Morphology, ordering, stability, and electronic structure of carbon-doped hexagonal boron nitride
Agnieszka Jamr\'oz, Jacek A. Majewski

TL;DR
This study uses theoretical methods to analyze the morphology, stability, and electronic properties of carbon-doped hexagonal boron nitride, revealing phase separation tendencies and conditions for uniform carbon distribution.
Contribution
It introduces a bond order valence force field and Monte Carlo simulations to explore atomic arrangements and electronic properties of CBN alloys, highlighting phase separation and electronic band bowing.
Findings
Phase separation into graphene and h-BN domains is common.
N-rich conditions can produce more uniform carbon distribution.
The energy gap exhibits strong bowing in stoichiometric alloys.
Abstract
We present theoretical studies of morphology, stability, and electronic structure of monolayer hexagonal CBN alloys with rich content of h-BN and carbon concentration not exceeding 50 %. Our studies are based on the bond order type of the valence force field to account for the interactions between atomic constituents and Monte Carlo method with Metropolis algorithm to establish equilibrium distribution of atoms over the lattice. We find out that the phase separation into graphene and h-BN domains occurs in the majority of growth conditions. Only in N-rich growth conditions, it is possible to obtain quasi uniform distribution of carbon atoms over boron sublattice. We predict also that the energy gap in stoichiometric C(BN) alloys exhibits extremely strong bowing.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1 Figure 2
Figure 2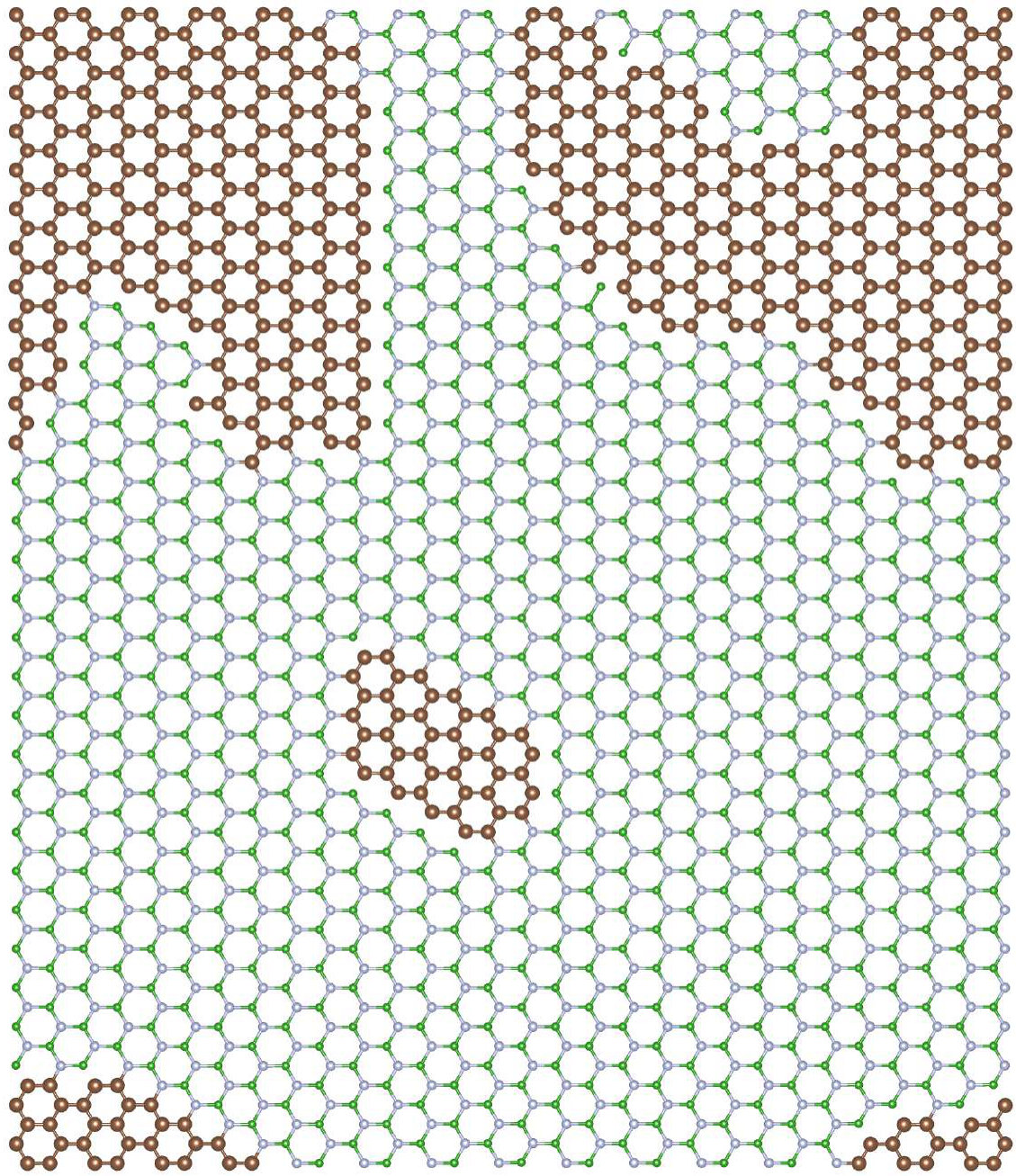 Figure 3
Figure 3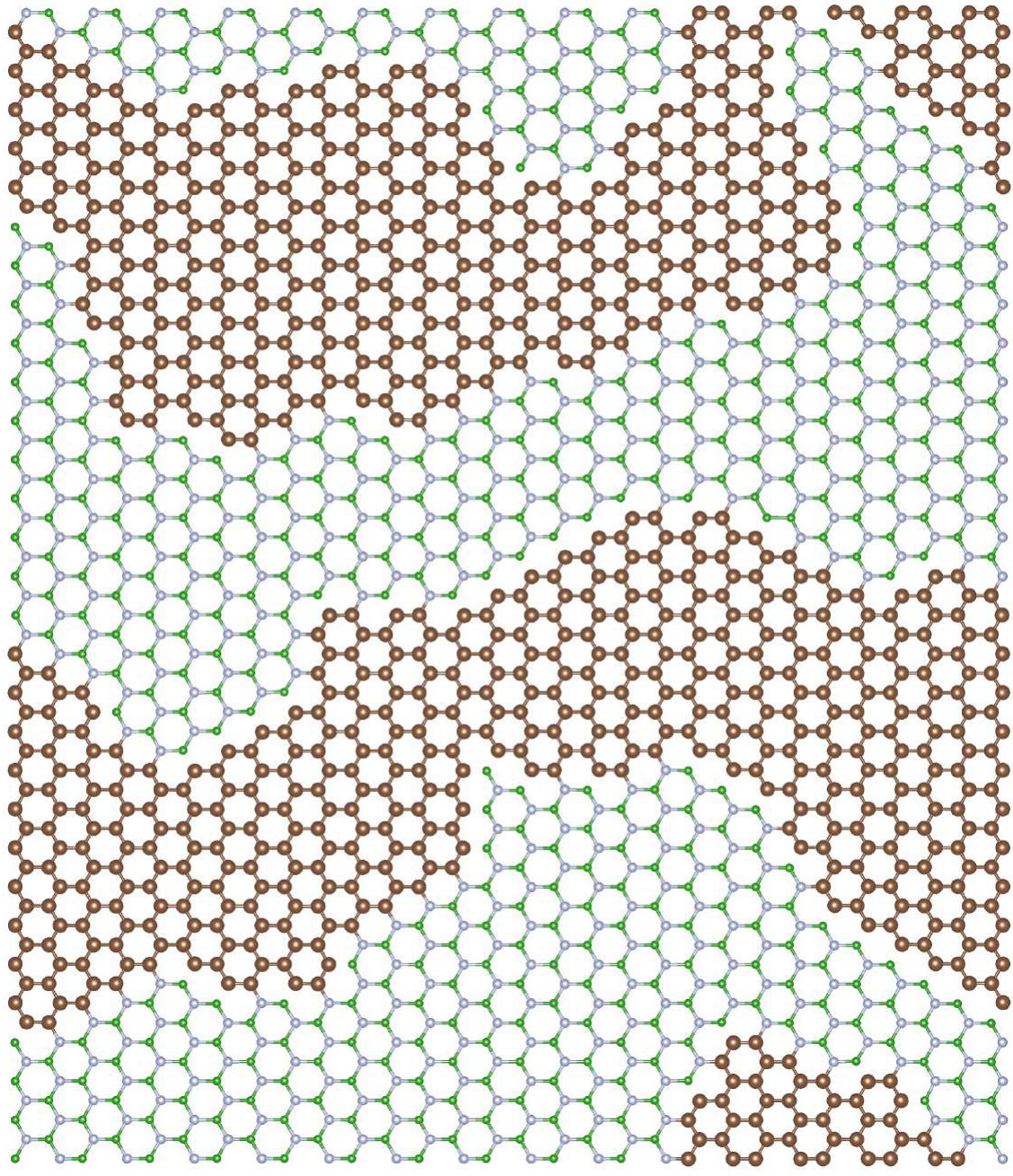 Figure 4
Figure 4 Figure 5
Figure 5 Figure 6
Figure 6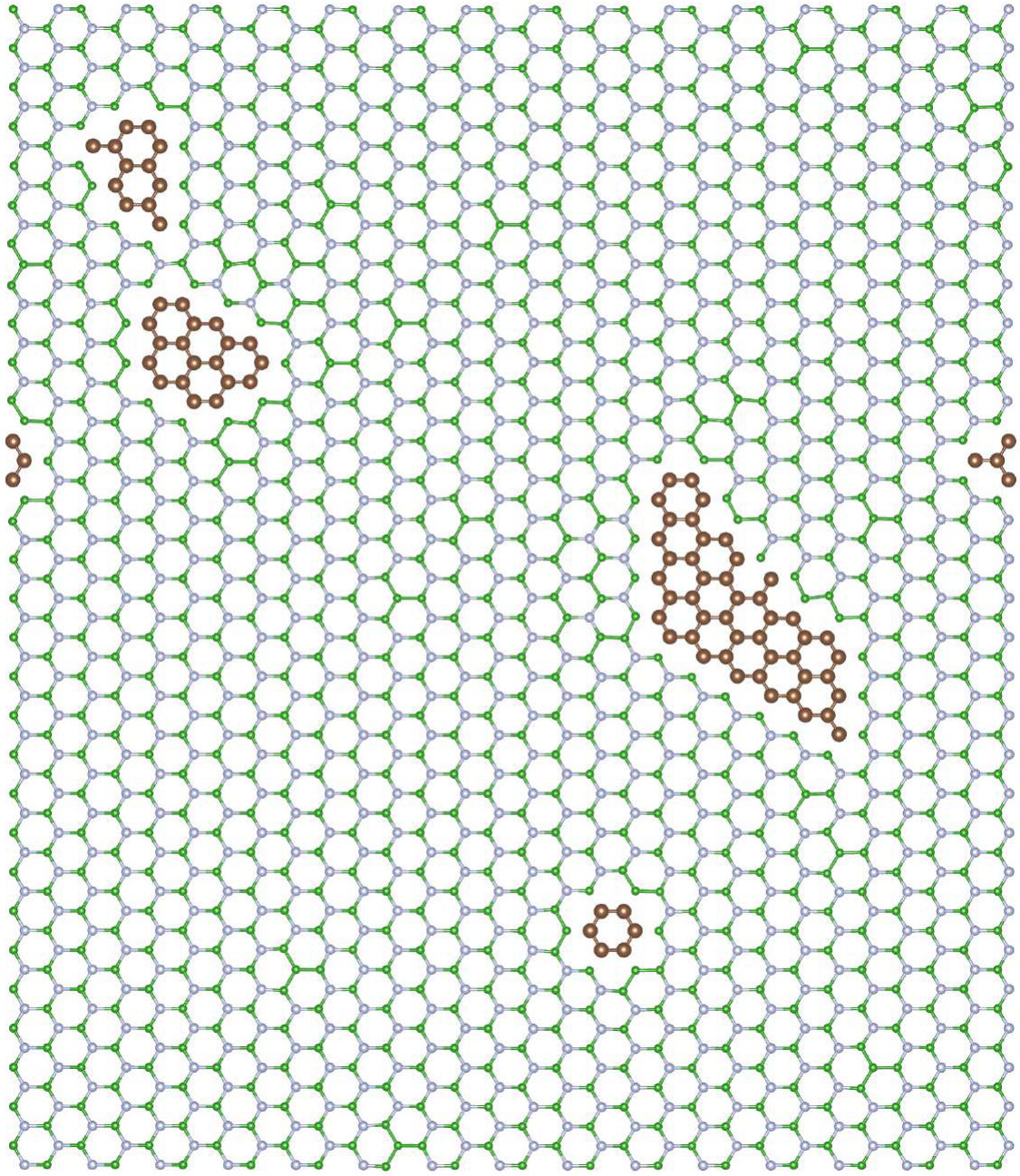 Figure 7
Figure 7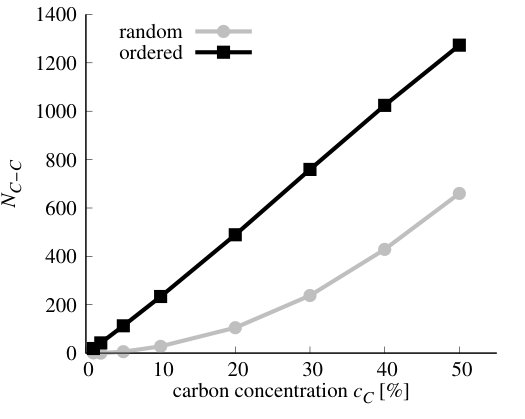 Figure 8
Figure 8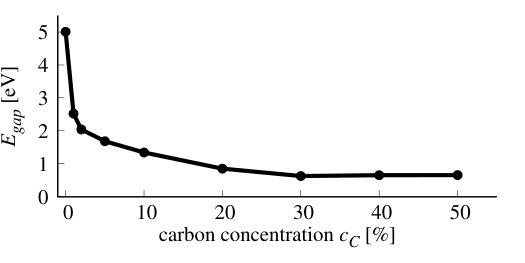 Figure 9
Figure 9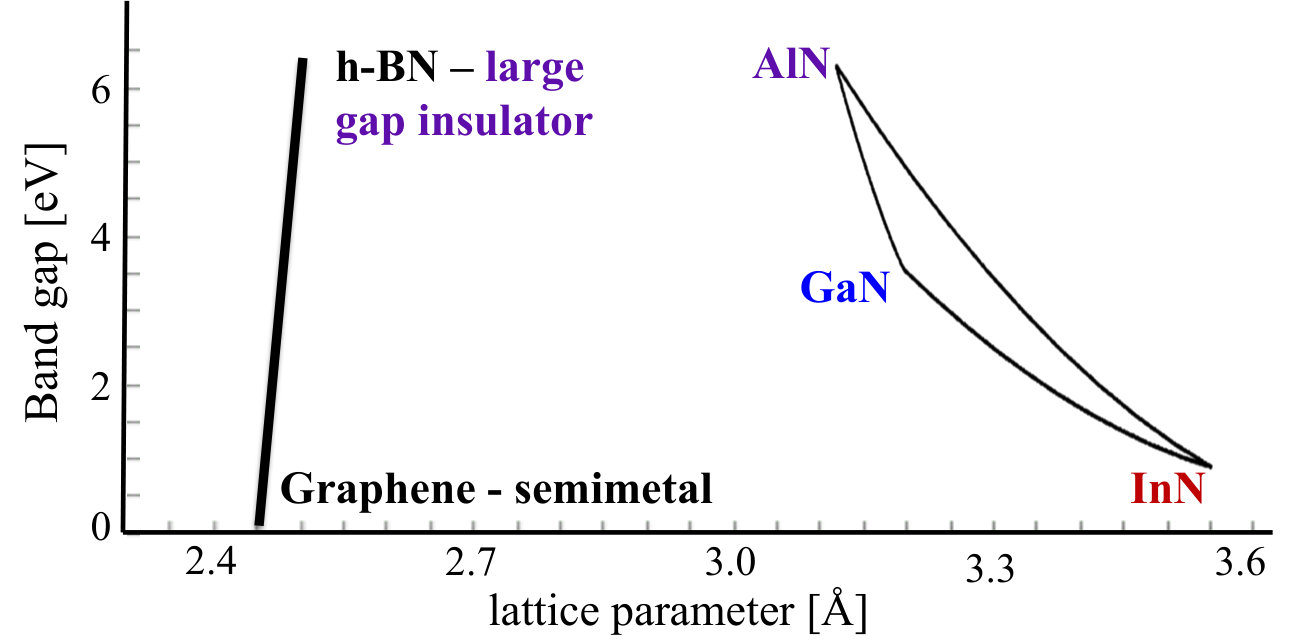 Figure 1
Figure 1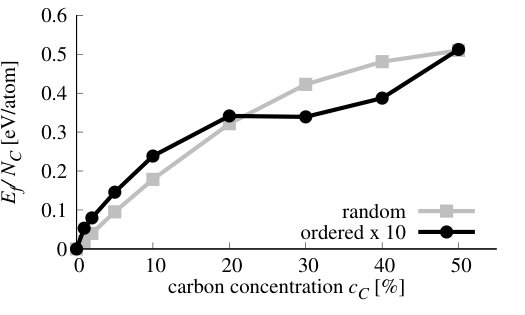 Figure 11
Figure 11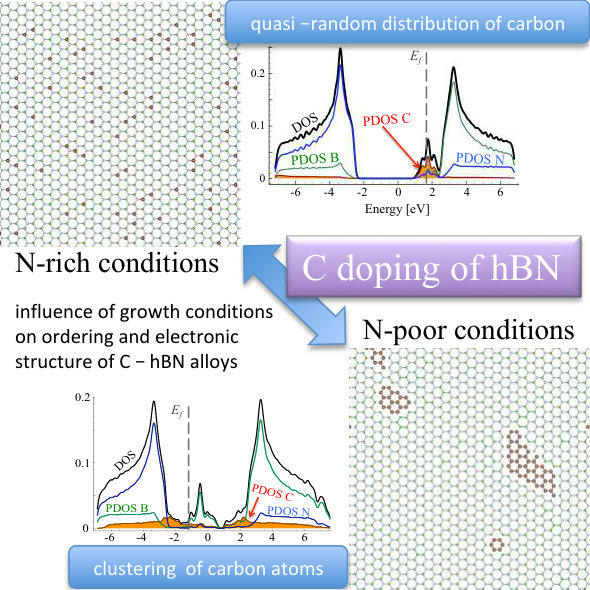 Figure 12
Figure 12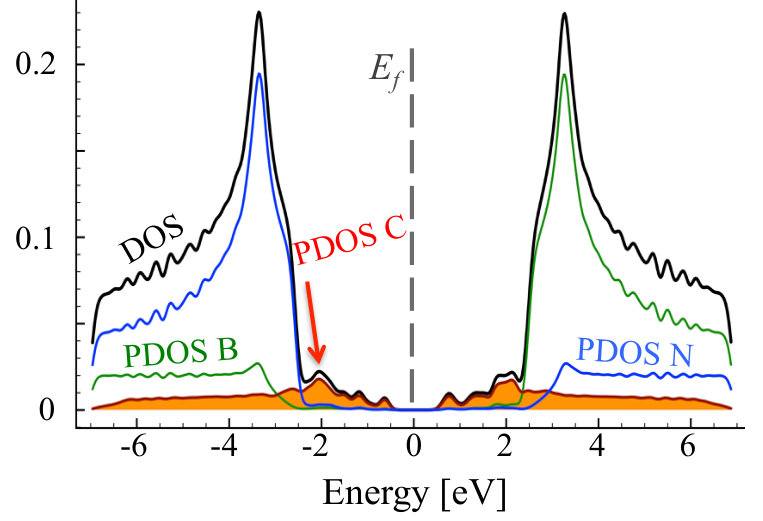 Figure 13
Figure 13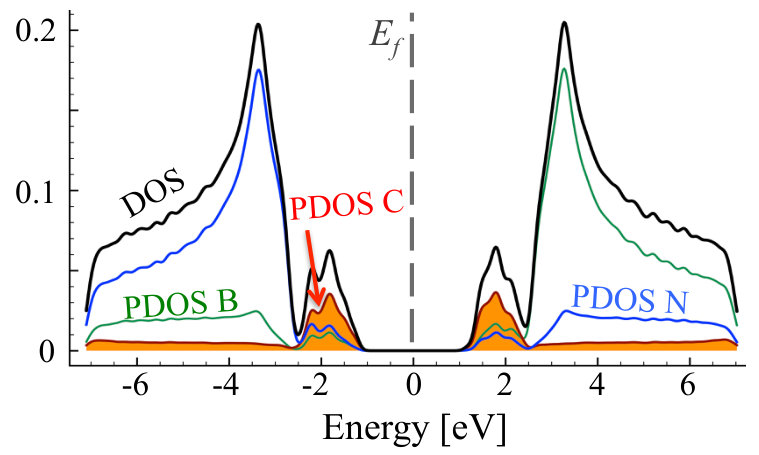 Figure 14
Figure 14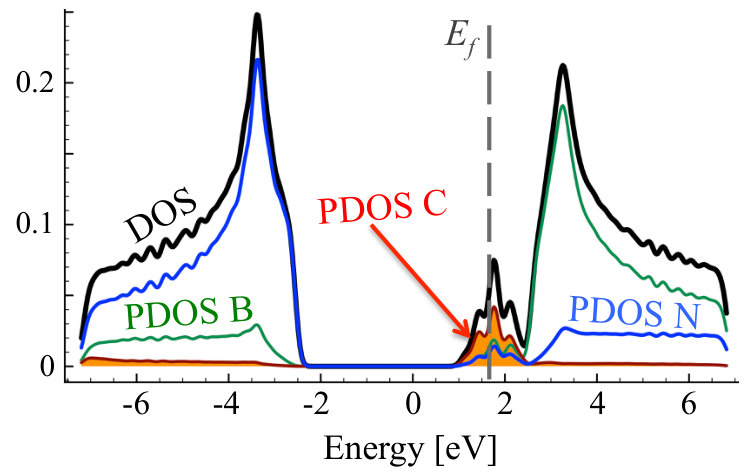 Figure 15
Figure 15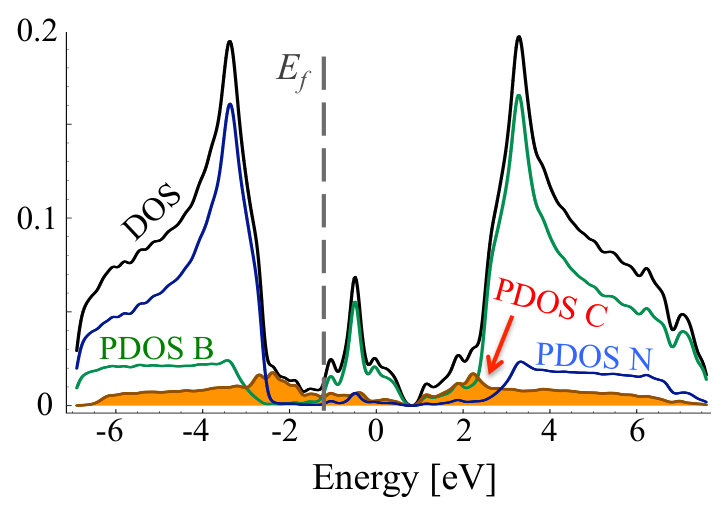 Figure 16
Figure 16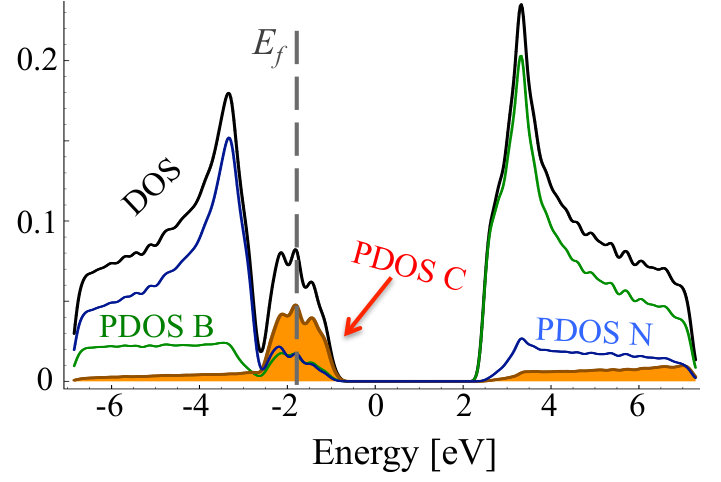 Figure 17
Figure 17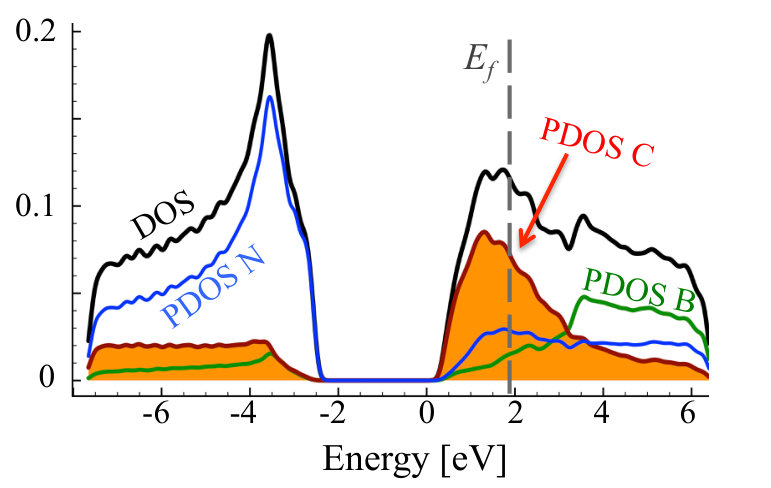 Figure 18
Figure 18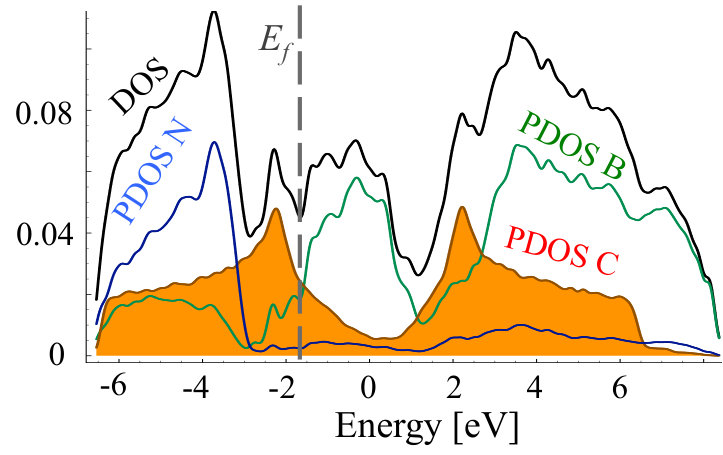 Figure 19
Figure 19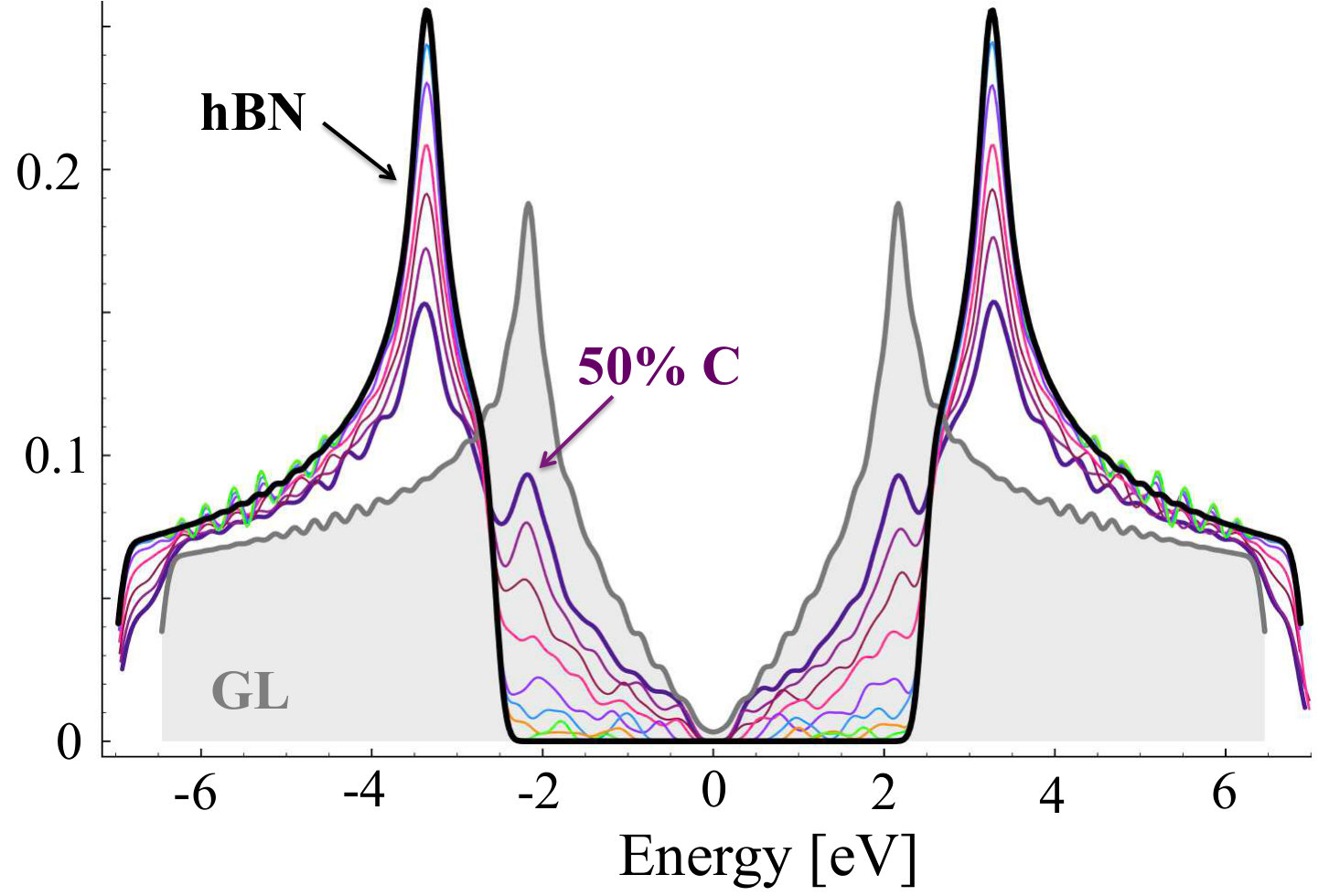 Figure 20
Figure 20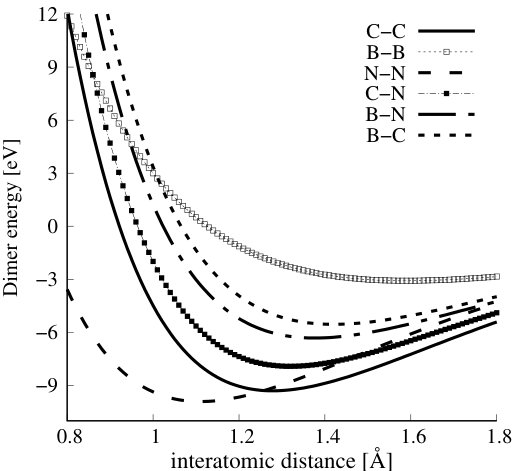 Figure 21
Figure 21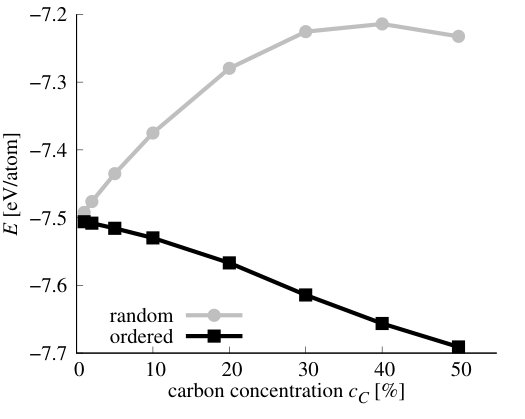 Figure 22
Figure 22Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
e-mail [email protected]
11institutetext: Faculty of Physics, University of Warsaw, Pasteura 5, 02-093 Warsaw, Poland
Morphology, ordering, stability, and electronic structure of carbon-doped
hexagonal boron nitride
Agnieszka Jamróz
Jacek A. Majewski
Abstract
We present theoretical studies of morphology, stability, and electronic structure of monolayer hexagonal CBN alloys with rich content of h-BN and carbon concentration not exceeding 50%. Our studies are based on the bond order type of the valence force field to account for the interactions between atomic constituents and Monte Carlo method with Metropolis algorithm to establish equilibrium distribution of atoms over the lattice. We find out that the phase separation into graphene and h-BN domains occurs in the majority of growth conditions. Only in N-rich growth conditions, it is possible to obtain quasi uniform distribution of carbon atoms over boron sublattice. We predict also that the energy gap in stoichiometric Cx(BN)1-x alloys exhibits extremely strong bowing.
keywords:
CBN lateral alloys, 2D hexagonal material, computer simulation, atomic scale structure, disordered systems, order-disorder effects, composition fluctuations
1 Introduction
Two-dimensional, layered, atomically thin systems play important role in contemporary materials science. Hexagonal layered boron-carbide-nitrides, i.e., nanosystems with boron, nitrogen, and carbon atoms distributed over the honeycomb lattice, constitute rich and interesting family of the 2D materials [1, 2, 3]. The two limiting cases of such systems are graphene and monolayer of hexagonal boron nitride, and these systems can be considered as building blocks of layered CBN alloys. Depending on the relative concentrations of C, B, and N, they might be on one hand referred to as boron- and/or nitrogen-doped graphene (sometimes called B-graphene, N-graphene, or, BN-graphene, respectively), or on the other hand, as carbon doped hexagonal boron nitride (h-BN), very often indicated as C-hBN, or Cx(BN)1-x. In addition, these layered CBN alloys can be combined into vertically stacked heterostructures [4], in a similar manner to fairly intensively studied nowadays the so-called van der Waals (vdW) heterostructures of graphene and h-BN. Such layered CBN alloys could definitely enrich the families of both vdW and also lateral heterostructures and are definitely worth of basic investigations. As it was mentioned above, the 2D layered CBN system can be considered as an alloy of graphene and h-BN. These two ingredients of the alloy are nearly lattice matched and exhibit the lattice mismatch of only 1% (the lateral lattice constants of graphene and h-BN are equal to 2.46 Å[5] and 2.504 Å[6], respectively). Simultaneously, their fundamental band gaps differ by 6 eV, ranging from semimetal to high band gap insulator. Therefore, the layered CBN alloys should allow for band gap tuning within this range. This uniqueness of CBN layered alloys is emphasized in Figure 1, where an illustrative comparison with the family of III-Nitrides (GaN, AlN, and InN as border materials) is given. It is, therefore, very natural that the layered CBN alloys are considered as obvious and promising candidates for numerous applications not only in the area of functional electronic and optoelectronic devices [7], but also in many other fields such as carbon capture [8], high energy density supercapacitors [9], Li-Ion batteries [10], metal-free photoredox catalysis [11], effective and highly selective H2 separation membranes [12], and many others.
In this paper, we focus on h-BN rich CBN alloys (i.e., with carbon content not exceeding 50%) and perform extensive theoretical studies of their morphology, ordering, stability, and electronic structure. These studies should shed light on the physical mechanisms leading to the synthesis of alloys with various phases and ordering patterns observed experimentally. Before we present the details of the employed methodology and the results of the studies, we would like to survey very briefly the basis experimental facts about the studied layered CBN alloys.
There are numerous approaches to synthesize carbon-containing hexagonal boron nitride, including CVD, MOVPE, or post-synthesis implantation of carbon into pristine h-BN layer. The possibility to synthesize one monolayer of hexagonal boron nitride was proved few years ago [13, 14]. In this pioneering studies, monolayers of h-BN were formed on the Ni(111), Pd(111), and Pt(111) surfaces, and the electronic dispersion relations of the monolayer were measured [13, 14]. Later on, the h-BN monolayer was fabricated by Chemical Vapor Deposition (CVD) and Metal Organic CVD (MOCVD) techniques [15, 16, 17]. Employing various growth techniques, synthesis of h-CBN graphetic but ternary monolayer was also successfully performed. For example, to synthesize h-CBN, the methane and ammonia borane were introduced at the same time to act as precursors for carbon and BN, respectively [2], or in other growth process the bis-BN cyclohexane, B2N2C2H12, was utilized for the synthesis of h-CBN [18]. Typically, the atomic ratio of B, C, and N can be tuned to some extent by changing the growth conditions, which leads to h-CBN samples of various homogeneity and morphology [18]. Electron-beam mediated post-synthesis doping of boron-nitride nanostructures with carbon atoms was also demonstrated [19, 20]. Generally, from experimental studies the following picture emerges: (i) the separation of phases occurs typically in growth conditions close to equilibrium; (ii) however, the ability in h-CBN alloys to form homophilic (C-C) as well as heterophilic (C-B, C-N, B-N) bonds generates a rich variety of polymorphic structures, making the precise control of their chemical stoichiometry and geometry formidable [21].
As a consequence, according to the authors of Ref. [21], detailed experimental insights into phase separation and ordering in such alloys are currently lacking. This strongly suggests the need of employing reliable theoretical modelling to increase understanding of these nano-structures, and some theoretical studies of the stability and electronic structure of the h-CBN alloys were already performed, both for ordered prototypes of alloys [22, 23, 24, 25, 26, 27, 28, 29] and taking configurational disorder into account [30, 31, 32].
In Ref. [30] phase stability for monolayer BNC ternary system was examined by Monte Carlo simulations and the cluster expansion effective Hamiltonian with parameters determined from the density functional theory (DFT) calculations. The studies concluded that all possible atomic arrangements exhibited positive formation energies, indicating phase separation into monolayer BN and graphene. The authors also concluded that only C-C and B-N bonds would be energetically favorable in the alloy, whereas all others, i.e., C-N, B-C, B-B, and N-N would be not. Also the optical absorbance and possibility of band gap engineering, phase separation, and composition fluctuations of Cx(BN)1-x alloys were investigated [31], where the DFT calculations combined with the generalized quasi-chemical approximation (GQCA) to account for disorder effects were employed. In that study the tendency to separation into h-BN and graphene was found to be strong, however, some mixing was allowed. In Ref. [32], an approach combining the DFT calculations and Monte Carlo was used to investigate the influence of interface geometry on phase stability and band gap engineering in boron nitride substituted graphene. The Monte Carlo sampling was performed with the model Hamiltonian defined on a bond basis with bond energies extracted out from DFT calculations. The authors came to similar conclusions as in the papers [30, 31], namely that the stability and properties of CBN alloys would be governed by the fact that C-C and B-N bonds are energetically more favorable than other bonds in the systems.
It is worth to note, that in all the studies of CBN alloys mentioned above, the DFT calculations that were used either directly to find out the physical mechanisms determining the stability of CBN systems or were used as a kind of intermediate procedure to determine parameters of model Hamiltonians utilized for further investigation of systems’ equilibrium configuration with Monte Carlo method dealt with rather small alloy structures, containing typically up to few dozens or sometimes hundreds of atoms [28]. In our methodology, we follow the direct approach, albeit empirical in nature, which allows us to treat systems consisting of thousands of atoms, either with or without periodic boundary conditions. This approach (named VFF-MC) is based on the valence force field (VFF) to calculate the total energy of alloys and Monte Carlo method to determine the equilibrium distribution of atoms over the lattice. We have used successfully the VFF-MC scheme (with Keating type of VFF) to investigate ordering issues in III-nitride ternary and quaternary alloys [33]. We have also investigated chemical order on fixed hexagonal lattice in B-, N-, and BN-doped graphene monolayer [34] with VFF of bond order Tersoff’s potential [35]. This potential has successfully history of implementations in huge number of studies dealing with systems composed of carbon, nitrogen, and also boron. In our previous studies [34], we considered carbon reach CBN alloys with concentration of dopants not exceeding 50%. In the present study, we address the h-BN rich side of mono-layer CBN alloys, with carbon part below 50%. This contributes to the hotly discussed issue of carbon doped nitrides. In addition, we have refined our VFF-MC approach: (i) allowing for lattice relaxation, and (ii) extending scheme for electronic structure calculations of alloys.
The paper is organized as follows. In Section 2.1 we describe first the specific implementation of VFF Tersoff’s potential and then MC-VFF simulation scheme in Section 2.2). Some kind of validation of chosen Tersoff’s potential is given in Section 2.4. The main results of this paper are discussed in Section 3, and finally the paper is summarized in Section 4.
2 Methods
2.1 Tersoff potential for simulating carbon doped
boron nitride systems
The CBN alloys as systems lacking long-range order and exhibiting short range order in turn are extremely computationally demanding for any atomistic scheme. To obtain reliable theoretical predictions of morphology and structural properties of these alloys, one needs typically to perform calculations for thousands of atoms, which is out of scope of presently so widely used ab initio methods. Therefore, the attractive alternative is to employ the VFF type of potential that makes the calculations feasible. Our previous studies for B and N doped graphene have confirmed that the semi-empirical Tersoff potential (T-P) [35], which belongs to the family of bond-order potentials and is fairly widely exploited in computational materials science, provides reasonable description of such systems. In contrast to classical two-body interatomic potentials (e.g., Lennard-Jones, Keating), the interaction strength between two atoms in the T-P depends not only on their mutual distance, but also on the closest environment (coordination number and types of neighboring atoms). This feature of the T-P allows even for the correct description of chemical reactions.
In the present work, we use T-P for multicomponent systems following the formulation of Kroll [36]. The total energy of the system is described as a sum over two center interatomic interactions between atomic pairs and separated by the distance
[TABLE]
where and constitute repulsive and attractive interaction, respectively, and is cut-off function, just to ensure potential’s short range. and are typically chosen to include only the first shell of neighbors 111In present research we used: Å, Å, Å, Å, Å, Å.
[TABLE]
[TABLE]
The bond-order term describes the influence of neighborhood on the binding strength of a pair of atoms by modifying the attractive part of the potential
[TABLE]
[TABLE]
[TABLE]
where is the angle between the atomic pairs and bonds. Altogether, there are 13 various parameters in the T-P: , , , , , , , , , , , , and . The parameters depend on the chemical nature of the atoms and , and their specific values for certain elements have to be established through the suitably performed fitting procedures.
In the present study, we use the following parameterisations of the T-Ps: the Lindsay and Broido parameters for the C-C interactions (they were originally developed for graphene and carbon nanotubes [37]); the Albe and Möller parameters for the B-B and N-N interactions (first introduced for description of the hexagonal boron nitride based systems [38]); and the parametrization of Kinaci et. al for the heteroatomic C-B, C-N, and B-N bonds (developed earlier for h-BN/graphene interfaces in lateral graphene/h-BN junctions [39]).
2.2 VFF-MC simulation scheme
In order to find equilibrium distribution of atoms over the lattice and atomic geometry of mono-layered hexagonal CBN alloys, we employ our own simulation technique that we refer to as VFF-MC scheme [33, 34]. We use Monte Carlo (MC) algorithm in Metropolis NVT ensemble to search the configurational space, and the Conjugent Gradient (CG) method to optimize geometry of the investigated structures. Computational details look as follows. Assuming certain concentration of carbon atoms, we distribute them randomly over the hexagonal lattice points according to the three patterns corresponding to the various growth condition: (i) over B sites, which corresponds to N-rich conditions, (ii) over the N sublattice, which corresponds to N-poor conditions, and (ii) evenly over N and B sublattices, which corresponds to the conditions inducing the growth of stoichiometric Cx(BN)1-x alloys. Then we perform Monte Carlo steps by swapping a randomly chosen pair of atoms and generating a new configuration. If the swapping procedure leads to the lower energy of the system the resulting configuration is unconditionally accepted, if not the new configuration is accepted with probability , where is the difference of the total energies of the system before and after the swap, is the Boltzmann constant, and is a temperature like parameter that determines only the swap probabilities in the Metropolis algorithm (T = 300K is used in simulations performed in these studies). The simulation continues for few millions of MC steps (however, the convergence is typically reached in MC steps).
The supercells used to study CBN alloys contain 1800 atoms, and have rectangular shape with both zigzag and armchair edges (’right/left’ and ’top/bottom’ edge in the figures presented later on, respectively). This gives us possibility to investigate not only periodic 2D systems (which is realized by assuming periodic boundary conditions in both x and y directions), but also 1D nanoribbons (periodicity only in one direction), and 0D flakes (no periodicity assumed), however, we focus on the 2D layered systems in the present paper.
2.3 Formation energy of defects and alloys
Having determined the alloy’s equilibrium distribution of atoms and geometry, we can calculate the formation energy (also called enthalpy of formation) of C-alloyed h-BN systems following the well established formula [40],
[TABLE]
where [CBN] and [h-BN] are the total energies of CBN alloy and pristine h-BN, respectively, and indicate the number of B and N atoms, respectively, which are substituted by carbon atoms. For example, for isolated carbon impurity substituted in place of B atom, = = 1, and = 0. Further, , , and are the chemical potentials of B, N, and C, respectively.
The chemical potentials represent the Gibbs free energy that isolated atoms exchange with the heat reservoir. Those variables should represent experimental conditions of alloy growth (growth conditions). The maximal possible values of the chemical potentials (with i = B, N, C) correspond to the most stable phases of the elements. Therefore, in this study, is referenced to as energy per atom in -rhombohedral phase of borophene, is energy per atom of nitrogen molecule, and is carbon energy in monolayer graphene, and all energies have been calculated employing Tersoff’s potential. In the real growth conditions, the chemical potential of element can differ from its maximal value , and this difference is commonly indicated as . In thermodynamic equilibrium [40], the differences in chemical potentials for two elements forming a chemical compound must be equal to the compound’s enthalpy of formation . For B and N pair, this rule reads , where is the enthalpy of formation of BN pair in h-BN, (BN) , with being the total energy of BN pair in h-BN. The values of the characteristic energies obtained with Tersoff’s potential employed in our studies are: = -15.01 eV, = -6.84 eV, = -4.96 eV, and = -7.98 eV, leading to the h-BN formation enthalpy of = -3.27 eV. This value agrees very well with formation enthalpies calculated in the framework of DFT for cubic and hexagonal bulk BN, and being equal respectively to -3.0 eV and -3.3 eV [41]. Computing formation energies of alloys, we will consider the N-rich conditions (i.e., when the system is in equilibrium with N2 atmosphere), with , which gives = 0 and , and also N-poor (B-rich) conditions, when = 0 and = . Before we turn to the discussion of predictions obtained within VFF-MC scheme for monolayer CBN alloys, let us present some test calculations for homovalent and heterovalent dimers involving B, C, and N, as well as for isolated carbon defects CB and CN, i.e., with carbon substituted in place of B and N, respectively. This should help to assess the quality of the Tersoff’s potential.
2.4 Validation of parameters set for 2D C-B-N systems
In the case of diatomic molecules, the Tersoff’s potential provides an analytic formula for equilibrium dimer bond length and dissociation energy. Table 2.4 summarizes equilibrium bond lengths and energies of all possible pairs of atoms forming dimer molecules out of atoms constituting the CBN alloys (C-C, B-B, N-N, C-B, C-N, and B-N), whereas in Figure 2, the dimers’ phase diagrams (the energy dependence on the distance between atoms) are depicted. The comparison with available experimental data for dimers, corroborates that the Tersoff’s potential provides overall reasonable description of the studied dimers.
[h]
Equilibrium bond lengths and energies of all dimers corresponding to all possible bonds in CBN alloys calculated with Tersoff’s potential, compared to experimental values, and , respectively.
[TABLE]
- •
(a) Ref. [45], (b) Ref. [46], (c) Ref. [47], (d) Ref. [48].
As a next step of Tersoff’s potential testing, we performed calculations for CB and CN point defects. For these calculations, relatively small supercells containing 200 atoms have been employed. The lattice relaxation around impurity has been performed with fairly standard accuracy for forces, which magnitudes in all directions have been required to be smaller than 0.01 eV/Å. Our calculations for CB impurity show that the nearest-neighbors C-N bond length is equal to 1.43 Å and compare very favorably with ab-initio calculations in the DFT framework that predict this distance to be equal to 1.409 Å [42] for bulk hexagonal BN. For CN defect, we observe the distance between C-B atoms equal to 1.55 Å in very good record with the DFT calculations that predicted this bond length to be equal to 1.509 Å [42]. In the case of N-rich conditions, the formation enthalpies obtained on basis of Tersoff’s potential are equal to -3.19 eV and 6.80 eV, for CB and CN defects, respectively. In qualitative agreement with ab initio calculations, our studies predict that CB defect has the lowest formation enthalpy in h-BN grown in N-rich conditions. However, the obtained magnitudes of the formation enthalpies for the CB and CN defects differ considerably from the DFT based values reported in the literature. Namely, for monolayer h-BN grown in N-rich conditions, the formation enthalpy of CB was calculated to be 1.8 eV (with PBE exchange-correlation functional) and 1.95 eV (with HSE exchange-correlation functional) [43], whereas the formation enthalpy for CN defect was found to be 4.1 eV (PBE) or 4.3 eV (HSE) [43]. Similarly, for hexagonal bulk BN, the formation enthalpies of CB and CN impurities in N-rich growth conditions were found to be equal to 1.7 eV and 4.3 eV [42], respectively. Also the very new DFT results for bulk hexagonal BN give the formation enthalpies of CB and CN defects as 1.5 eV and 4.2 eV [44], respectively. We ascribe these discrepancies to the nature of Tersoff’s potential, where charge transfer effects are not taken into account, and, plausibly, they might be relevant in the presence of heterovalent C-N and C-B chemical bonds.
To summarize, the Tersoff’s potential satisfactorily describes geometry and energetics of C, B, and N mutual dimers and carbon defects in h-BN, corroborating that the VFF-MC scheme based on Tersoff’s potential may provide valuable predictions for morphology, ordering, and energetic stability of CBN alloys. However, before we turn to the discussion of this issue, we would like to provide a short description of the tight-binding formalism for the low-energy electronic structure of CBN alloys, which definitely increase our understanding of these systems.
2.5 Tight Binding approach for Density of States
For the description of the electronic structure of CBN alloys, we use simple empirical tight-binding (T-B) theory considering only -type orbitals. We consider just first neighbor hopping characterized by single hopping parameter .
[TABLE]
where are -states centered on hexagonal lattice sites. We would like to stress that such simple T-B method describes very well the low-energy spectrum around -point in graphene and h-BN [49, 50]. Numerous parameterizations of the T-B Hamiltonian in Eq.8 are used in calculations of electronic structure for CBN systems. In our calculations, we employ the parameters obtained through fitting the T-B electronic structure of h-BN to the predictions of the density functional theory [51], and specifically = -2.16 eV, and the on-site energies of -2.55 eV, 0.0, and 2.46 eV, for nitrogen, carbon, and boron, respectively [51]. We have also used another set of T-B parameters [52] to calculate the density of states for few test structures and observed that it leads only to insignificant changes.
The T-B Hamiltonian of the size , where is the number of lattice points considered within the VFF-MC formalism, can be easily diagonalized by any method for lattices considered in this paper. The projected density of states (PDOS) on the state and density of states (DOS) are defined as follows
[TABLE]
[TABLE]
For practical computations, we substitute delta function by the Gaussian function of a given width
[TABLE]
.
3 Results
Now we turn to the discussion of the morphology, ordering, stability, and electronic structure of monolayer honeycomb CBN alloys, which emerges from the calculations within the developed VFF-MC + T-B scheme. In our calculation the content of carbon atoms does not exceed 50%. As it was described in the introduction, the morphology of alloys depends very strongly on the growth conditions. Here, we would like to present results of our calculations performed in such a way that they mimic various growth conditions.
3.1 CBN alloys in N-rich growth conditions
We have performed calculations for carbon concentrations ranging up to 30%. Figure 3 depicts the distribution of C atoms at exemplary concentration of 10% in CBN alloy after VFF-MC simulation.
As one can see, in the equilibrium structure C dopants are distributed quasi-randomly within B sublattice, strongly indicating that C-N bonds are rather energetically favorable, and no clustering happens in N-rich growth mode. The formation energy of a CBN alloy, as calculated according to Equation 7, is very close to sum of formation energies of isolated CB defects, i.e., eV, where is the number of C atoms in the system, and exhibits extremely weak dependence on the concentration of C atoms. Figure 4 depicts density of states for optimized geometry of C0.1B0.4N0.5 alloy. One can observe impurity band originating from carbon states around the Fermi energy, i.e., the typical donor like band characteristic for carbon substituting boron. Note that in the case of C atoms distributed over the B sublattice, the initial and optimized atomic configurations exhibit very similar DOS.
Even in the case of 30% of B substituted by C atoms, there is no clustering, but the density of states is significantly modified, especially in the region of conduction band, as shown in 5, and the Fermi level is shifted towards higher energies.
3.2 CBN alloys in N-poor growth conditions
Now we turn to the case of CBN alloys grown in the N-poor conditions. It corresponds to the situation when carbon atoms are substituted initially on nitrogen sites (randomly), and the number of N atoms in the system is reduced. The distribution of atoms in the energetically optimized configuration for the carbon concentration of 10% is depicted in figure 6.
It can be clearly seen that we have different behavior of the alloys in N-rich (as described above) and N-poor conditions. In N-rich conditions, C dopants are distributed quasi-randomly within B sublattice, while in N-poor case - clustering of C atoms is observed for all investigated carbon concentrations. The formation energy in systems with C substituted on N sublattice is very high eV in all cases, with values slightly higher for smaller concentrations of C. Thus we can conclude that in N-rich growth conditions, there is possible to synthesize C-B-N alloy with quasi-random distribution of C atoms, and such alloy should be more stable than in the case of conditions with deficiency of N. It is clearly seen that after the VFF-MC simulation carbon atoms form clusters of graphene in C0.1B0.5N0.4 alloy, and also some B-B bonds appear. This causes significant differences in DOS for structures: (i) with initial random distribution of C over the N sublattice, and (ii) with final energetically optimized morphology. This is depicted in Figures 7 and 8. With C atoms randomly distributed over N sublattice, one observes states in DOS corresponding to individual CN impurities, whereas the energetically favorable structure exhibits following features in its electronic structure, such as widen owing to the C-C interaction carbon PDOS, and appearance of states of boron origin in the band gap of h-BN. This effect is even more pronounced for C concentration of 30%, as illustrated in Figure 9, where C states seem to form graphene like DOS, and B states largely modify area of h-BN gap.
The cases discussed so far reflect two extreme cases that can emerge during growth of C-doped h-BN systems, for example, within a CVD process. In the following section, we present the result for intermediate case, i.e., when C atoms are distributed equally between B and N sublattices. This corresponds to the situation when we have proper concentrations of gases providing both N and B to the system in equal amounts.
3.3 Ordering and density of states in CBN stoichiometric alloys
We have performed simulations of stoichiometric CxByNz alloys, i.e., when . Henceforward, we will refer to them as Cx(BN)1-x systems. We have performed VFF-MC simulation to obtain equilibrium distribution of atoms for x in the range of 1-50%. The resulting distributions of carbon atoms over the lattice are presented in Figure 10 for the exemplary concentrations cases of x = 1, 5, 10, 30, 50 %. It is clearly seen that, for all concentrations of carbon, separation of graphene (GR) and h-BN phases appears. This result is in accordance with experimental results, that frequently report clustering of GR and h-BN in ternary alloys [21, 53, 54, 55]. On the basis of our theoretical investigation, this result can be explained by the fact that C-C and B-N bonds are much more energetically favorable in comparison to C-N and particularly to C-B bonds, thus during MC simulation system tends to minimize number of the latter. We have compared energy per atom and number of C-C bonds in the system at non-equilibrium (i.e., random in our case) and in the system corresponding to the thermodynamic equilibrium (i.e., after MC procedure) for all investigated concentrations of C. They are shown in Figure 11.
As can be seen in Figure 11 b), the number of C-C bonds in equilibrium structure deviates strongly from number of these bonds in the case of randomly distributed C atoms. This happens in the whole range of concentrations. The formation energies of the stoichiometric Cx(BN)1-x alloys, both with random and optimized distribution of atoms are depicted in Figure 12. It is worth mentioning that in the case of these alloys = = , and the alloy formation energy in Eq.(7) may be expressed in terms of the carbon concentration = (with being the total number of atoms in the system) as follows
[TABLE]
where the is the so-called mixing energy commonly used in the literature as the indication of the alloy formation energy, and , , and are the energies per atom for Cx(BN)1-x alloy, h-BN, and graphene, respectively. As one can see, the alloy formation energies for the Cx(BN)1-x alloys with energetically optimized distribution of C atoms (i.e., exhibiting short range order) are generally an order of magnitude lower (maximal energy is of order 0.04 eV) than the alloy formation energies for random alloys (maximal energy is of order 0.4 eV). However, they are both positive, which indicates the tendency for the phase separation between h-BN and graphene. In the case of alloy with short range order, one observes also a minimum in for around 30%, whereas for the random alloys, exhibits monotonic (nearly parabolic) dependence on the carbon concentration .
Qualitatively, the similar behavior of alloy formation energies for Cx(BN)1-x systems was observed in Refs.[27, 32], where stability of nanoribbons with zig-zag and armchair interfaces between h-BN and graphene was investigated. The maxima of alloy formation energies lie there by 0.15 eV and 0.25 eV for structures with zig-zag and armchair interfaces, respectively. Also reaches there some plateau for carbon concentration above 30%, however, there is so pronounced minimum of as observed in the present study for Cx(BN)1-x alloys with short range order see Figure 12.
Having calculated the mixing energy , it is very easy to calculate free energy defined as
[TABLE]
[TABLE]
where is the Boltzmann’s constant, and entropy of mixing. Such procedure was performed in the Refs. [30, 31] and the critical temperatures for the miscibility of carbon in h-BN were determined. Our estimations lead to the conclusion that the full miscibility could be achieved only above the melting temperature of h-BN. Taking into account that we (in analogy to Refs.[30, 31]) do not consider vibrational entropy contribution to and also configurational part of the entropy should be modified in the case of the short range order present in the alloy, we will perform the discussion of the phase diagrams for CxByNz alloys elsewhere.
The results for density of states for stoichiometric compounds are presented in Figure 13, whereas, the dependence of band gap on C concentration is shown in Figure 14. The band gap decreases very fast with increasing concentration of C in the system, and is below 1 eV for concentrations higher than 20%, just exhibiting drop by 4 eV. This would suggest possible way for tuning the band gap in CBN systems. Similar non-linear absolute decrease of the band gap was previously observed in several experimental [28, 53] and theoretical studies [27, 28, 32, 56], however, the band gap bowing observed in the bulk h-BN [53] is much smaller than the value reported in this paper. In other studies, the decrease of energy gap in Cx(BN)1-x alloy with C concentration is very abrupt in full analogy to our findings.
Figures 15 and 16 show TDOS and PDOS for exemplary case of C0.1(BN)0.9 before and after MC simulation, i.e., for non-equilibrium random and equilibrium distribution of C atoms exhibiting short range order, respectively. One can notice significant difference of Projected Density of States connected with carbon atoms. For random distribution of C, one can notice significant contributions of carbon states in the area of h-BN band gap. They are connected to not clustered CB and CN impurities in h-BN lattice which act like donors and acceptors, respectively. The structure is different in the system with short range order. Carbon states are more uniformly distributed, and the band-gap is much smaller due to the presence of graphene areas in the system.
4 Summary
In the summary, we have performed calculations of morphology, ordering, stability, and electronic structure of h-BN rich monolayer CBN alloys with carbon concentrations below 50%, paying particular attention to accounting for the various growth conditions in experiments. In agreement with some experimental and theoretical studies, we find that in the most cases h-BN and graphene segregate building domains or precipitates. However, we predict that the CBN alloys with the homogeneously distributed carbon atoms could only form during growth process with the N-rich conditions. Such alloys have metallic character with Fermi level lying in the carbon induced impurity band. In the Cx(BN)1-x stoichiometric alloys, the band gap exhibits very fast non-linear decrease with growing carbon concentration.
{acknowledgement}
This work was supported by the National Science Centre in Poland (NCN) through the grants PRELUDIUM (No. UMO-2017/25/N/ST3/00660) and OPUS-12 (UMO 2016/23/B/ST3/03567).
Graphical Table of Contents
GTOC image:
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1[1] Y. K. Yap editor, B-C-N Nanotubes and Related Nanostructures , Lecture Notes in Nanoscale Science and Technology (Springer, Heidelberg, 2009).
- 2[2] L. Song, Z Liu, A.L.M. Reddy, N. T. Narayanan, J. Taha‐Tijerina, J. Peng, G. Gao, J. Lou, R. Vajtai, P. M. Ajayan, Adv. Mater. 2012 , 24 , 4878.
- 3[3] N. Kumar, K. Moses, K. Pramoda, S. N. Shirodkar, A. Kumar Mishra, U. V. Waghmare, A. Sundaresan, C. N. R. Rao, J. Mater. Chem. A 2013 , 1 , 5806.
- 4[4] B. Sachs, T. O. Wehling, M. I. Katsnelson, A. I. Lichtenstein, Phys. Rev. B 2016 , 94 , 224105.
- 5[5] D. R. Cooper, B. D’Anjou, N. Ghattamaneni, B. Harack, M. Hilke, A. Horth, N. Majlis, M. Massicotte, L. Vandsburger, E. Whiteway, V. Yu, ISRN Cond. Matt. Phys. 2012 , 501686; https://dx.doi.org/10.5402/2012/501686.
- 6[6] R.W. Lynch and H.G. Drickamer, J. Chem. Phys. 1966 , 44 , 181.
- 7[7] M.R. Uddin, S. Majety, J. Li, J.Y. Lin, H.X Jiang, J. Appl. Phys. 2014 , 115 , 093509.
- 8[8] S. Chen, P. Li, S. Xu, X. Pan, O. Fu, X. Bao, J. Mater. Chem. A 2018 , 6 , 1832.