, , of an infinitesimal vacuum sphere immersed into a homogeneous medium with the Fermi refraction index, . The vacuum part in (18) is thus composed of a contribution due to a homogeneous medium and another contribution due to the radial neighbor-shell order which is represented by .
Similar to the structure factor (8), the latter contribution is reinforced on matching the shell sequence to the interference function, , in (18). The interference function offers two extreme alignments: On the one hand, the shell radii can follow the imaginary part, , which implies that the real part, , is suppressed in the integral (18). This applies to a Friedel-like sequence, (). On the other hand, the shell radii can follow the real part, , with the corresponding suppression of the imaginary part which applies to the sequence (10).
Kroha [27] have analyzed this -shift between the alternative shell sequences by field-theoretical means. It turned out that the sequence (10) should be favored even in common amorphous metals where the quasiparticle life time is large in comparison with the transport relaxation time. In the present approach, due to non-zero phase shifts, a small admixture of the imaginary part besides the dominating real part may support the relaxation towards the neighbor-shell sequence with the lowest electronic band energy.
We conclude that the radial interference conditions (11) favor neighbor-shell sequences around (10) which enable joining radial order to planar order with notable consequences for the AS-DOS at the center of the shell sequence and hence for the electronic band energy.
Hybridization in electron eigenstates, , of a large atom cluster appears already in the incident vacuum fields at the AS, . They are self-sustaining because it takes no initiating source: . Hybridization results from the structure-dependent mixing of the atomic-site angular-momentum components of by powers of . Hence, enhanced radial interference is generally accompanied by enhanced on-site hybridization. The conditions (11) for radial interferences indicate on-site hybridization of active orbitals, too. The first part of (11) ensures that all inward-backscattered waves from the -spaced neighbor shells interfere constructively. It depends on how this interference acts on the AS-DOS and hybridization at the central atom of the shell sequence.
. A stack of (hkl) planes is barely stabilized against lateral shifts of single planes if the interplanar links have the length . This is evident for simple geometrical reasons and moreover from the electronic point of view, because the planar and the radial interferences would occur at the notably different momentum transfers (6) respectively (5/4) (11). If, on the contrary, both interferences occur at the same momentum transfer, such as proposed by (14), the planar-radial coupling must be strong.
The stabilizing interplanar links, , of existing systems should thus obey the relation
[TABLE]
because small lateral shifts around this equilibrium must cause restoring forces. Suppose and a small shift which changes the links into and (Figure 3). The attractive coupling will be enhanced along respectively weakened along which drives back to the equilibrium distances . The key argument is that the local radial interference increases upon approaching due to joining the planar interference . Starting with , on the contrary, causes the opposite assignment and hence destabilization.
In bcc systems, e.g., where the strongest x-ray peak is due to the planar interference (110), the stacks of (110)-planes are stabilized against shifts along a cubic axis by joined interferences [,] = [,]. The condition (19) can be reduced to read as , i.e. . Lateral stability is achieved at nearly the highest level of planar-radial coupling.
Summarizing this planar-radial cooperation, the Bragg effects in the true backscattering support stable interplanar distances, . Radial interference and hybridization in pairs of atoms which link successive lattice planes can support the missing lateral stability.
2.5 Virtual valence
Table 1 presents planar and radial length scales of bcc, fcc, and diamond lattices together with the virtual valences according to (7) and (12). The indicated interplanar distances correspond to strong diffraction peaks. For the short interatomic distances we refer to Appendix 6.1. Joined interferences, e.g. of the diamond lattice, are [, 2] and [, 1]. The atoms in the SC unit cell form a tetrahedral arrangement (scale ) of centered tetrahedra (internal scale ) (Appendix 6.1). Hence, both joined interferences have specific stabilization missions.
We deal with systems of SC translation symmetry where sublattices close to n$$\times$$n$$\times$$n-bcc are inscribed into the SC unit cells. Table 2 characterizes the reference systems with true n$$\times$$n$$\times$$n-bcc substructure where the bcc subcells per SC unit cell are stabilized by joined interferences around the planar interferences with .
The indicated virtual valences (7,12) do not depend on , provided that all the sublattice sites contribute to the MS (). If part of them is inactive, the active sites must compensate for the missing contributions.
Joined interferences act without perturbation by the next higher planar interferences if the virtual valence is well above . Table 2 reveals that the structure types = 1 (CsCl) and = 2 (diamond, zinc blende) can easily meet this demand. The -brasses ( = 3), however, have only slightly above = 1.574. Planar interferences = 19 may thus affect the radial interference in the joined interferences . Hence, -brasses exist at the transition to structure stabilization by blocks of interacting planar interferences which generally prevails in approximant phases ().
Electronic stabilization of the examined systems relies mainly on joined interferences because they drive towards bcc-like subcells on the scale . To ensure just these joined interferences the active MS should enclose between 2 * 1.481 and 2 * 1.574 states, i.e for = 3 between 80 and 85 states.
The empirical Hume-Rothery rule for -type alloys rests on the assumption that the allocation of partial charges by the effective atoms of the crystal is not too much different from the free atoms. For estimation purposes, the effective atoms are thus replaced by free atoms. This way, supposing only = 52 sites per SC unit cell, the empirical Hume-Rothery rule predicts alloys to form -brasses ( = 3) if the free atoms contribute on the average e/a = 21/13 1.615 -electrons per site. At the Fermi energy, the MS should thus enclose 52 * 21/13 = 84 states, just in the expected range between 80 and 85 states. Perturbing planar interferences = 19 occur somewhat above where 54 * 1.606 86.7 states are enclosed by the MS.
Generalizing to arbitrary -brasses ( = 3, = 52, -electrons possible), we suggest that the MS of any active subspace at any energy gives rise to stabilizing joined interferences provided that 21/13 states are contributed per site. This applies to the MS, calculated from first principles and regardless of the -affiliation of the contributions. In the following, the label HRR will indicate the energies where the active MS achieves this critical size.
3 Diamond and zinc blende phases
This section examines a group of 18 phases with structure (number 227, C, Si, Ge, and Sn) respectively structure (number 216, zinc blende phases). They have in common that a 222-bcc sublattice is inscribed into the SC unit cell and that the effective atoms arise from -type free atoms.
The whole crystal can be seen as a superposition of four interpenetrating fcc-sublattices or of two diamond-sublattices (Appendix 6.1). Only one diamond-sublattice is occupied with atoms. In the LMTO-ASMT calculation, we put empty spheres (ES, no nuclear charge) to the other one. Studies on the role of vacancies in stable structures deserve high interest since a long time. For just this purpose we let all the 16 AS in the SC unit cell carry only - and -orbitals. As a consequence, besides the redistribution of the -charges in the AS with atoms, charging the ES is the only additional freedom for adjusting to the interference conditions. Up to 45760 special -points [28, 29] are included in the irreducible wedge of the SC reciprocal lattice. Besides several projections of the electronic density of states we gather information on individual band states.
The diamond phases of carbon (dia-C, = 3.567 Å[30]) and cubic boron nitride (cub-BN, = 3.615 Å[31]) can be regarded as prototypes of group IV diamond phases respectively of III-V zinc blende phases. The atomic spheres have nearly equal diameters, and the average Pauling electronegativity of B(2.04) and N(3.04) is close to C(2.55) [32, 33].
One may expect comparable trends towards charging the ES on the expense of the AS with atoms. However, the electronegativities of B and N are notably different which suggests a quite complex behavior of the average effective atom.
3.1 Stability of the essential length scales
Essential length scales of crystals are the large interplanar and the short interatomic distances. The corresponding lattice planes are densely occupied which results in strong Bragg reflections, and close pairs of atoms allow for pronounced electronic multiple scattering and hybridization. As shown in Table 1, the essential length scales of alloys with diamond structure give rise to joined interferences [,1] and [(3),2].
. The Figures 4 and 5 show the calculated densities of states (DOS) together with site-related projections and the corresponding integrated DOS. Note that the -space is the total EQS of the present approach. Each DOS is upscaled by a factor of 10 for reasonable appearance besides the integrated DOS.
The panels a) present the total DOS where no distinction is made between AS with atoms and ES ( = 16, the total EQS). The integrated DOS at reaches 2 states/AS. The panels b) and c) show the projections to the AS with atoms ( = 8) respectively to the ES ( = 8). Averaging b) and c) provides a).
Vertical dotted lines (labels , increment 1 of ) denote the energies (3) of planar interferences. The lower parts of the dotted lines show where the interference conditions are fulfilled with the MS, i.e. the integrated DOS passes the virtual valences (7) (small bullets, for -brasses cf. Table 14). After kinks the upper parts continue at those energies where the interference conditions are fulfilled with the integrated free-electron DOS. These energies are somewhat larger than the equally-spaced energies, , due to the full free-electron -space sphere,
[TABLE]
The symbol denotes the energy at the bottom of the valence band with reference to . (20) results from replacing .
Radial interferences on the scales = and = (Table 1) are indicated by magenta lines and bullets in the Figures 4 and 5.
. Both panels a) suggest that the wide gaps above are associated with the joined interferences [d(8),R1] which control the internal stability of the centered tetrahedra (length scale 1). Depending on the applied -space spheres we obtain different energies of the planar interferences (220). The free-electron -spheres predict them inside the gaps where no electron is affected because spectral weight is fully removed starting with the first scattering event (Figure 68). The self-consistently determined MS, on the contrary, predict them at lower energies in the hat-shaped features where spectral weight is accumulated. Enhanced fluctuations of the DOS reveal that certain waves are amplified by constructive interference whereas a great amount of smoothing weak waves is missing due to the intentionally sparse -space sampling. Hence, the assignment (3) of interferences to band states is realistic.
Joined interferences [,2] in the lower valence band support the tetrahedral arrangements of the two types of centered tetrahedra, one with atoms at the center - the other with ES (Figure 66). Cub-BN (Figure 5a) generates a wide gap whereas dia-C shows only a weak signature (Figure 4a), despite of the higher bulk modulus (C(442 GPa), BN(369 GPa), [34], Table 10). Both systems differ at least regarding the stabilization of the arrangement of the centered tetrahedra in the SC unit cell.
Note that the joined interferences [,] in the hat-shaped features (panels a) act fairly deep below the Fermi energy. Hence, it seems reasonable to ask whether the stabilization processes so far assigned to interferences in the total EQS do really emerge in subspaces.
. We decompose the total EQS into two subspaces, namely that of the AS with atoms ( = 8) and that of the ES ( = 8). Suppose, on approaching the band states are more and more confined to the subspace of the atoms because the joined interferences [,2] generate a separate occupation edge in the subspace of the ES fairly below . Hence, for the interference in band states at , the whole spectral weight in the ES is hidden, interference conditions include only the spectral weight on the sublattice of the atoms.
In that case essential length scales are stabilized in separate subspaces on approaching . No stabilizing gap is required down in the low valence band. Vice versa, such a gap due to the interferences [,2] is required for a stable arrangement of the centered tetrahedra if the complete separation of both subspaces below is not achieved.
It turns out that this separation towards is almost perfect in dia-C but incomplete in cub-BN and the other 16 examined phases. For proof we analyze the compositions of the band states. In Figure 6a each band state of dia-C contributes two dots which represent the parts of the norm One on the sublattices of C respectively of the ES. Approaching , the band states are more and more confined to the
sublattice of C and, moreover, to the C orbitals (Figure 6b). Hence, the states towards form almost pure C-networks.
Figure 8a applies to cub-BN. The symbol AT designates both atoms, B and N. Contrary to Figure 6a the band states towards are less clearly confined to the atoms, a small coupling to the ES remains. The -orbitals are again excluded (Figure 8b). In both phases, certain branches of band states are thinned out by the joined interferences [,]. Building -dominated electron networks in band states towards must be an integral part of stabilization.
The Figures 6 and 8 show the areas which are occupied by band states, it is not intended to characterize individual band states. Just the latter is aimed at by the visualization suggested in Appendix 6.6. Employing the data set of Figure 6 this method is now specified to the decomposition of the EQS into the three subspaces of C, C, and ES. By means of the PGPLOT routines [35] an animation is obtained which displays, with stepwise rising energy, red dots for all band states in an energy interval around the actual energy and the black tracking curve of the average (Figure 7). Note that the tracking curve can also be derived from the relative contributions made by the projected densities of states to the total DOS (cf. Appendix 6.6).
Approaching in Figure 7, the fluctuations of the red dots cease down gradually, and the tracking curve reaches its topmost point with the coordinates (C,C,ES) = (0,1-,) where 0.005. The norm One is almost fully deposited to the subspace C, and the band states form nearly pure C-networks. A green arrow in Figure 7 indicates the onset of the plateau of the hat-shaped feature in Figure 4a. At this energy the tracking curve turns clearly towards the point (0,1,0) due to enhanced hybridization in absence of perturbing planar interferences = 7 (Figure 4a).
Above the Fermi energy a straight line bridges over the gap. At the upper bound of the gap, the fluctuation patterns, \Delta$$pC = -2ES = -2\Delta$$sC, reveal that equal losses of ES and C enable gains of C twice as large and vice versa (cf. Appendix 6.6).
Figure 9 shows the corresponding for cub-BN. There are two straight lines across two gaps. Different to Figure 7, the tracking curve reaches only the point (0,0.976,0.024) at , i.e. the band states are not completely confined to the atoms.
. At this point some information is available on the composition of band states which can support the analysis of the site-projected DOS curves in the Figures 4 and 5.
The panels b) reveal interference in the subspace of the atoms. Hybridization along the hat-shaped features takes advantage of missing planar interferences = 7. In both phases, an interesting transition appears between the upper edge of the plateau and the the point of inflection below . The dominating interference changes from the planar (220)-type to the radial 1-type with the assigned hybridization. Obviously, the latter processes in the subspace of the atoms ensures the internal stability of the atom-centered tetrahedra.
The panels c) indicate interference in the subspace of the ES. Contrary to the panels b) the kinks of the vertical lines point towards lower energies. This means that the deposition of electrons in the charged ES acts anti-bonding. Charge is moved to the ES for a resulting bonding effect in the subspace of the atoms.
Joined interferences [,] in the subspace of the ES control both the size of the ES-centered tetrahedra and the arrangement in the SC unit cell. In dia-C (Figure 4c), fairly below , the joined interferences [,] generate a soft occupation edge and enable this way increasingly C-confined band states towards (Figure 7). Approaching , joined interferences [,] are confined to the C-sublattice, and the interference conditions refer only to the occupation of the C-sublattice ( = 8). Hence, in the panels b) and c), the required contributions per site are twice as large as in panel a).
Different cub-BN (Figure 5c) with the same structure but two inequivalent effective atoms. In the subspace of the ES, radial interference and hybridization on the scale occur at very low spectral density immediately below . No separate occupation edge is generated in the ES-subspace. The arrangement of the ES-centered tetrahedra may thus be less efficiently stabilized than in dia-C. However, down in the valence band opens a wide gap which can be assigned to the joined interferences [,] in the subspace of the atoms.
3.2 Stabilizing -networks
Essential radial length scales of dia-C are stabilized below by interferences and hybridizations in separate subspaces. In that case the 32 valence electrons per SC unit cell which are contributed by the 8 free C-atoms should be allocated to these sublattices as to ensure at equal deviations from the respective virtual valences. In the group of the 18 examined IV-IV, III-V, and II-VI phases this expectation may be violated the more the wider a gap opens down in the valence band. A wide gap indicates preference for the stabilization of the scales and in the subspace of the atoms deep in the valence band.
. For proof we display the charges (ES) of the ES versus the charges (AT) of the AS with atoms in a figure like Figure 10. The dashed lines indicate the virtual valences after (7) and (12) which are significant to the respective AS in view of the dual stabilization close to the Fermi energy.
The diamond phases and the average effective atoms of the zinc blende phases must occupy the line (ES) + (AT) = 4. Colored symbols (+) on this line highlight the positions of equal distances to both planar respectively to both radial virtual valences. Regarding the radial interferences the conditions read as (cf. Table 1) together with (ES) + (AT) = 4 which is solved by = 3.213 and = 0.787.
We expect that the charge partition is guided by the radial interferences because they open the door to -hybridization. The dashed box encloses the positions of the 18 average effective atoms along the line (ES) + (AT) = 4, obviously well correlated with interference aspects.
Figure 11 shows the content of the box together with the effective atoms of the components. Dia-C and dia-Si are very close to the optimum charge partition with respect to both radial interferences.
Two systems in Figure 11 are tuned to only one radial interference immediately below . Cubic BN has (ES) = 0.723 and BP (AT) = 3.149. In both alloys, the subspace-projected DOS at the radial interference is very low (BN: Figure 5c, BP: similar, not shown). In the case of cub-BN the size of the ES-centered tetrahedra is concerned (scale 2, cf. discussion on Figure 5c) and in BP the internal stability of the atom-centered tetrahedra (length scale 1). With P at the corner and B at the center (distance 1) this link is tuned to perfect radial interference and hybridization. As a result, the ES and, to minor extent, B are charged on the expense of P until the effective atoms of B and P reach almost the same -configurations of the valence electrons (Figures 11 and 12). The final state may be described by 8 dimers in the SC unit cell which have almost identical -configurations but different total charges. Four dimers (B+ES) carry 0.925 excessive electrons and the other four (P+ES) the neutralizing positive charges.
All systems have the red component with lower electronegativity ([32, 33], Table 10) than the green component. One expects that the ES are charged mainly on the expense of the component with the lower electronegativity. Separately for III-V and II-VI the colored full lines indicate where both components have equal charge losses. Outside the strips between these lines the less electronegative component contributes more and inside vice versa. Besides BP only AlSb, GaSb, InSb, and CdTe do not meet the expectation.
- . The valence charge of the effective C-atoms in dia-C is 3.233. The close-fitting value 3.213 ensures equal distances to the fulfilled interference conditions of both essential radial interferences (red cross in Figure 11). Furthermore, 3.233 is very close to 2 * 21/13 3.231 which invokes a link to the Hume-Rothery condition for -brasses. For explanation we remember that the electron states at are confined to the sublattice of the C-atoms. The size of the MS at is due to the contributions 3.233 of the 8 C-atoms per SC unit cell. The same size of the MS must arise if all the 16 sites of the SC unit cell would contribute each 3.233/2 = 1.6165 which is close to 21/13 1.6154. In dia-C, the interference at fulfils the Hume-Rothery condition for -brasses.
This guides back to the Table 2. For an arbitrary value of , contributions around 1.6 by each of the sites per SC unit cell must allow for joined interferences which drive towards bcc-like structure on the length scale . Diamond (dia-C) is the case = 2. Upon creating empty sites (ES) which are hidden to the electrons at the polyvalent carbon atoms build stable structure elements (the centered tetrahedra) which are reserved to lower valence.
-- . The analysis above suggests that phase stabilization attempts to transform -weight into -weight. The former stands for ductility, the latter for hardness. We follow this idea and describe the SC unit cell again as composed of 8 dimers, each ES joins an AS with atom. and in Figure 13 (dia-C) denote the cumulated weights on the respective AS-orbitals in the dimer (symbols , ).
We start from the hypothetical situation (symbol , dimer = = 2) where the ES are really empty and the AS with atoms contain free atoms. In the final state (symbol , dimer + = 4) the contribution of the ES (arrow) has been added to the contribution of the effective C-atom (, (C), (C)). The effective C-atom ends up close to the dotted line + = 3.213 where the sublattice of the atoms is equally far away from = 3.149 as the sublattice of the ES is away from = 0.723. The line + = 3.149 implies perfect adaption only to .
The dimer has increased its -weight by = 0.650 on the expense of former -weight. Charging the ES creates new centers for electrons on directed orbitals which makes the -network more space-filling, and the mechanical stability should be enhanced. Other diamond phases of elements are less successful in this respect: Si( = 0.386), Ge( = 0.216), and Sn( = 0.136).
Cubic BN ( = 0.660, Figure 14) compares with dia-C. However, estimates only the average enhancement of the -occupation per dimer. The -occupation in the sublattice of B is much lower (cf. Figure 14) which acts as a weak chain link. We conclude that the mechanical stability of cub-BN must be less than that of dia-C.
-- . Bulk moduli, , characterize the mechanical stability of materials. Note that dia-C with the highest bulk modulus reveals two peculiarities in the employed approximation: (i) Band states at put almost the whole weight to -orbitals. (ii) Among the diamond phases of elements, dia-C has the highest -to- transfer starting from free atoms. One may presume that a general correlation exists between the bulk modulus and the -to- transfer.
Table 10 shows the measured bulk moduli [34] of the 18 phases. The expected correlation proves valid for diamond phases (Figure 15) where the bulk moduli rise steeply with increasing . Zinc blende phases, are not completely characterized by , because one of both AS with atoms has the weight on -orbitals less than the average. Hence, at equal the bulk moduli of zinc blende phases are generally smaller than those of the diamond phases.
BP looks like an exception which fits rather into the sequence of the diamond phases. This is proved valid by Figure 12 where the effective atoms of B and P have almost equal -configurations as a result of the strong radial interference on the scale 1. Starting from the free atoms, B even acquires some charge (Figures 8 and 12) whereas P looses electrons, despite of the higher electronegativity.
3.3 Summarizing
Structure stability of 18 crystalline phases has been analyzed within the concept of joined planar-radial interference and hybridization where the tight connection is emphasized between the medium-range neighbor-shell order and the extended planar order. Hybridization is controlled by radial interferences which are in momentum contact with certain planar interferences.
Stabilizing interferences can be confined to subspaces of the total electron quantum space. In particular dia-C reveals somewhat below that the internal stability of the C-centered tetrahedra arises in the C-subspace from radial interference and hybridization along the links 1 between the vertices and the center. At slightly lower energy, joined interferences [(3),2] in the ES-subspace stabilize the arrangement of the ES-centered tetrahedra. Hence, dia-C stabilizes both essential length scales close to in separate subspaces. The other 17 members of the group tend to fail the separation of the subspaces below . Instead, gaps open down in the valence band to ensure the stable arrangement of the centered tetrahedra.
Hybridization prefers spectral ranges without perturbing planar interferences, such as along the hat-shaped feature of the DOS of dia-C around = 7 (Figure 4a).
Stabilizing charge redistribution aims at space-filling networks made of equally stiff interatomic links. Relaxation steps towards structure stability transform former -weight into -weight. The ranking of the 18 examined phases due to the measured bulk moduli follows this concept.
Three stabilizing contributions can be ascribed to the empty spheres: (i) They absorb the electron charge which is released by the atoms for optimizing both above interference. (ii) They complete the electron network by new centers for directed -orbitals. (iii) They enable polyvalent atoms to build structure elements which require lower valences.
4 Electronic interference in -brasses
In the following we expand on three phases of the space group I3m (number 217), namely - [36], - [37], and - [38]. The references specify the origins of the applied structure data. Powder diffraction reveals very strong reflexes (330,411) in all cases. Only - exhibits moreover a similarly strong reflex (211). Mott and Jones [5] reported that the stability of - must be ascribed to the dominating planar interferences = 18 where the active -space sphere contains 1.538 electron states per atom (Table 14).
Within the empirical Hume-Rothery concept one estimates the -ratio from the -electron configurations of the free atoms. For the prototype alloy, -, one obtains 21/13 1.615. This value is generally considered characteristic of -brasses. The alloys - and - do not meet this expectation. For - one obtains 1, and - results 34/13 2.615 with plenty VV′-loaded band states close to . The present study has a special focus on the role of the Ag-band in the lower valence band, on the V-band around the Fermi energy, and on the interacting -bands of Zn in the lower and Cu in the upper valence band.
4.1 -
At given energy, the FLAPW-Fourier method [7, 21, 9] extracts a representative -space sphere from the FLAPW wave functions between the muffin-tin spheres. The diameter of this sphere around decides on the possibility of planar interferences = 18. Application to - (Mizutani [11]) confirms the estimated ratio of 1 which proves the alloy an exception to the empirical Hume-Rothery rule. Instead, they announce hybridization-based stabilization in the bonding AgAg′-band.
The present approach considers pronounced hybridization as due to pronounced radial interference which joins a strong planar interference, e.g. = 18. Such joined interferences drive the content of the SC unit cell towards 27 nearly bcc-like subcells. This is common to -brasses (cf. Table 2). However, the stabilizing joined interferences = 18 do not necessarily act in an -dominated subspace close to as supposed by the empirical Hume-Rothery rule. In the case of - it is foremost the activity in the total EQS at the upper edge of the bonding AgAg′-band.
333- One can describe the structure of - with reference to the SC lattice. Two 26-atom clusters are located at the center respectively a corner of the SC unit cell [39]. As the center of the 26-atom cluster is not occupied, the resulting model of - carries no atoms at the bcc-positions on the scale of the SC unit cell. Furthermore, the atoms in the SC unit cell are close to the sites of an inscribed 333-bcc sublattice [39]. The decoration of this sublattice is a simple task (Appendix 6.4).
We use the structure model as proposed by Noritake [36] with 52 atoms in the SC unit cell, side length = 9.6066 Å. The 26-atom cluster is a concentric arrangement of four subclusters, namely the inner tetrahedron IT(4 Li), the outer tetrahedron OT(4 Ag), the octahedron OH(6 Ag′), and the cuboctahedron CO(12 Li′) (Appendix 6.4). Partial occupancies as reported by Noritake [36] are not considered. Mizutani [11] employ the same structure model in their FLAPW-based study.
Suppose that the transition from 333-bcc to the -phase is the final step in a relaxation sequence. What is changed in this step and what is improved? Except for the IT(Li) the subclusters are less modified (Appendix 6.4). However, the diversity of the interatomic distances grows (Table 11) which indicates that mainly the radial interferences and hybridizations are optimized in this step. We focus on just this aspect.
Figure 16 compares the 10 shortest interatomic distances between Li, Ag, Ag′, and Li′ in the -phase with their former values in 333-bcc (Table 11). Interatomic links between the 26-atom clusters are distinguished from those inside by red color. To the right the equivalent radii (14) together with the assigned interplanar distances are shown, including the radii and where no planar interferences belong.
The most obvious result of the hypothetical final relaxation step is the separation of the IT(Li) from the OT(Ag) which is supported by joined interferences [,9(Li-Li)]] and [,10(Ag-Ag)] with =14 respectively =6. This follows from Figure 16 on applying (19) which suggests the interatomic distances (Li-Li) and 10(Ag-Ag) between and respectively and . Note that in particular 10(Ag-Ag) is close to where no additional planar reflexes acts. This ensures freedom for radial interference and hybridization. The joined interferences [,10(Ag-Ag)] control the bcc-like arrangement of the OT(Ag) in the SC unit cell together with the size of the OT(Ag).
As the second remarkable result of the hypothetical final relaxation step we announce the formation of the planar-radial interference block around the planar interference = 18. This concerns the length scale /3 of the 27 bcc-subcells in the SC unit cell (cf. Table 2). Obviously, the radial interferences and hybridizations along interatomic distances close to (18) are tuned to the requirements of optimized hybridizations. Hence, in particular the joined interferences [,4(Ag′-Ag′)] and [,6(Li′-Li′)], both red in Figure 16, exhibit a strong planar-radial coupling. This way the short-range contacts between the 26-atom clusters via 4(Ag′-Ag′) and 6(Li′-Li′) can respond to the long-range arrangement of the clusters.
Invoking only structure parameters and the conditions of planar and radial interferences we have shown that certain joined interferences are formed in - which improve the interference status. Two questions arise: (i) How does the spectral distribution of the valence states reveal the improved interference status? (ii) Is the search for ”maximum interference“ equivalent to the search for ”lowest band-structure energy“? In the following we will examine both issues.
Besides the Noritake model of - [36] we use two modified trial models, namely 333-bcc (equal decorations) and the model --ITOT which is obtained upon interchanging the decorations of IT and OT in the Noritake model, i.e. Ag on IT and Li on OT (equal structures). Three systems are thus available which resemble each other regarding the planar interferences (cf. Appendix 6.4) but differ regarding the radial interferences and hybridizations.
The AS-models for application in the LMTO-ASMT calculations are obtained via Voronoi tessellation of the structure models. Each Voronoi polyhedron is replaced by an AS of the same volume, and the radius of the inscribed muffin-tin sphere is the largest to avoid overlaps with neighboring muffin-tin spheres. This way we prepare effective four-component systems. Valence orbitals up to l = 2 are considered in each AS. In the -space treatment we confine to the 6th special -set [28, 29] in the irreducible wedge of the reciprocal SC lattice (5984 -points). A local exchange-correlation potential [40] is used.
We search for parts of the EQS where the interference-controlled interplay in the individual band states is confined. Hence, the band states have fluctuating projections to parts of this active subspace whereas the projections outside the active subspace are almost constant, such as fluctuating - and -projections at nearly constant -projections. In that case, the MS collects only the active -weights.
For proof we proceed in two steps. In a first step
we evaluate only the average compositions of band states in small energy intervals around given energies whereas the second step examines band-state resolved information. Projected densities of states are the most prominent quantities which provide the required information for the first step. The small and flat DOS around (Figure 17) indicates absent -based activities. Vertical dashed lines in the AgAg′-band refer to the -resonances. They denote the spectral range of the non-bonding states which separates the bonding states below from the anti-bonding states above.
The Figures 18 to 20 use the visualization technique of Appendix 6.6. We decompose the norm One of each band state into the -parts. The averages of small energy intervals provide the tracking curves shown. At the bottom of the valence band the tracking curves start in the -dominated corners, then roam in the -dominated corners (AgAg′-band), and roughly from -1.5 eV to +1.5 eV around they join lines of nearly constant average -weight where -weight is continuously transformed into -weight. Similar to dia-C, -dominated networks evolve within 3 eV around in the three systems due to the planar relationship. Differences occur in the AgAg′-bands and around the topmost points close to +1.4 eV where both -related systems reach maximum -contents of 0.78 whereas 333-bcc is lacking a small additional -excess. Without resolving individual band states we assume that the -subspace separates from the active -subspace between -1.5 eV and +1.5 eV.
In the second step we check this conjecture using the fluctuation patterns (red dots) which visualize band-state resolved information. Preferences of the fluctuation patterns prefigure restrictions of the -interplay (Appendix 6.6). Along the tracks of nearly constant average -weight the fluctuation patterns start around -1.5 eV without preference. At rising energy two preferences appear, namely linear patterns perpendicular to the -axis of the triangle (p = -2s, d = s) and linear patterns along the tracking curve (p = -s, d = 0). The first mode allows for maximum -gain on the expense of equal - and -losses whereas the second mode separates the -subspace from the -interplay. Above the -related systems tend towards the second mode whereas 333-bcc follows less strictly. Finally the second mode dominates all the three systems (cf. Figures 18 to 20). In the first 1.5 eV above , at least - and --ITOT reveal the -subspace active.
-. The Figures 21 to 23 show the DOS, the integrated DOS, and the energies of interferences due to the MS of the -subspace (the lower parts of the vertical lines) respectively to the free-electron -sphere (the upper
parts). Upwards pointing green arrows designate the energies HRR where the integrated DOS reaches the value of 21/13 1.615. According to the standard Hume-Rothery rule for -brasses this should happen at , just between the planar interferences = 18 below and the resulting pseudogap above.
The system 333-bcc (Figure 21) follows a less restrictive version which does not insist on the coincidence of HRR with the Fermi level. As the spectral signature of the radial interferences 1 is very weak, we conclude that the linear descent of the DOS around HRR collects the bonding states of a wide splitting due to planar interferences. The center of this wide splitting is close to the planar interference (421) ( = 21) near +3.5 eV where the kinks of the dotted lines change from pointing upwards to pointing downwards. Above this energy, the population of the -subspace acts anti-bonding in comparison with free electrons. The other two systems resemble 333-bcc with respect to planar interferences (cf. Appendix 6.4). This explains in Figures 22 and 23 the corresponding wide splittings centered close to +3.5 eV. On this background they evolve additional redistributions due to radial interferences and hybridizations.
As discussed in connection with Figure 16 a block of planar-radial interferences and hybridizations forms around the planar interference = 18. All the members interact via a common momentum pool. This block acts bonding, i.e. high DOS is accumulated below HRR, and the assigned pseudogap appears above.
In the anti-bonding AgAg′-band, the -subspace adopts starting information from the -dominated processes. This is accomplished via joined interferences [,3] (Figure 21) respectively [,10] (Figures 22 and 23). The strong planar interference (211) ( = 6) controls the bcc-like arrangement of the OT on the scale of the SC unit cell, and 10(Ag-Ag) describes the size of the OT. Well-tuned systems should concentrate high spectral -weight to the interference energies. In particular - (Figure 22) meets this demand with respect to both components of [,10]. Note that = 7 (no SC reflex) above this range ensures the necessary freedom for local processes.
The Fermi energy just between the joined interferences [(14),9(Li-Li)] below and = 15 (no SC reflex) above suggests that the radial interference and hybridization 9(Li-Li) attempts joining exclusively the planar interference = 14. Inspecting the course of the DOS around the Fermi energy in the Figures 21 to 23, we find that - (Figure 22) replaces the upwards pointing course of the competing systems (Figures 21 and 23) by a plateau, i.e. spectral weight has been moved downwards, a stabilizing contribution. This plateau disappears in Figure 23 after interchanging the decorations of IT and OT. The compressed weakly scattering IT(Li) are thus required to leave the strongly scattering OT(Ag) centered at the bcc-positions on the scale of the SC unit cell.
. We focus on processes in the lower left corner of Figure 24 which represents the bonding AgAg′-band. All band states are clearly -type, and the fluctuating red dots indicate -interplay at small -content.
The Figures 25 to 27 show the DOS of the systems together with the integrated DOS. The interference energies (vertical lines) result from (7,12) using the integrated DOS. Green upwards pointing arrows (labels HRR) show where the integrated tDOS reaches the level of 21/13 1.615.
Suppose, - is tuned to stabilizing -dominated interference and hybridization in the bonding AgAg′-band. In that case, the interference block around = 18 should act in the bonding AgAg′-band at high DOS, followed by a distinct pseudogap. Inspecting the Figures 25 to 27 we find that the -band splitting of - meets just the above expectation. Different 333-bcc (Figure 25) where the radial interference 1 occurs at very low DOS in contrast to the planar interferences = 18. Consequently, the radial effects on the scale 1 are suppressed, just as observed in the -subspace (Figure 21), too. Also different --ITOT (Figure 27), the interference block around = 18 acts in the non-bonding range where spectral weight should be removed rather than piled up.
Down in the bonding AgAg′-band, the joined interferences [(6),3(Ag-Ag)] (Figure 25) and [(6),10(Ag-Ag)] (Figure 26) are well centered at the respective lowest pronounced peaks of the DOS. This equivalence manifests the memory of the bcc-like planar order in -. With OT(Li) and IT(Ag), on the contrary, --ITOT (Figure 27)
suffers from the decay of the bonding AgAg′-band with lower spectral weight at the joined interferences around = 6.
Otherwise than in the -subspace of - (Figure 22) where the joined interferences [(14),9(Li-Li)] cause no striking spectral features they accumulate notable spectral weight in the total EQS (Figure 26).
As to -, we can answer the first question posed above in this Subsection 4.1: Structure relaxation aims at increasing electronic influence due to enhanced electronic interference, i.e. spectral weight in the active space is allocated to the essential planar-radial interferences and hybridizations.
- . We calculate the band-structure energy per atom (AS),
[TABLE]
as a function of the energy throughout the valence band where is the total density of states per atom (tDOS), and denotes the bottom of the valence band.
The results are shown in Figure 28. Colored dotted lines indicate the values of and . The edges of the AgAg′-bands in the DOS as well as the lower -resonances are marked by colored vertical dashed lines, and the shaded area above remembers the pseudogap of - (Figure 22).
Despite of different interference tunings of - and 333-bcc in the bonding AgAg′-bands (Figures 25 and 26) the band energies respond notably not until the anti-bonding range (Figures 28 and 29). The trial system --ITOT (Figure 27) with OT(Li) has the bonding range heavily distorted. The system leaves the AgAg′-band with the highest band energy, and this stays valid up to the Fermi energy. Hence, the ranking of the systems results mainly from different stacking modes of band states in the AgAg′-band.
For evidence we show the integrated DOS of the systems in Figure 30. The systems agree in that = (511+81)/13 4.846 states/AS are reached up to the respective Fermi energy and nearly 4 states/AS up to the upper edge of the AgAg′-band. Apart from a tiny exception at the lower -band edge, the -phase has the largest integrated DOS and thus the most efficient stacking mode of band states throughout the occupied valence band.
Figure 28 allows only for a limited insight into the competition between 333-bcc and - at given energies. For improved insight we show in Figure 29 the distances of the band energies from the average, . This retains the true differences. Except for a small environment of 1.7 eV, the band energy of the -phase is the larger
one throughout the AgAg′-band. This changes to the opposite at the upper edge of the AgAg′-band, because high spectral weight around the planar interferences (440) and (441) (Figures 25 and 26) acts in the -phase at lower energy above the bottom of the valence band.
The final ranking of the systems at the respective Fermi energies is derived from equivalent situations, both systems occupy equal numbers of band states. To compare the systems in equivalent situations throughout the valence band we replace by , i.e. by the band-structure energy up to included valence band states per atom. The energies are implicitly determined by
[TABLE]
Figure 31 shows the band energies versus the relative valence band filling, /. At the resolution of this plot the upper AgAg′-band edges of the systems coincide. Starting around the AgAg′-resonances the band energies grow without change of the ranking.
Figure 32 applies the presentation mode of Figure 29. The advantage of - over 333-bcc at equal band fillings grows monotonously up to -0.45 eV/atom at .
To answer the second question at the top of Subsection 4.1 we argue as follows: - achieves the lower band energy due to the concentration of spectral weight to joined interferences in the narrow AgAg′-band. This configures the stacking of band states up to and beyond . Enhanced electronic interference causes reduced band energy.
- . The FLAPW-based analysis of interference in - [11] starts with the inspection of the Fourier representation of the FLAPW wave functions in the interstitial between the muffin-tin spheres,
[TABLE]
The key argument is that large are due to the coupling with certain fulfilled Bragg conditions at symmetry points, such as a point N where .
The favored Bragg conditions specify constructive interference in the true backscattering of plane waves, , by appropriate (hkl) lattice planes, i.e. + = -. As depicted in
Figure 1, this can be seen as a transition on the surface of a sphere in the extended -space, diameter . Following this concept the authors extract a continuous function (the Hume-Rothery plot, HRP) from the dominating spectral components throughout the Brillouin zone which defines the diameter, , of a representative -space sphere. From 13.4 they deduce 1 inside the sphere and conclude that - is an exception to the empirical Hume-Rothery rule because it fails 21/13 1.615 .
Figure 33 shows the lower part of the Fourier spectrum at symmetry points N which has been extracted from results published by Mizutani and coworkers [41, 42]. Three groups are formed depending on the cumulated weights (short weights) of the with . Big circles indicate weights 0.5, the medium-sized circles 0.5 weights 0.1, and the small circles including the elongated boxes in the AgAg′-band refer to weights 0.1. Big bullets highlight the largest weights along the respective lines . The full curves show the Hume-Rothery plot (red, HRP) and the continuous functions of the present VEC-based approach applying (7,3) combined with the respective integrated partial DOS of the subspaces (blue), (green), and (cyan). For comparison we add the blue dashed curve labelled as FE which is obtained upon using in each AS the integrated free-electron DOS above the bottom of the valence band (symbol B, = -5.62 eV, LMTO-ASMT). Note that the energies indicated by the upper parts of the vertical dotted lines in Figure 22 are just the energies where FE passes integer values of .
Several conclusions can be drawn from Figure 33:
(i) Around the dominating planar interferences = 18 the HRP matches the blue curve derived from the DOS, i.e. the HRP extracts the total accumulated -weight including the contributions of radial interference and hybridization. Both approaches are thus equivalent with respect to the internal length scale of the 27 bcc-like subcells.
(ii) The bullets at = 2, 10, 14, and 18 are close to the blue full curve (label ). Even = 6 may be added because the DOS slightly below -4 eV is small (Figure 17). Note that the bullets indicate vicinity to planar interferences in the true backscattering at symmetry points N. The interference energies are thus determined by the MS of the -subspace. The HRP, on the contrary, matches the blue dashed curve FE where no spectral charge redistribution is considered. At least regarding = 6 one may doubt because of the strong x-ray peak (211).
(iii) The planar interferences (211) (no common measure) control the bcc-like arrangement of the subclusters OT(Ag) on the length scale of the SC unit cell. Bullet and big circle at = 6 around -3 eV close to the intersection with the green -derived curve suggest that the HRP is really -based. For verification we refer to Figure 34 where a wide b/ab-splitting of the DOS (center close to -2 eV) accumulates bonding weight around -3 eV, supported by the joined interferences [(6),10].
(iv) The intersection between the - and the -related full curves slightly above the Fermi level in Figure 33 allows for enhanced -interplay which promotes the hybridization around = 15 as mentioned above in connection with Figure 22.
. We start from a hypothetical initial state where the AS contain free atoms (configurations Ag(1 0 10), Li(1 0 0)). The 20 AgAg′ contribute 220 valence electrons and further 32 are added by the 32 LiLi′. In the final state, the charges inside the AS result from the LMTO-ASMT calculation. We find that 5.3 valence electrons move from AgAg′ to LiLi′, i.e. 2.1 % of the total 252 valence electrons per SC unit cell.
As shown in Section 3, the -to- transfer is sensitive to structure stability. Table 3 gives a compilation of the charge transfers between the involved AS-orbitals. The last two lines refer to the average changes in the first step from free atoms to the perfect planar order and in the second step to the adjusted radial order.
Starting from free atoms, on the average 0.527 + 0.005 = 0.532 -states per AS are occupied in two steps, mainly on the expense of decreasing -occupation. The chief contribution is due to bonding into planar order (step 1), the adjustment of the
radial order (step 2) adds only a small enhancement of the network-forming -weight (cf. Figures 18 and 19). Note that this charge partition at represents an intermediate state of the growing -network along the line in Figure 19.
The ratio is intended to describe the size of the active -space sphere at . Mizutani [11] derive from the HRP (cf. Figure 33, red curve) and insert it into (/156)( = ((7) with = 52). The result is = 0.99 (Table 4).
Using the integrated free-electron DOS (Figure 33, the blue dashed curve, label FE), we obtain = 0.98. Except for the AgAg′-band the Hume-Rothery plot proves FE-like up to . This disregards the wide splitting of the -band which is centered close to +3.5 eV.
With the integrated DOS of the present approach (Figure 33, the blue full curve, label ) one obtains = 14.46 and hence = 1.11, just between 1.0549 ( = 14) and 1.1699 ( = 15) (Table 14). Obviously, the joined interferences [(14),9(Li-Li)] in the -subspace are addressed. They act in Figure 19 around along the line of nearly constant average -weight. Hence, the ratio = 1.11 is significant as it refers to the enhancement of the -network around . Rather than a pseudogap the spectral signature is a plateau of the DOS (Figure 22).
** -**. The alloy is electronically stabilized in the extended -space by joined planar-radial interferences around = 18, 14, and 6 which is common with other -brasses.
Joined interferences = 18 around the planar interference (330) include the radial interferences and hybridizations on the shortest interatomic distances. This interference block keeps the content of the SC unit cell close to 27 bcc-related subcells.
The joined interferences = 6 and = 14 with the guiding planar interferences (211) respectively (321) keep the OT(Ag) nearly conform to the bcc-positions on the scale /3 whereas the IT(Li) are notably compressed.
As the planar interferences = 7 and 15 are not excited in SC lattices the energy range for radial adjustments in the joined interferences = 6 respectively 14 is enlarged.
In the atomic-site angular-momentum representation the electronic stabilization of - arises from interferences in various parts of the electron quantum space at various energies. The empirical Hume-Rothery rule, on the contrary, supposes that the main contribution, the joined interferences = 18, arise in the -subspace close to . This way, the empirical Hume-Rothery rule selects a subclass of -brasses which achieve structure stability mainly in the -subspace below , - does not belong to this subgroup. However, the chief information of the rule stays valid, namely that the atoms in the SC unit cell are driven towards the sites of a 333-bcc sublattice if e/a = 21/13 is achieved in an active electron space.
The systems - and 333-bcc are nearly equivalent regarding planar interferences, differences occur in the radial interferences and hybridizations. Hence, a spectral DOS-feature of - which does not occur in 333-bcc must arise from the readjustment of the radial order, i.e. from radial interference and hybridization. This applies to the peak/dip-feature (width 2 eV) in the DOS of - around the joined interferences = 18 above . The planar interference = 18 links efficiently the local radial processes.
Similar to dia-C (Figure 7) the band states de-
velop -dominated networks. However, at , the -dominated networks of - are neither strictly -type nor fully developed.
Enhanced spectral density at fulfilled interference conditions indicates improved interference and the band energy is reduced.
4.2 -
The early transition metal vanadium in - contributes occupied -orbitals around the Fermi energy which must affect the structure stabilization. For this reason, Mizutani [12] have examined - by means of the FLAPW-Fourier method employing the structure model of Brandon [37] with the lattice constant = 9.223 Å.
They extrapolate the Hume-Rothery plot outside the VV′-band to the Fermi energy and conclude from = 21 that - must be stabilized at = 1.94 (cf. Table 14). This is clearly above = 21/13 1.615 and therefore in contrast to the empirical Hume-Rothery rule.
However, the structure of -brasses suggests that above all the interplanar length scale and the assigned radial length scales must be stabilized. This requires joined interferences around = 18 at = 1.538 (Table 14). Two questions arise: In which active space? At which energy?
** 333-bcc**. We use the Brandon model [37] just as done by Mizutani [12], i.e. = 9.223 Å. No mixed occupations of the subclusters IT(Al), OT(V), OH(V′), and CO(Al′) are considered. The shortest interatomic distances among the four components are listed in the Table 12. For comparison we add the reference system 333-bcc.
Joined planar-radial interferences of - (Figure 35) arise by analogy with - (Figure 16). Main processes are again the compression of IT(Al) and the formation of the interference block around = 18. Different from -, the distance 8(Al-Al) (size of the IT(Al), cf. Table 12) now joins the interference block. Only (V′-Al′) remains assigned to the planar interference = 14. The radial interference and hybridization along 6(V′-V′) (OH-OH, link between 26-atom clusters) joins again closely the planar interference = 18.
We conclude that - and - are almost equivalent regarding the essential length scales which must be stabilized by joined planar-radial interferences.
** **. Figure 36 compares the interference status due to the present VEC-based approach with results of the FLAPW-Fourier analysis. We use the same three classes of circles and bullets as in Figure 33 to reproduce the FLAPW-based weighting of planar interferences at symmetry points N [42, 21].
The continuous curves are derived from (7,3) with different choices of the integrated projected DOS, in detail the int-DOS (black, label ), the int-DOS (blue, label ), the int-DOS (cyan, label ), and the int-VV′DOS of the vanadium subsystem (dark violet, label VV′). The blue dashed curve (label FE) is derived from the integrated free-electron DOS (symbol B, bottom of the valence band, - 10.01 eV).
Magenta horizontal lines in Figure 36 result from the inversion of (14),
[TABLE]
which defines a hypothetical planar interference (non-integer) with the same momentum transfer as the radial interference along n. The interference block around = 18 between 1 and 9 in Figure 36 is replaced by a magenta shaded stripe.
Suppose slightly above an integer . A joined interference [,n] arises. This enables the interplay between the radial order in the SC unit cell and the global planar order. On the one hand, at given size of the SC unit cell (side length ) certain radial interferences are selected for global support. The global order acts on the local order. On the other hand, a given strong radial interference n, e.g. for chemical reasons, may cause the size of the SC unit cell to adjust for slightly above an integer . The local order acts on the global order. Hence, along the magenta horizontal lines in Figure 36, appropriate conditions can emerge for the interplay between the local radial order and the global planar order.
All subspace-related curves have intersections with the magenta lines at certain energies where the above mentioned joined interferences may be active in the chosen subspace. The question arises whether the FLAPW-derived weighting of interferences in the extended -space follows the expectation, i.e. whether the large circles and the bullets are close to the intersections.
Up to the lower bound of the VV′-band in Figure 36, the FLAPW-derived weighting reveals indeed large circles and bullets close to the intersections between magenta lines and the blue curve with the label . This means that each strong planar interference in the -subspace indicates strong radial interference and hybridization on an assigned radial scale, and this is relevant to the charge redistribution.
Inside the VV′-band, however, the -type radial interferences and hybridizations on the scales 1 - 9 are represented by wide flat spectra of planar interferences in the extended -space, starting from = 18 up to large in the total EQS (Figure 36, label ). Different from below the VV′-band, no radial interferences are assigned to individual planar interferences via (24) because there are no interatomic distances which are short enough. Individual planar interferences are thus less reliable indicators regarding hybridization on the atomic scale. Hence, the information content of seems problematic if derived from a single interference .
To analyze the processes on the shortest radial scales 1 - 9 we follow in Figure 36 the shaded stripe around = 18 towards increasing energies, knowing that the interference block gets active in successively reduced subspaces. Starting at the lower bound of the VV′-band with the total EQS (label ) the maximum weight of the Fourier component = 18 (bullet) occurs at the intersection between the blue curve (-subspace) and the line = 18. We conclude that an important stabilizing contribution must arise from the interference block around = 18 in the -subspace at the lower bound of the VV′-band. The question is how such essential interferences clearly below the Fermi energy can support the stabilization of the structure.
The DOS of - (Figure 37) offers an answer to this question. Joined interferences = 18 around -2.1 eV, the lower bound of the VV′-band, support the opening of a wide -pseudogap throughout the VV′-band. This kind of pseudogaps is commonly ascribed to the displacement of spectral -weight by -states. The complete spectral feature is centered at = 23, again a gap in
the sequence of the SC planar interferences. Comparison with 333-bcc (Figure 38) confirms the enhanced concentration of -weight at the interference energies in -. Even the joined interferences = 14 and = 6 on larger length scales are included. Hence, the radial adjustment in the hypothetical final relaxation step improves the interference status.
Figure 39 reveals the view of the band states. We show the -decompositions. Starting at the lower bound of the VV′-band around -2.1 eV, the band states get increasingly -dominated. Towards , the -subspace is nearly excluded, only a small interplay of the network-forming -components remains. This way, joined interferences = 18 in the -subspace fairly below the Fermi energy enable nearly undisturbed -networking towards the VV′-resonances.
If we reduce to the subspace of VV′ (Figure 40, in Figure 36 the dark violet full curve) the planar interference = 18 must be active at the energy where the 20 VV′ contribute each (52/20) 1.538 4 VV′-states on the average. This happens at +0.118 eV, just below +0.187 eV where the (52/20)(21/13) = 4.2 VV′-states are are reached due to the Hume-Rothery rule. In the VV′DOS (Figure 40) a deep pseudogap opens above the lowest VV′-resonance at +0.318 eV. Such pseudogaps are known to result from bonding/anti-bonding splittings of transition metal -bands. Again, the joined interferences around = 18 support the formation of this hybridization phenomenon.
The band states close to the lowest VV′-resonance at +0.318 eV prove -type with moderate V-V′ interplay and minor participation of the residual EQS (Figure 41). This confirms that the wide -pseudogap (Figure 37) facilitates just this kind of nearly undisturbed -networking.
The situation around proves intermediate on the way to the interference-supported -band splitting (Figure 42). The cyan full curve (label ) in Figure 36 passes the line = 18 (1.538 per site) at -0.144 eV whereas 1.659 per site are accumulated up to . This is close to 21/13 1.615 as supposed by the empirical Hume-Rothery rule at the occupation edge. Hence, along with the growing VV′-network, joined interferences = 18 arise in the total -subspace close to . The Hume-Rothery rule applies to the total -subspace (cf. the comments on Table 2). Figure 42 shows that the interference block around = 18 of - acquires more spectral weight than [(18),1] in 333-bcc (Figure 43).
Summarizing joined interferences = 18 we note activities at the bottom of the VV′-band in -dominated subspaces and towards the VV′-resonances in -dominated subspaces. The former open a wide -pseudogap for low -presence in the band states towards the VV′-resonances, the latter act in the VV′-subspace for -band splitting and in the total -subspace below the Fermi energy for bcc-related subcells on the scale /3.
** **. The Figures 44 to 47 use the same presentation modes of the band energies (21) of - and 333-bcc as demonstrated with -. Vertical dashed lines denote the lower bounds of the VV′-bands and the lower VV′-resonances whereas dotted lines highlight the Fermi energies and the resulting band energies. The shaded areas remember both pseudogaps of the -phase.
The plots versus the energy above the bottom of the valence band (Figures 44 and 45) compare the systems in non-equivalent situations due to different band fillings . Large differences arise in particular in the VV′-band due to specific b/ab-splittings on small energy scales. To get rid of such details we plot the band energies in the Figures 46 and 47 versus the relative band filling, . Similar to - (Figure 32) a monotonous increase of the band-energy difference remains.
With the improved interference status, the -phase ends up with the lower band energy at the Fermi energy (26.366 eV/atom versus 27.196 eV/atom, Figure 44). The difference of -0.830 eV/atom results mainly from the -subspace (-0.683 eV/atom) whereas the -subspace contributes only -0.147 eV/atom. Nevertheless, the stable radial order of - results from the cooperation of joined interferences = 18 in both the - and the -subspaces.
** -**. We proceed as with -. In the initial state free atoms are supposed in the AS (configurations V(2 0 3),Al(2 1 0)). The 20 VV′ contribute 100 valence electrons, and the 32 AlAl′ add further 96. Inspecting the
final state shows that 0.18 valence electrons per SC unit cell have moved from VV′ to AlAl′, only 0.1 % of the total 196 valence electrons per SC unit cell.
Table 5 shows the changes of the partial AS-charges in the transition from free atoms to the -phase. Enhanced - and -occupations support the opening of the stabilization-relevant pseudogaps in the respective subspaces (cf. Figures 37 and 40).
The last two lines present the average changes due to bonding into the perfect planar order (line 5) and due to readjusted radial order (line 6). Hence, 1.129 = 1.136 - 0.007 -electrons per AS change into 0.624 = 0.610 + 0.014 -electrons and 0.505 = 0.526 - 0.021 -electrons with the partitions according to the two steps mentioned. The chief contributions arise from bonding into the perfect planar order.
Table 6 displays -ratios of - which result from the FLAPW-Fourier method [12, 9] respectively from the present approach. The top two lines (label HRP) refer to extrapolations of high-energy Hume-Rothery plots to the center of the VV′-band. The former one [12] provides = 21 in perfect agreement with the free-electron value (label FE) of the present approach. A subsequent attempt [9] results = 22.7 but this extrapolation suffers from wide fluctuations. Without arbitrariness the present VEC-based approach (label ) derives = 22.17 from the integrated DOS (Figure 36, blue full curve).
However, none of the offered -ratios in Table 6 is really significant regarding the electronic stabilization of -. As pointed out in connection with Figure 36, one cannot deduce the status of charge redistribution on existing interatomic length scales from an individual planar interference 20. Individual planar interferences 19, on the contrary, are indicators of stabilizing charge redistributions on the shortest radial scales via (24).
The alloy - prefers stabilization in the total -subspace close to . By analogy to the empirical Hume-Rothery rule one could try to estimate from free atoms the capacity to accumulate -weight. This results e/a = 60/52 = 1.154 21/13 1.615, not sufficient for planar interferences = 18 in the total -subspace. Thanks to the considerable -to- transfer of +0.505 (Table 5), the calculated occupation of the total -subspace up to is 1.659 close to 21/13 1.615. The corresponding demands the empirical Hume-Rothery rule regarding planar interferences = 18 in the -subspace slightly below the Fermi energy. However, one cannot estimate the -to- transfer by simple means. Hence, a practicable prognostic tool is not obtained this way.
** -**. The alloy is electronically stabilized by joined planar-radial interferences and hybridizations = 18, 14, and 6.
The interference block around = 18 contributes threefold: (i) = 18 acts in the -subspace at the lower bound of the VV′-band which ensures low -presence around the VV′-resonances. (ii) = 18 acts in the VV′-subspace below the VV′-resonance for -band splitting. (iii) = 18 acts in the total -subspace below in an intermediate state of the growing -network.
The interference block = 18 in the -subspace configures favorable conditions for the interference block = 18 in -dominated subspaces towards the VV′-resonances.
Two findings regarding the electron-per-atom ratio are announced in view of -: (i) Estimating e/a due to the -configurations of the free atoms is not significant because the stabilization is rather -like. (ii) Estimating e/a due to an individual planar interference is not significant because the shortest interatomic distances couple to = 19 at the most.
Joined interferences = 6 stabilize the OT(V) which is common with -. Different from -, the compression of the IT(Al) is assigned with joined interferences = 18 instead of = 14.
The band-energy differences at given band fillings grow monotonously throughout the valence band. This is common with -.
4.3 -
The alloy is the prototype of the group-one -brasses in the classification scheme after Mizutani [9]. Generic properties are the estimated ratios e/a = 21/13 1.615 and distinct pseudogaps around the Fermi energy. This is confirmed by the FLAPW-Fourier method, in particular by the Hume-Rothery plot [9]. Powder diffraction experiments show dominant peaks (330,411) and much smaller peaks nearby. Paxton [8] have performed the first LMTO-ASA calculations. They ascribe structure stability mainly due to the planar interferences (330,411) whereas deviations from bcc enter via the weak planar interferences (420), (332), and (422). Reinvestigation by Mizutani [43], again using the LMTO-ASA, reveals a moderate contribution only by (420) in addition to the dominating contributions by (330,411). More important, they demonstrate that the Hume-Rothery stabilization is efficiently assisted by - hybridization.
In the following we focus on both above topics: (i) We analyze the relaxation of the properly decorated 333-bcc substructure to the experimentally verified structure. (ii) We merge planar interference and hybridization to result in joined planar-radial interferences.
The alloy explains how two interacting -bands control the environment of the Fermi energy.
333-. Gourdon [38] compare different stoichiometries around the ideal system -. We employ the ideal structure due to neutron diffraction with the side length = 8.866 Å of the SC unit cell.
By analogy with Figure 16 (-) and Figure 35 (-) we characterize in Figure 48 the relaxation of the shortest interatomic distances on going from 333-bcc to - (cf. Table 13). Note again the compression of the IT(Zn) and the formation of a block of joined planar-radial interferences around = 18, all in close analogy with - (Figure 35).
. We search for joined interferences = 18 in the momentum space, i.e. for the active subspaces in the atomic-site angular momentum representation and for the interference energies.
The Figures 49 and 50 show the total DOS of 333-bcc and - with some indicated interferences. In both cases, the joined interferences = 18 act in the bonding ZnZn′-band. Different from 333-bcc, the -phase has the -resonances of Zn (-7.14 eV) and Zn′ (-6.97 eV) close together. The width of the ZnZn′-band shrinks and distinct bonding and anti-bonding peaks appear, separated by a narrow pseudogap around the -resonances. Note that the joined interferences = 18 act rather far below the -resonances. Similar to - (Figure 40), one may suppose active subspaces where they get closer to the ZnZn′-resonances such as the ZnZn′-subspace (Figure 51).
Going into detail we reduce the observed space in the Figures 50 and 52 to 54 from the total EQS to
the subspaces ZnZn′, Zn′, and finally to Zn. The latter three subspaces contain only 32, 24, respectively 8 of the 52 AS in the SC unit cell. At interference the MS requires contributions per active AS which are enlarged by factors of 52/32, 52/24, respectively 52/8 in comparison with Figure 50. Hence, joined interferences = 18 in the subspaces of ZnZn′ (Figure 52) and Zn′ (Figure 53) dominate the lower respectively the upper subpeak in the bonding ZnZn′-band. The small spectral feature at the upper edge of the anti-bonding ZnZn′-band results from joined interferences = 18 in the Zn-subspace (Figure 54). We conclude that joined interferences = 18 in particular in the Zn′-subspaces support the -band splitting.
Contrary to the ZnZn′-band the CuCu′-band (Figure 55) appears quite strange. Joined interferences = 18 at low spectral density and high spectral density at the CuCu′-resonances are just the opposite of the expected allocations. Obviously, the zinc -band preconfigures the copper -band for low efficiency of joined interferences = 18 in the CuCu′-subspace. Instead, apart from the CuCu′-resonances, a deep pseudogap opens around -2.8 eV. Moreover, centered just around -2.8 eV, a wide spectral depression appears in the DOS of - (Figure 57) after radial adjustment starting from 333-bcc (Figure 56). Hence, - coupling must be important, and the stabilization of - is neither simply NFE-like nor is it confined to the pseudogap around the Fermi level.
Searching for the origin of the deep pseudogap around -2.8 eV (Figure 55), we inspect the partial weights of the band states in the subspaces ZnZn′, Cu, and Cu′. Figure 58 applies to the peak below the pseudogap and reveals extensive Cu-Cu′ interplay, well separated from the ZnZn′-subspace. However, the MS of the total CuCu′-subspace is too large for joined interferences = 18 which occur even in the CuCu′-subspace clearly below -2.8 eV (Figure 55).
To find the really active subspace we follow Figure 69c. Like the subclusters OH(Ag′) of -, the OH(Cu′) occupy sites in the SC unit cell close to the (330)-planes. Joined interferences = 18 must thus be possible provided the MS of the Cu′-subspace is large enough. Figure 59 confirms this conjecture. Hence, the deep pseudogap around -2.8 eV arises from the total Cu′-subspace due to joined interferences = 18 in the spectral peak around -2.9 eV.
What is the most essential process above the copper -band? The trajectory in Figure 60 shows the average -decompositions of the band states up to the upper bound of the pseudogap above . Band states above the upper bound of the copper -band (-2 eV, bullet 6) form -dominated networks supported by joined interferences = 18 in the -subspace around -1.42 eV (Figure 57). The fluctuation patterns get -type (red dots). Approaching the Fermi energy, the main process is the formation of -dominated networks, just as observed in the cases of dia-C (Figure 7) and - (Figure 19). The DOS of 333-bcc (Figure 56) and - (Figure 57) indicate joined interferences = 18, 14, and 6 as stabilization-relevant. Around = 18 the situation resembles - (Figures 21 and 22). However, planar interferences = 18
in - occur in the occupied valence band at -1.42 eV. Hence, structure stabilization can benefit from the fully pronounced -dominated network (Figures 60 and 19). Note that the spectral appearance resembles dia-C (Figure 4), too, but without a point of inflection towards a gap because there is no critical multiple scattering like C-C.
Planar interferences = 14 join the radial interferences (Cu′-Zn′) along links to neighboring 26-atom clusters in adjacent SC unit cells. The radial adjustment benefits from the absence of planar interferences = 15.
Joined interferences [d(6),R10(Cu-Cu)] with = 6 act in the -subspace around -7.8 eV at enhanced spectral density (Figure 61) just in the bonding range of a -band splitting centered somewhat below -5 eV. A related spectral feature with the joined interferences = 6 appears in the -subspace (Figure 57) at slightly lower energy which confirms the -character. All that remembers - (Figure 34) where the size and the arrangement of the subclusters OT are stabilized by similar means.
The planar interference (420) with = 20 is the first one above the interference block around = 18 which includes the planar interferences = 17, 18, and 19 together with the radial interferences on the shortest interatomic distances. Hence, no radial interference and hybridization joins = 20 via (24). = 20 is the first lonely planar interference with limited impact on structure stabilization. This explains the identical appearance in 333-bcc (Figure 56) and - (Figure 57) which differ mainly by radial adjustments.
. The Figures 62 to 65 (-) use the same presentation modes of the band energies (21) as the Figures 29 to 32 (-) and the Figures 44 to 47 (-). Vertical dashed lines denote the lower -resonances whereas dotted lines indicate the Fermi energies and the resulting band energies. Shaded areas around refer to the pseudogap of the -phase. With the specified partitions into - and -contributions we obtain = (52.05 + 10.77) eV/atom for 333-bcc and = (51.45 + 10.60) eV/atom for - (Figure 62). The step from 333-bcc to the -phase lowers the band energy by -(0,60 + 0.17) eV/atom, mainly -type due to the short-range character of the charge redistribution.
Band-energy differences at given energies above the bottom of the valence band are much better evident from Figure 63 where the band energies are plotted with reference to the common averages. The excessive band-energy of the -phase which emerges in the zinc -band is removed again in the copper -band. For this purpose both -bands couple across the interstitial -type spectral range. The Figures 64 and 65 compare the band energies of the systems in equivalent situations at equal valence-band fillings, . Figure 62 changes into Figure 64
which reveals a fundamental difference to - (Figures 31 and 32) and - (Figures 46 and 47). In both cases the band-energy differences grow monotonously towards the Fermi energy in favor of the -phase whereas - starts at low valence-band filling with the larger band energy. This turns into the opposite around the Cu′-resonance. For further analysis we show in Figure 65 besides the total band energies (full curves) the -parts (dashed curves) and the -parts (dotted curves), too, each with reference to the respective average, . The superscript ”“ specifies optionally to , , or . Both partial band-energy differences add up to the total difference, . Dashed lines denote the -resonances of the -phase. The planar interference (620) at occurs in the total DOS of the -phase just at the main peak of the anti-bonding zinc -band close to the upper bound (Figure 50).
With emphasis on the band energy, Figure 65 gives a detailed description of the transition from 333-bcc to -. Two items are evident: (i) Due to appropriate stacking of band states in the zinc -band, the -phase accumulates band energy over 333-bcc. (ii) After removal of the excess between the upper bound of the zinc -band and the Cu′-resonance the band-energy splitting acts in favor of the -phase.
The removal of the band-energy excess of the -phase occurs on large and short length scales. For separate access to large respectively short length scales we dissect the band-energy splitting into -
and -parts. Hence, band-energy differences on large length scales start acting in favor of the -phase just beyond the upper bound of the zinc -band and continue acting in the upper 50 of the valence band. Short length scales, on the contrary, contribute this way only in the upper 10 of the valence band. The electronic stabilization of - thus depends critically on the - interplay throughout the valence band. Only the short-range support is confined to the environment of the Fermi energy.
. In the initial state we suppose free atoms inside the AS (configurations, Cu(1 0 10), Zn(2 0 10)). The 20 CuCu′ contribute 220 valence electrons, and the 32 ZnZn′ add further 384. In the final state, due to the LMTO-ASMT calculation, 2.2 valence electrons per SC unit cell have moved from ZnZn′ to CuCu′. This makes 0.36 % of the 604 valence electrons per SC unit cell.
Table 7 shows the changes of the partial charges in the transition from free atoms to the -phase. The charge redistribution aims at a pronounced continuous -network around the Fermi energy which is evident from Figure 60. The last two lines present the average partial changes in two steps: The first step (to 333-bcc) concerns bonding into perfect planar order whereas the second step (to -) adjusts the radial order. Hence, 0.772 + 0.003 = 0.775 -electrons per AS and 0.213 - 0.005 = 0.208 -electrons per AS change into 0.985 - 0.002 = 0.983 -electrons per AS. This forms the stabilizing -network. In the final state, due to the LMTO-ASMT
calculation, 2.2 valence electrons per SC unit cell have moved from ZnZn′ to CuCu′. This makes 0.36 % of the 604 valence electrons per SC unit cell.
By means of , Mizutani [9] extract = 18.55 0.55 from the Hume-Rothery plot and obtain via (7) = 1.61 0.05 in perfect agreement with the estimated 21/13 1.615. One may expect the planar interference = 18 immediately below .
In the present approach, the equivalent of the -ratio is the average -occupation up to . We obtain (1.615 - 0.775) + 0.983 = 1.823 and via (7) = 20.16 which explains the planar interference (420) slightly below (Figure 57, = 20, = 1.8012, Table 14).
** -**. Joined interferences = 18 play a major part in the electronic stabilization of -. They support the resonance splitting of the zinc -band and the formation of the deep pseudogap in the upper copper -band as well as the growing -dominated network below the Fermi energy.
The ratio e/a = 21/13 = 1.615 of -, estimated from the free atoms, suggests the joined interferences = 18 to occur in the -subspace closely below the Fermi energy. However, due to -to- transfer (Table 7) the total content of the -subspace amounts to 1.823 which allows for the planar interference = 20 at -0.187 eV whereas the joined interferences = 18 occur at -1.42 eV. Planar interference = 20 have no direct access to the radial interference and hybridization on the atomic scale because no existing interatomic distance is short enough to form joined interferences.
The spectral signature of the joined interferences = 18 in the DOS resembles - and dia-C, except for a pseudogap above instead of a gap. The three systems support the formation of stabilizing -dominated networks by joined interferences = 18 respectively = 8.
The transition from 333-bcc to - adjusts the radial order (short-range) whereas the planar order (long-range) is less affected. The interference status is improved upon concentrating spectral weight at fulfilled interference conditions, and the total band energy decreases.
In the zinc -band, the -phase accumulates excessive total band energy over 333-bcc which is removed again up to the -resonance of Cu′(OH). For specific access to the short-range respectively the long-range stabilization, we subdivide the total band energy at given valence-band filling into the -part and the -part. It turns out that the -part of the band-energy difference acts in roughly the upper half of the valence band for stability of the -phase and the -part only in the top 10 . Nevertheless, the -part dominates the resulting band-energy difference of -0.77 eV/atom at the Fermi energy.
5 Conclusions
This study examines cubic crystals where the content of the simple cubic (SC) unit cell (side length ) is close to a n$$\times$$n$$\times$$n-bcc substructure. We confine to = 2 (diamond-structures: number 227 and zinc-blende structures: number 216) and to = 3, (-brasses: number 217). Using published structure models in LMTO-ASA calculations with inscribed muffin-tin potentials we obtain various projected densities of states together with the compositions of the band states in the atomic-site angular-momentum representation.
The main purpose of the study is the detailed analysis of the first-principle results within the framework of a VEC-based concept after Hume-Rothery, Mott, and Jones (HMJ-concept). At each energy, the size of the active momentum sphere (MS) in the extended -space is derived from the calculated integrated density of states in atomic-site angular-momentum representation, projected to the active electron space. The stabilization-relevant interferences occur in transitions on the surface of the MS which involve the largest momentum transfers (along the diameter of the MS, ”true backscattering“). We derive the active electron space from restricted compositions of the electron band states in atomic-site angular-momentum representation.
Interference and hybridization determine the charge distribution on the atomic scale and control this way the formation of low-temperature equilibrium structures. To surmount the usual contrasting treatment of interference and hybridization we use ”joined planar-radial interferences“ where hybridization results from well-tuned radial interference due to the sequence of neighbor shells around reference atoms. Separate interference conditions for planar and radial interferences select momentum transfers as close together as to allow for interplay via the common momentum pool in the extended -space. This ensures the balance between the local radial order inside the SC unit cell and the global planar order outside. With reference to the SC unit cell certain inside-outside relations arise such as the empirical Hume-Rothery rules which predict crystal structures on the basis of the electron-per-atom ratios (e/a-ratio) in the SC unit cell, estimated from the -configurations of free atoms.
Drawing on the example of bcc-related crystals, the present study demonstrates a general principle of electronic structure stabilization: The accumulated charge in an active electron quantum space determines the length scale of stabilizing interferences in this space and consequently the formation of the quantum states. This can be seen as a consequence of the Hohenberg-Kohn theorem [44].
The examined systems reveal several structure-relevant planar and radial length scales. Most prominent, despite of non-uniform decoration, the subcells in the SC unit cell must be kept close to bcc on the scale . This is achieved by joined interferences = 2 which describe the interplay between the planar interferences and the radial interferences (hybridizations) on the shortest interatomic links. Hence, the leading interferences in diamond- and zinc-blende phases are the joined interferences = 8 and = 18 in -brasses.
A different kind of essential joined interferences connects planar interferences (no common measure) with radial interferences along interatomic links which characterize the size of certain subclusters in the SC unit cell. This way both the size and the arrangement of such subclusters in the SC unit cell are controlled by electronic interference. In diamond- and zinc-blende phases this applies to the planar interferences (111) which act on the length scale of the elementary tetrahedron (interatomic link ). In .brasses the planar interference (211) and (321) act on the subclusters OT respectively IT. Starting from the sites in the 333-bcc sublattice, the IT(weak scatterers) are notably compressed whereas the OT(strong scatterers) almost keep their bcc-positions on the scale /3.
The predictive power of the empirical Hume-Rothery rules rests on the reasonable assumption that the allocation of partial charges to the effective atoms of the crystal is not too much different from the free atoms. The rules estimate the size of the MS in the -subspace at and compares with the required sizes at the main planar interferences of crystals. Stabilizing interference in the extended -space is thus supposed to occur around and, after projection to the atomic-site angular-momentum representation, in the -subspace.
The present study demonstrates that the essential stabilizing interferences are neither confined to the environment of nor are they confined to the -subspace. They occur in various parts of the total electron quantum space including -related subspaces In view of the long-lasting discussions concerning the electron quantum weight which should be considered in the HMJ-concept [2, 21] the present study has demonstrates that the stabilization of the real-space structure is due to interferences in the extended -space regardless of the assignment to partial charges after projection to the atomic-site angular-momentum representation.
On this background, in systems with nearly n$$\times$$n$$\times$$n-bcc sublattices in the SC unit cell, electron-per-atom ratios close to e/a = 1.6 turn out to indicate active joined interferences in the concerned subspace just below the respective energy which drives towards 2 bcc-subcells. The planar and the radial components of these joined interference act at e/a = 1.481 respectively 1.574 (cf. Table 2), in the case of -brasses at e/a = 1.538 respectively 1.635 due 52 instead of 54 occupied states in the SC unit cell.
Regarding -brasses, the ratio e/a, estimated from the -configurations of free atoms, predicts significant joined interferences = 18 in the -subspace in any case: (i) They must act clearly above if the estimated e/a 21/13, such as -. Note that the assigned -dominated network acts already around . (ii) They must act clearly below if the estimated e/a 21/13, such as -. Note that favorable conditions are adjusted for joined interferences = 18 in -type subspaces around . (iii) They may act rather around if the estimated e/a 21/13, such as -. Note that the true interference energies depend on the actual -to- transfer.
Two different concepts are usually employed as guides along paths to low-temperature equilibrium structures, namely decreasing electronic band energy and increasing electronic interference. We analyze hypothetical relaxation steps from properly decorated n$$\times$$n$$\times$$n-bcc systems to the respective -phases which improve in particular the radial interferences and hybridizations. In each case, improved interference manifests by enhanced spectral weight at fulfilled interference conditions, and the electronic band energy decreases.
Vacancies (in the atomic-sphere approximation: empty spheres ES) are created in order to enable polyvalent atoms to build structure elements in the SC unit cell which are reserved to lower valences. Only half the lattice sites in the SC unit cell are occupied with atoms in dia-C. We show that the ES acquire charge from the atoms for separate joined interferences close to the Fermi energy in the sublattice of the atoms ( = 8) and in the sublattice of the ES ( = 3).
Band states with internal -dominated networks dominate towards the Fermi energy, The transfer of -weight into -weight turns out a universal process in structure formation. We show for the examined = 2 systems that the measured bulk moduli follow this trend.
The existing medium-range radial order of amorphous phases is included as the limiting case where global planar order has not yet developed, e.g. for time-scale reasons. As yet, sufficiently detailed structure information is missing.
As an outlook, we note that the concept of joined planar-radial interferences allows for treating several issues of electronically supported structure formation. The size and the shape of the unit cell of a stable crystal which both determine the translation symmetry must act on the content of the unit cell and vice versa depending on strengths of the radial respectively the planar processes. Even quasicrystalline order in two and three dimensions is involved where other global planar arrangements develop for partnership of local radial order.
Acknowledgements
The author gratefully acknowledges the exchange of ideas with Prof. P. Häussler, Dr. R. Arnold, and Dr. C. V. Landauro during many years at the Chemnitz University of Technology. Also gratefully acknowledged, the financial support by the Deutsche Forschungsgemeinschaft during this period.
6 Appendices
6.1 Diamond, zinc blende, and 222-bcc
We assign the 16 lattice sites in the SC unit cell (side length ) of 222-bcc to four fcc-sublattices F1 - F4 (Figure 66). F1 belongs to the depicted SC unit cell. Shifting F1 by along a space diagonal guides to F2. F3 and F4 are obtained on shifting F1 respectively F2 by along cubic axes. F1 with F2 and F3 with F4 form two interpenetrating diamond lattices. The fcc-sublattices are linked by 1 = /4, 0 = /2, and 3 = /2 as shown by Table 8. 2 = /2 are interatomic links inside the fcc-sublattices. Table 9 shows decorations of the fcc-sublattices in essential cases.
6.2 Single-scattering approximation to the atomic-sphere density of states
Within the muffin-tin scattered-wave (MT-SW) concept, we calculate an approximate partial DOS, , of the AS number (ASs) which includes only scattering paths with one scattering in the environment of the ASs. The energy and refer to the muffin-tin zero (MT0). In this approximation to the back-scattered field, one can estimate the consequences of fulfilled interference conditions.
The MT orbitals are used in the straightforward representation with complex spherical harmonics, , where is the unit vector
along . Outside the MT spheres, the regular basic solutions and the singular outgoing ones are chosen as respectively . The and (asymptotic radial part ) are vacuum solutions. The complex scattering amplitudes of the MT potentials, , depend on the real scattering phase shifts, .
In this framework, the representation (16) of the partial DOS of the ASs is available which derives from the DOS of the isolated frozen ASs without environment, which depends only on the , and the diagonal elements, , of the scattering-path matrix (15). collects all scattering paths through the system which start and terminate inside the ASs at the valence orbital with the angular momentum (Figure 67). The exponentials with the scattering phase shifts result from the transitions between muffin-tin orbitals inside and vacuum orbitals outside the AS. The elements of the propagation matrix describe the propagation of vacuum waves from (s^{\prime}$$L^{\prime}) to (s$$L) and the elements of the diagonal matrix the scatterings by the spherically symmetric AS.
We obtain the envisaged single-scattering approximation to the partial DOS of the ASs upon terminating the multiple scattering in (15) after one scattering in the environment,
[TABLE]
This way a single-scattering approximation to the AS-DOS, , is obtained for the central AS of a big cluster. The real phase shifts, , at energies above the muffin-tin zero are calculated from the self-consistent LMTO-ASMT potentials.
For dia-C, Figure 68 compares this single-scattering DOS with the LMTO-ASMT calculation and with the bare-AS DOS (frozen ASs without environment). The energy scale refers to the MT0, .
Active interference in the interatomic free propagation can be expected at those energies where interference conditions (6), (11) are fulfilled with the momentum transfer on the left-hand side. They are indicated by vertical lines with labels [(220),1] and [(111),2]. The interferences [(220),1] remove spectral weight from the bare-AS DOS starting at the upper edge of the gap. The bonding component of the b/ab-splitting appears at the upper edge of the hat-shaped feature. Including higher terms of the multiple-scattering expansion of (15) causes the suppression of spectral weight in the gap to moves downwards towards the Fermi energy.
In the ES the interferences [(111),2] act at the upper edge of the spectral range which has been attributed to the stabilization of the arrangement of the centered tetrahedra. Zinc blende phases open an additional gap in this energy range.
6.3 Diamond- and zinc-blende phases
Measured bulk moduli and Pauling electronegativities of the examined alloys are shown in Table 10. The sequence of the systems follows growing size of the SC unit cell. EN(A) and EN(B) refer to the original respectively the corrected electronegativities.
6.4 The -brasses and 333-bcc
Green bullets in Figure 69 denote the atomic sites of - whereas the red bullets belong to 333-bcc. The following shifts of the atomic sites occur in the transition from 333-bcc to -: Li(IT) and Ag(OT) move along the space diagonals, Ag′(OH) along the cubic axes, and Li′(CO) towards the space diagonals.
Most remarkable, the compression of the IT whereas the OT stay very close to the sublattice sites of 333-bcc. As the bcc-sites on the scale of the SC unit cell are empty, the OT preserve the memory of bcc-structure on the scale .
Inspecting the relaxation step more in detail (here not shown) reveals that pairs of atoms on strongly reflecting lattice planes of 333-bcc are moved to symmetric positions with respect to the former planes. We conclude that the planar interferences must be less affected by the step from 333-bcc to the -phase.
The Tables 11 to 13 show the shortest interatomic distances n in and between the subclusters of the
-phases respectively the n in the reference structure 333-bcc. The order of the interatomic links between and inside the subclusters IT, OT, OH, and CO is taken from the sequence of growing interatomic distances n of -. The Tables 12 and 13 use the same order despite of different sequences of growing interatomic distances n.
Table 14 is a list of the required virtual valences for excitation of SC Bragg reflections (7). All the 52 sites in the SC unit cell of -brasses are supposed to participate. If only sites participate, the required virtual valences increase by factors of . enclosed in braces indicates a gap in the sequence of SC Bragg reflections.
6.5 The -resonances of the -phases
The spectral positions of -resonances prove important to the electronic stabilization of -brasses. The following compilation of materials-related data is derived from muffin-tin scattered-wave calculations on the basis of the calculated LMTO-ASMT potentials. Small spectral shifts arise from different environments of the atoms (AS) in the respective structure model. So, up to four different resonance energies can arise. Note that the assignment of atoms with subclusters IT, OT, OH, and CO applies only to the -phases. For the properly decorated 333-bcc systems we take the atoms prior to the relaxation step. The following symbols are used: Fermi energy (F), muffin-tin zero (MT0), and the bottom of the valence band (B).
6.6 Visualization of the composition of band states
Each band state distributes the norm One throughout the EQS. We decompose the total EQS into any three subspaces with non-negative partial weights where = 1 - . e.g. the decomposition into the -subspaces. Phase diagrams of three-component systems demonstrate how such normalized decompositions are visualized by means of equilateral triangles (Figure 70). All the marked triangles are equilateral triangles.
A Cartesian coordinate system supports the navigation inside the big triangle. We put the origin in Figure 70 to the intersection of the -axis with the -axis and obtain the position of the bullet,
[TABLE]
Suppose a small energy interval centered at a given energy . All the band states inside generate a distribution of bullets which is poorly represented by one bullet at the average position. To utilize the full information content we plot the tracking curve of the average together with all bullets of the actual energy interval. The resulting animation reveals preferences of the fluctuation patterns which indicate the favored as well as the avoided interplay between subspaces of the total EQS.
Let us now replace the along the sides of the big triangle by where and are the subspace-projected respectively the total densities of states. The result will be a position very close to the average bullet above. Deviations are due to the special smearing of the energy eigenvalues in the LMTO-ASMT calculation. Hence, with rising energy, the average bullet follows the DOS-based curve.
The Cartesian coordinates are very useful in order to analyze the fluctuations. If, e.g., the fluctuation patterns point perpendicular to the W1-axis we have x = 0 which provides 1 = -2/2. Furthermore, the norm One provides 3 = -(1 + 2) = -2/2. This mens that equal losses of 1 and 3 result in a gain of 2 twice as much. The opposite applies, too.
This technique has the advantage that the distribution of quantum weight throughout the total EQS is visualized by the geometry of equilateral triangles.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1[1] W. Hume-Rothery. J. Inst. Met., 35 (1926) 295.
- 2[2] T. B. Massalski. Materiels Transactions, 51[4] (2010) 583.
- 3[3] G.-V. Raynor. Prog. Met., 1 (1949) 1.
- 4[4] G. Trambly de Laissardiére, D. Nguyen Mahn and D. Mayou. Prog. in Mater. Sci., 50 (2005) 679.
- 5[5] N. F. Mott and H. A. Jones. The Theory of the Properties of Metals and Alloys. Clarendon Press: Oxford, UK, 1936; Dover Publicatiions Inc.: New York, NY, USA, 1958.
- 6[6] W. Hume-Rothery. J. Inst. Met., 9 (1962) 42.
- 7[7] R. Asahi, H. Sato, T. Takeuchi, and U. Mizutani. Phys. Rev. B, 71 (2005) 165103.
- 8[8] A. T. Paxton, M. Methfessel, and D. G. Pettifor. Proc. Roy. Soc. London, Ser A 453 (1997) 1493.