Universality of the collapse transition of sticky polymers
Aritra Santra, Kiran Kumari, R. Padinhateeri, B. Duenweg, J. Ravi, Prakash

TL;DR
This study demonstrates that sticky polymers exhibit a universal swelling behavior similar to homopolymers during the thermal crossover from $ heta$ to athermal conditions, despite the presence of specific sticker interactions.
Contribution
It introduces a polymer model with sticky monomers and shows that their collapse transition is second-order and exhibits universal swelling behavior, extending the understanding of polymer universality.
Findings
Sticky polymers undergo a second-order $ heta$ collapse.
Swelling behavior matches universal homopolymer swelling in crossover regime.
Kuhn segment length remains unchanged with stickers under $ heta$ conditions.
Abstract
The universality of the swelling of the radius of gyration of a homopolymer relative to its value in the state, independent of polymer-solvent chemistry, in the crossover regime between and athermal solvent conditions, is well known. Here we study, by Brownian dynamics, a polymer model where a subset of monomers is labelled as "stickers". The mutual interaction of the stickers is more attractive than those of the other ("backbone") monomers, and has the additional important characteristic of "functionality" , i.e., the maximum number of stickers that can locally bind to a given sticker. A saturated bond formed in this manner remains bound until it breaks due to thermal fluctuations, a requirement which can be viewed as an additional Boolean degree of freedom that describes the bonding. This, in turn, makes the question of the order of the collapse transition a…
Click any figure to enlarge with its caption.
 Figure 1
Figure 1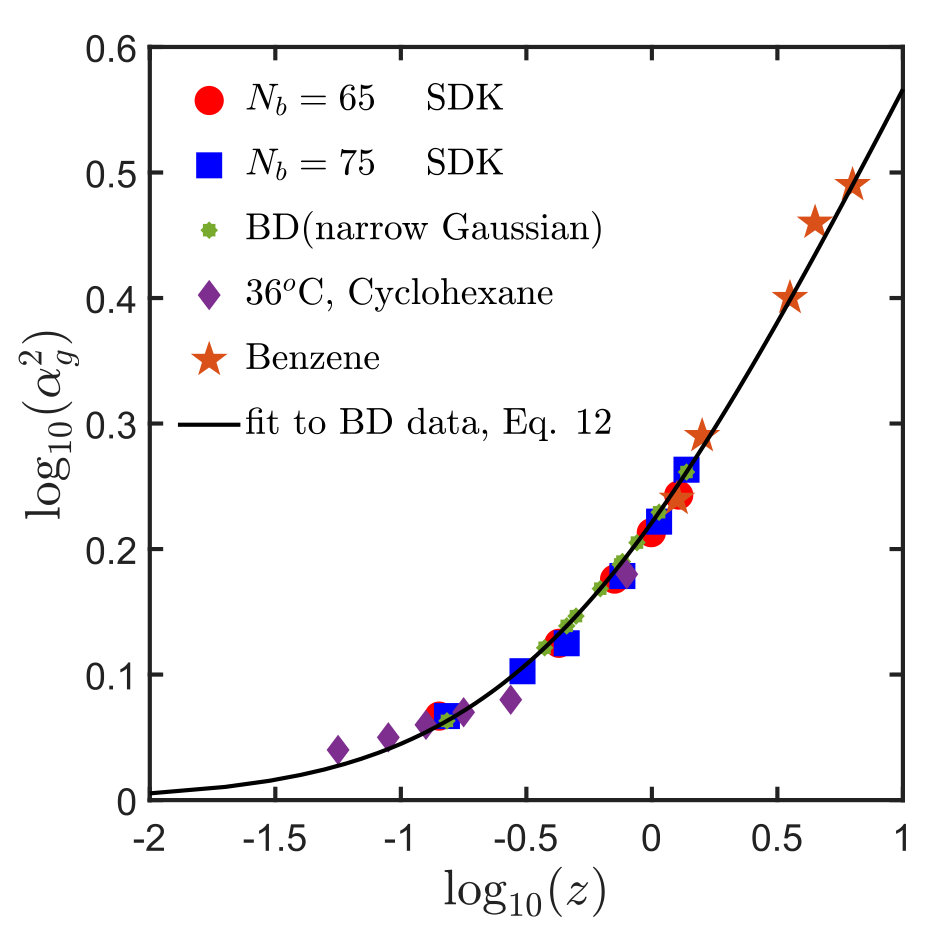 Figure 1
Figure 1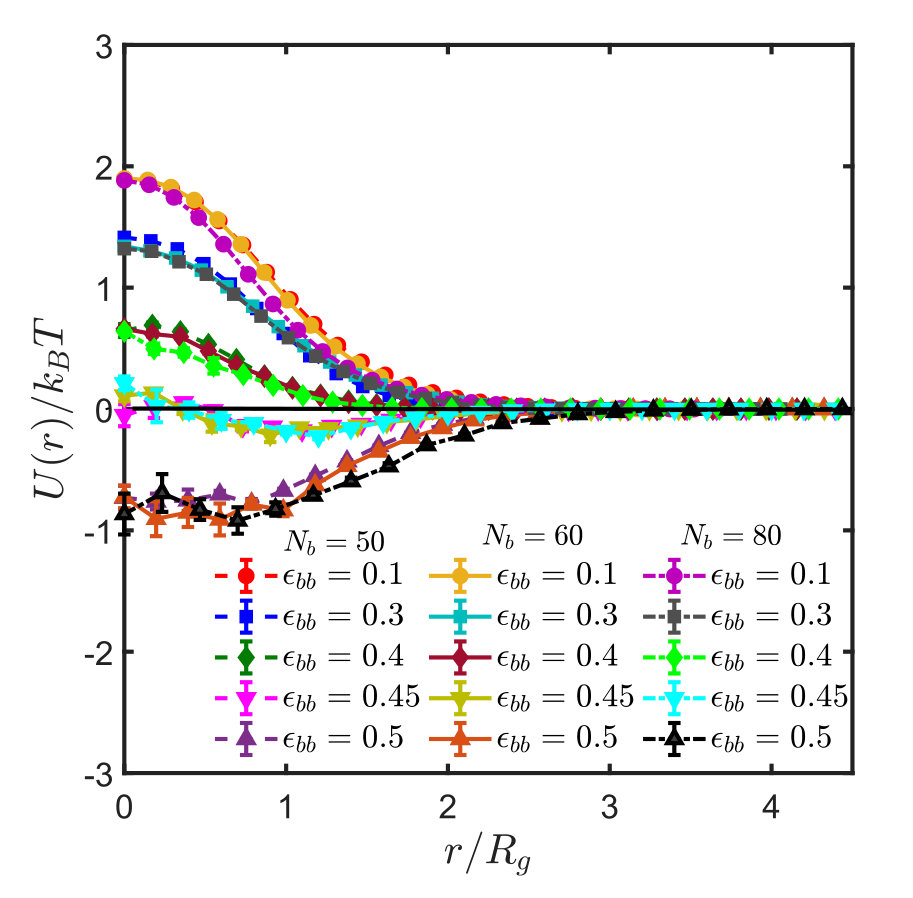 Figure 10
Figure 10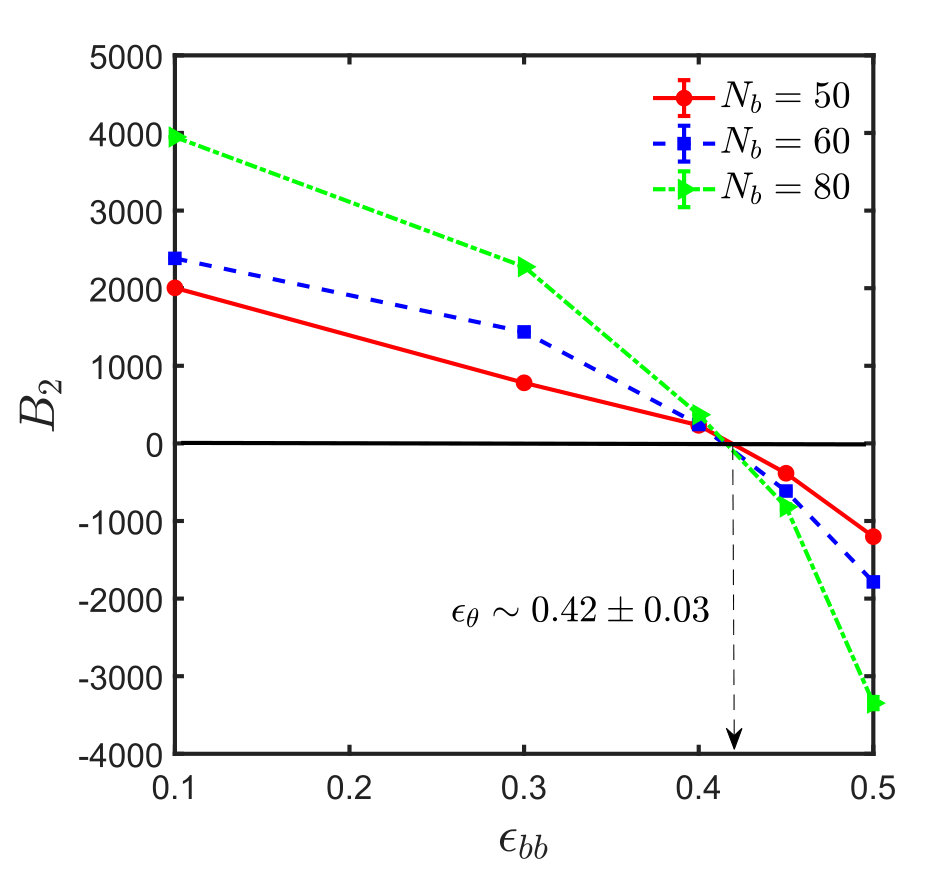 Figure 10
Figure 10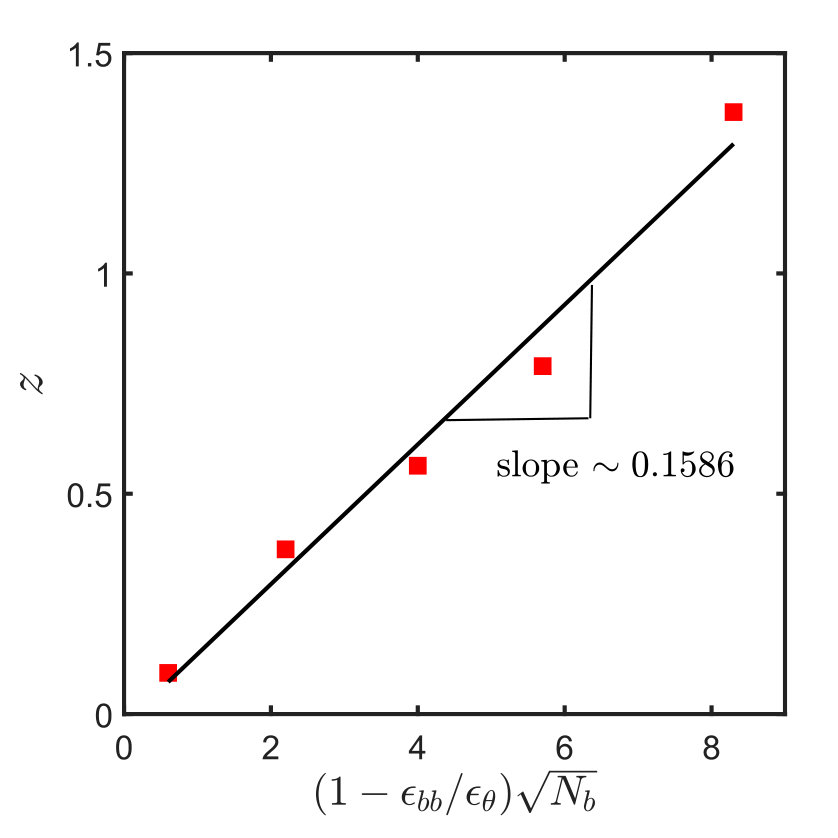 Figure 2
Figure 2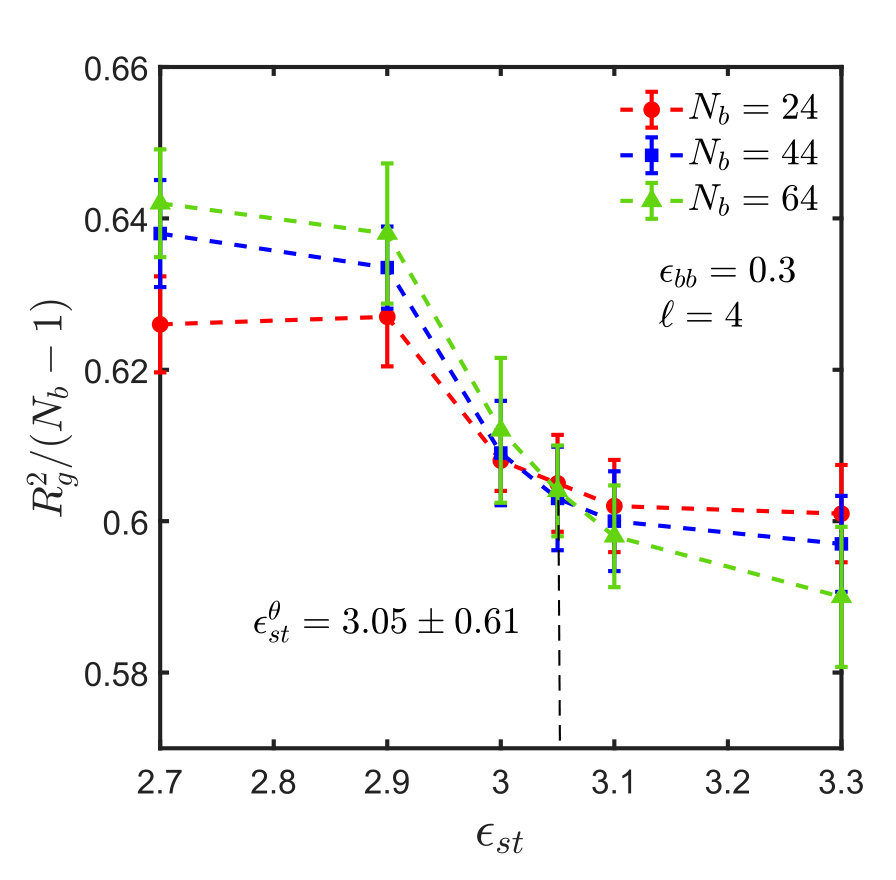 Figure 3
Figure 3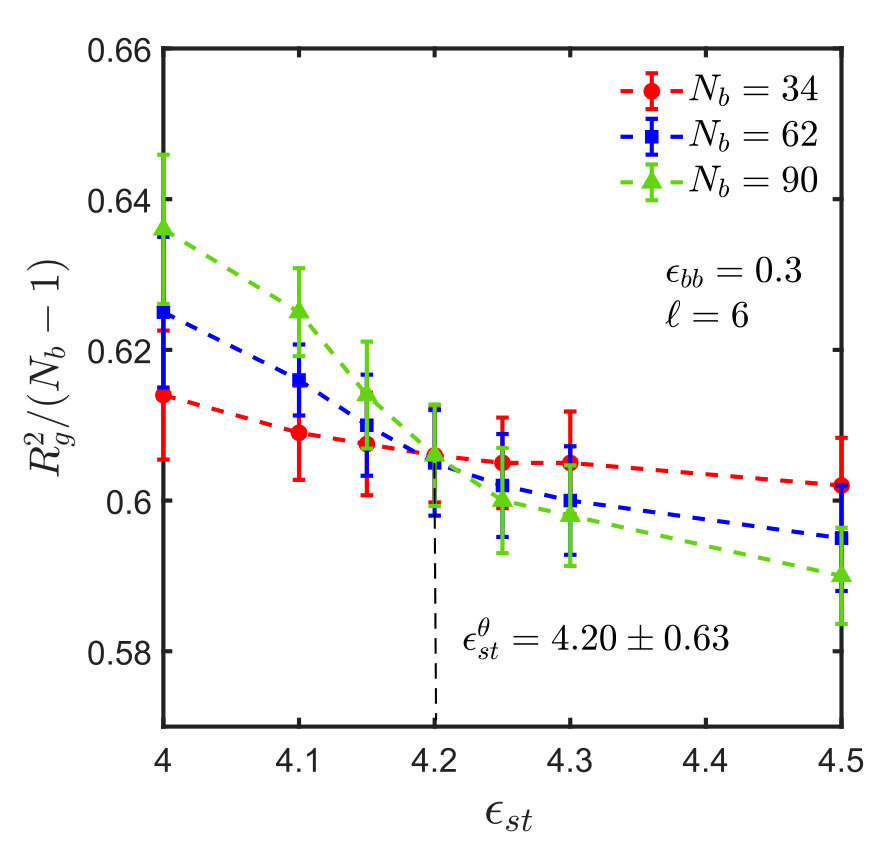 Figure 3
Figure 3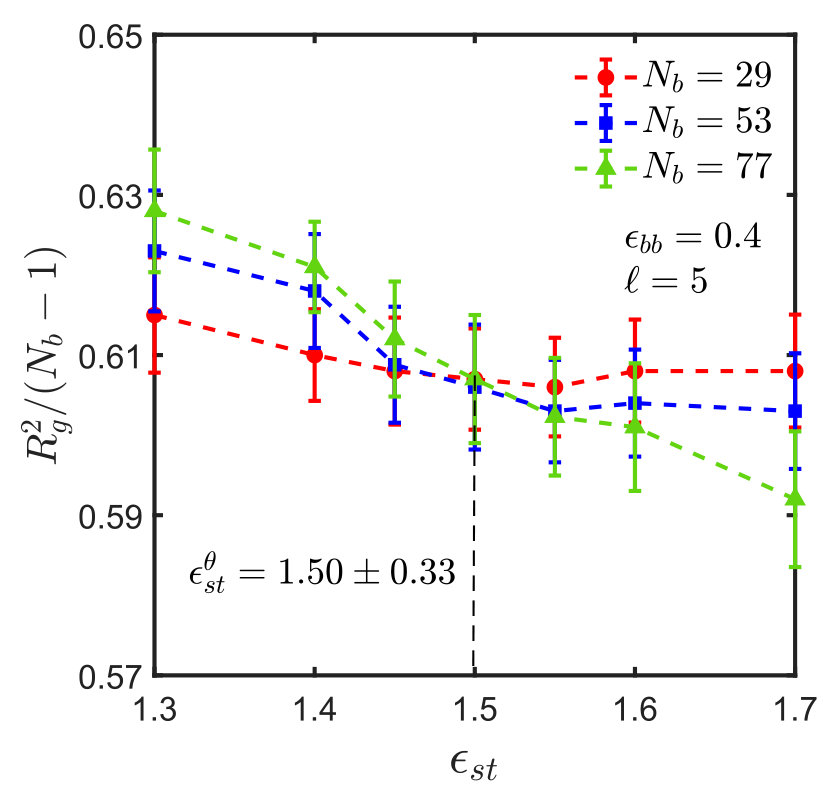 Figure 3
Figure 3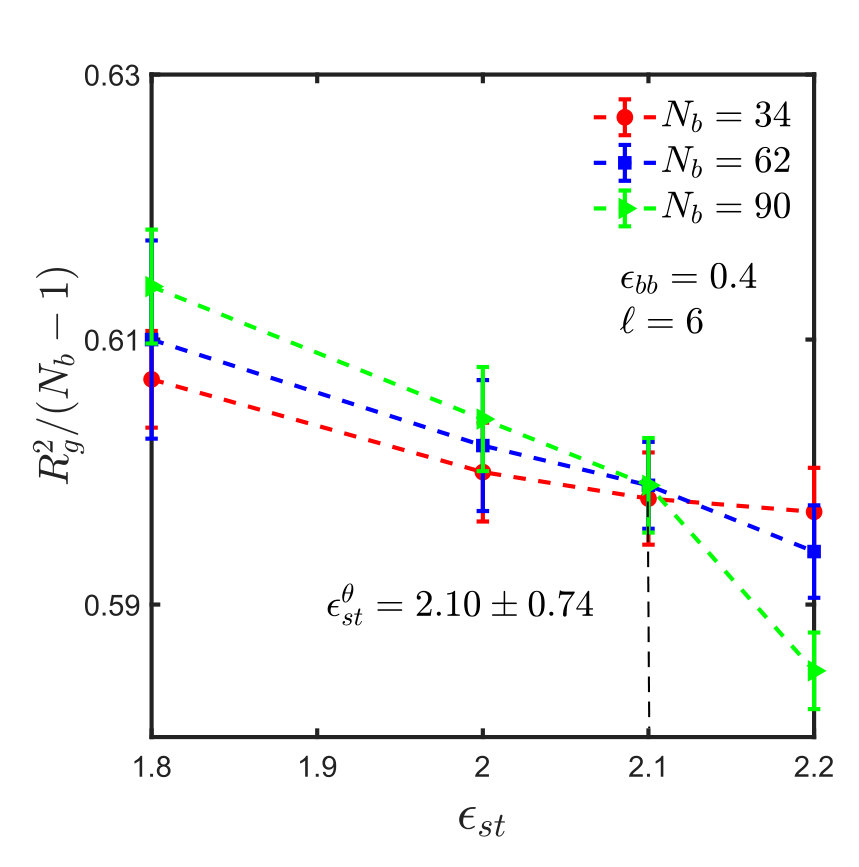 Figure 3
Figure 3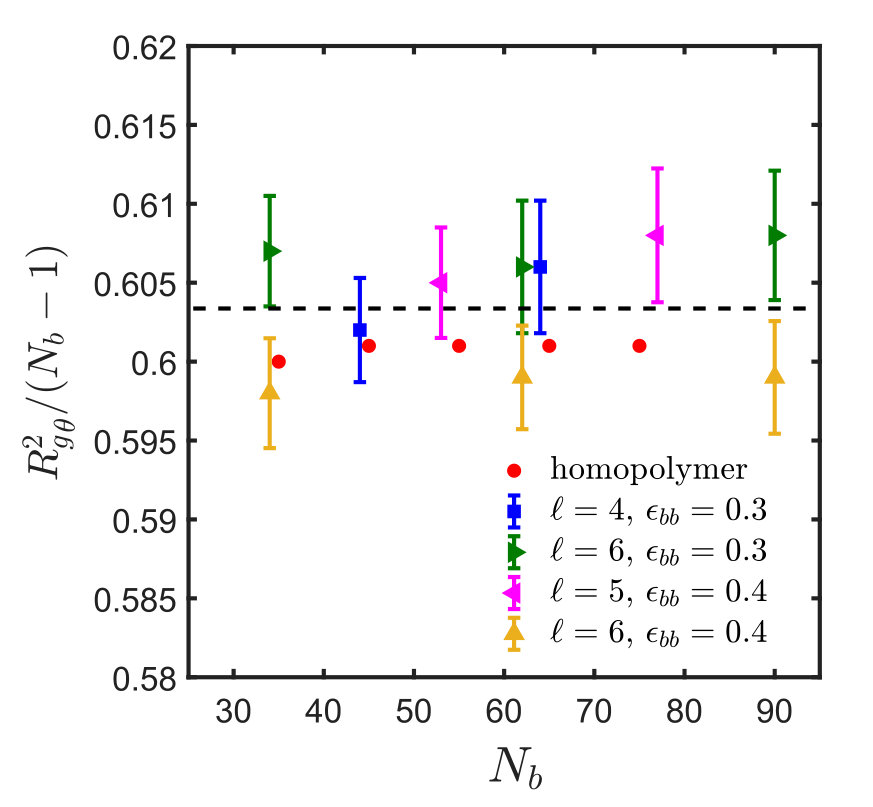 Figure 4
Figure 4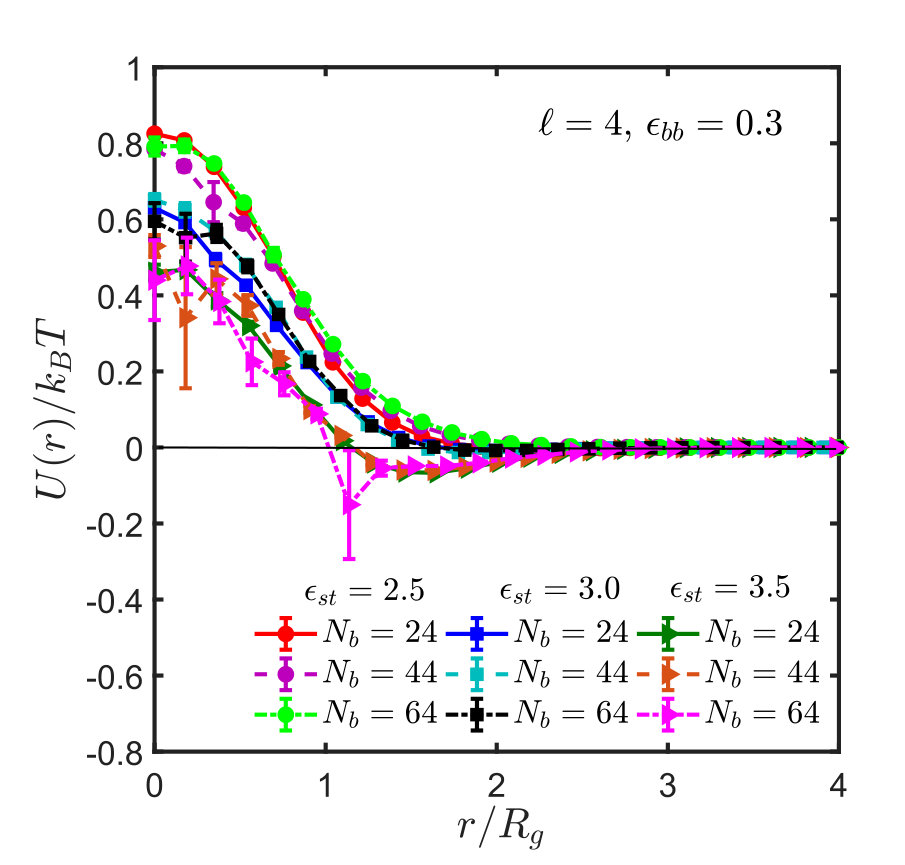 Figure 5
Figure 5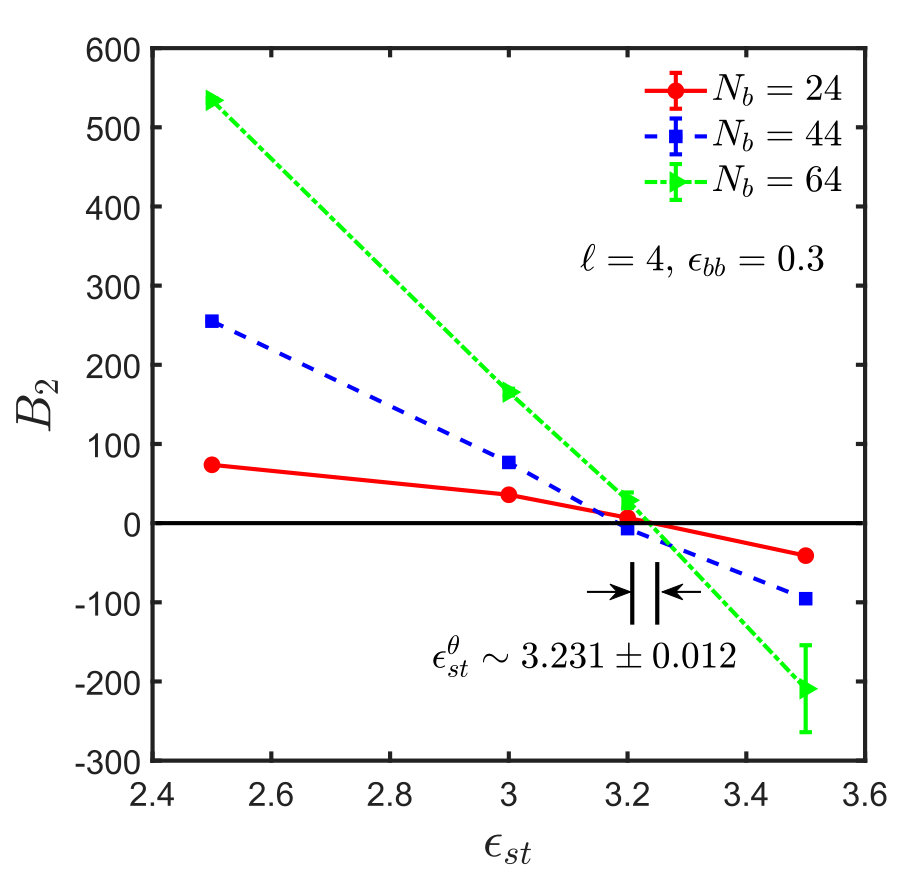 Figure 5
Figure 5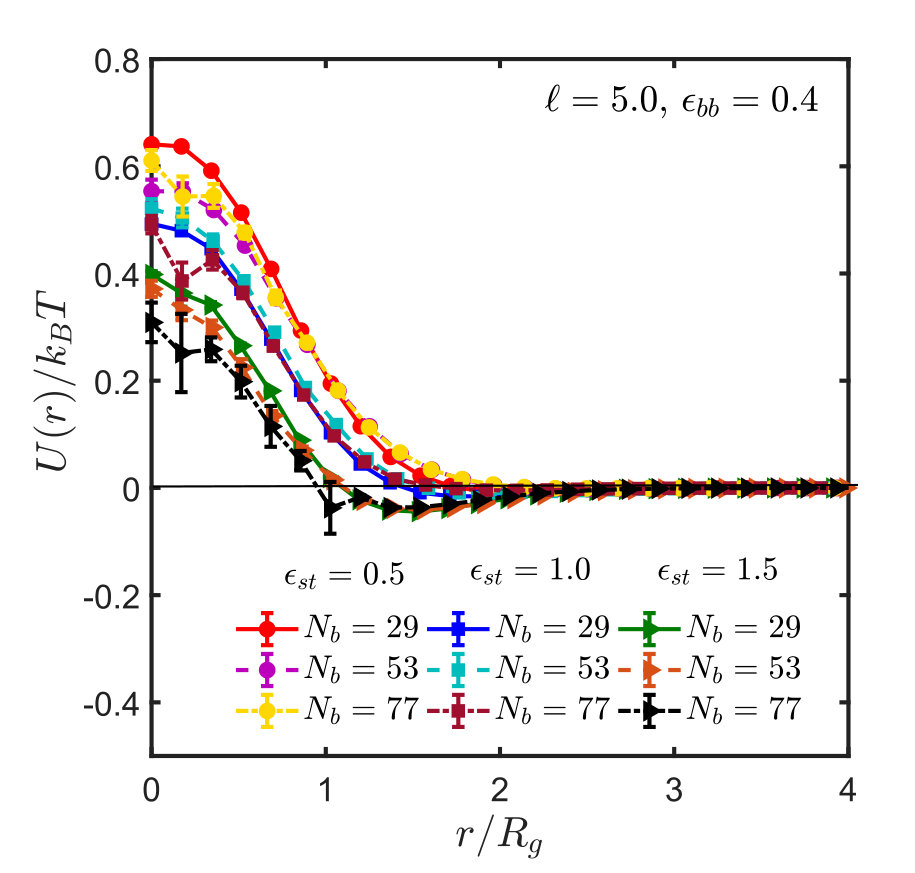 Figure 5
Figure 5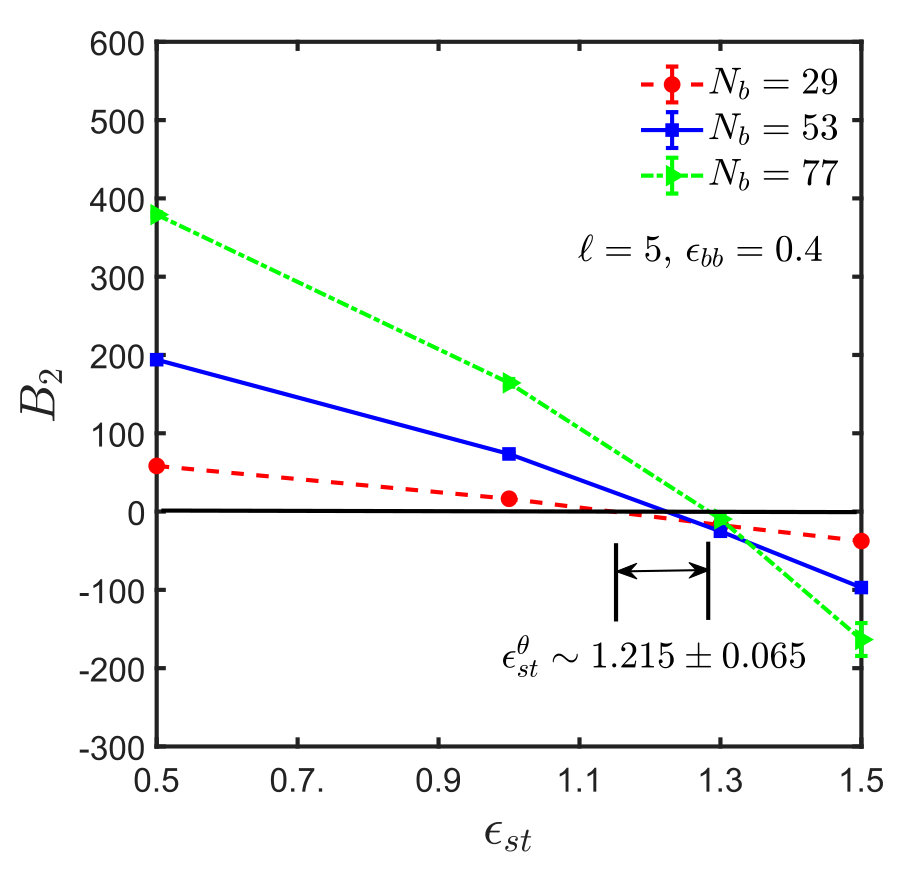 Figure 5
Figure 5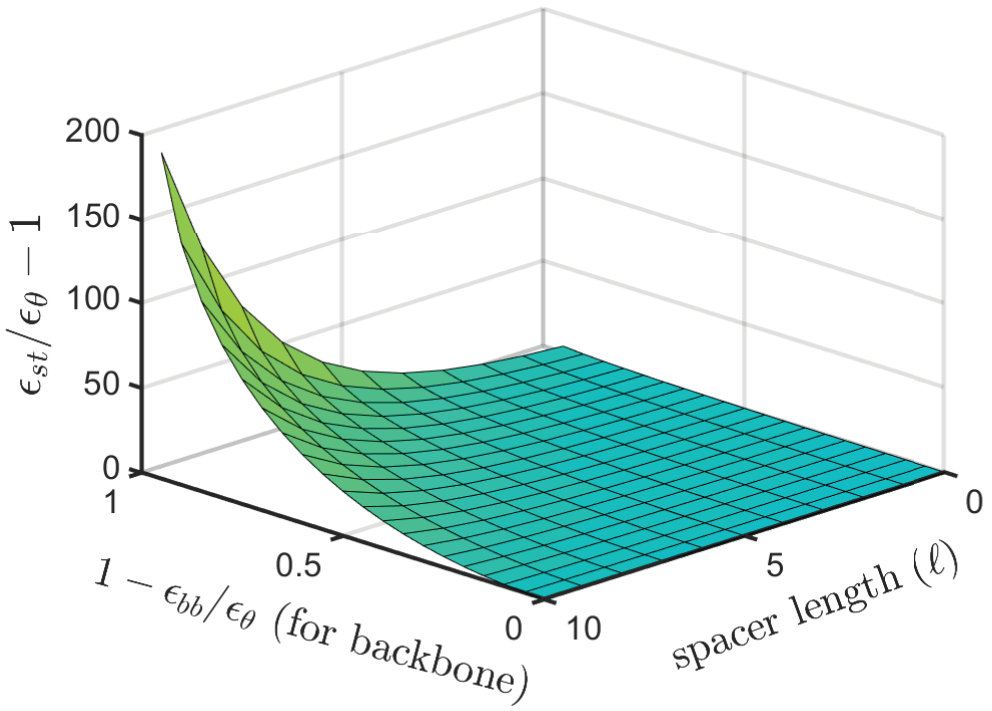 Figure 6
Figure 6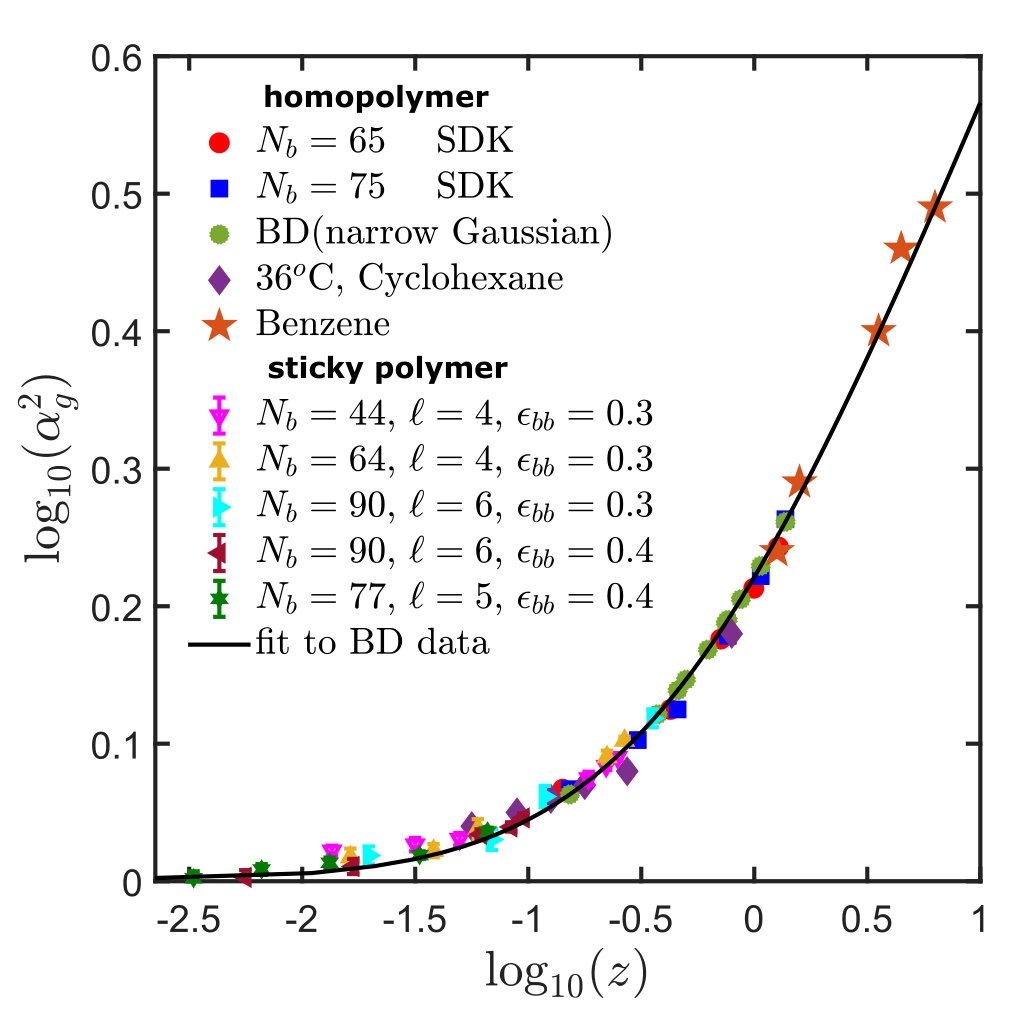 Figure 7
Figure 7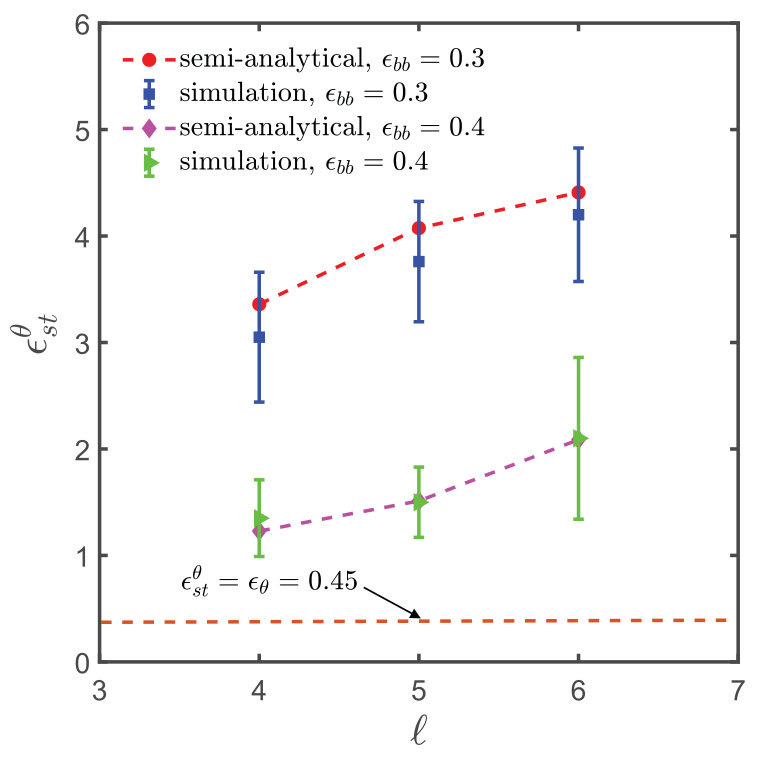 Figure 8
Figure 8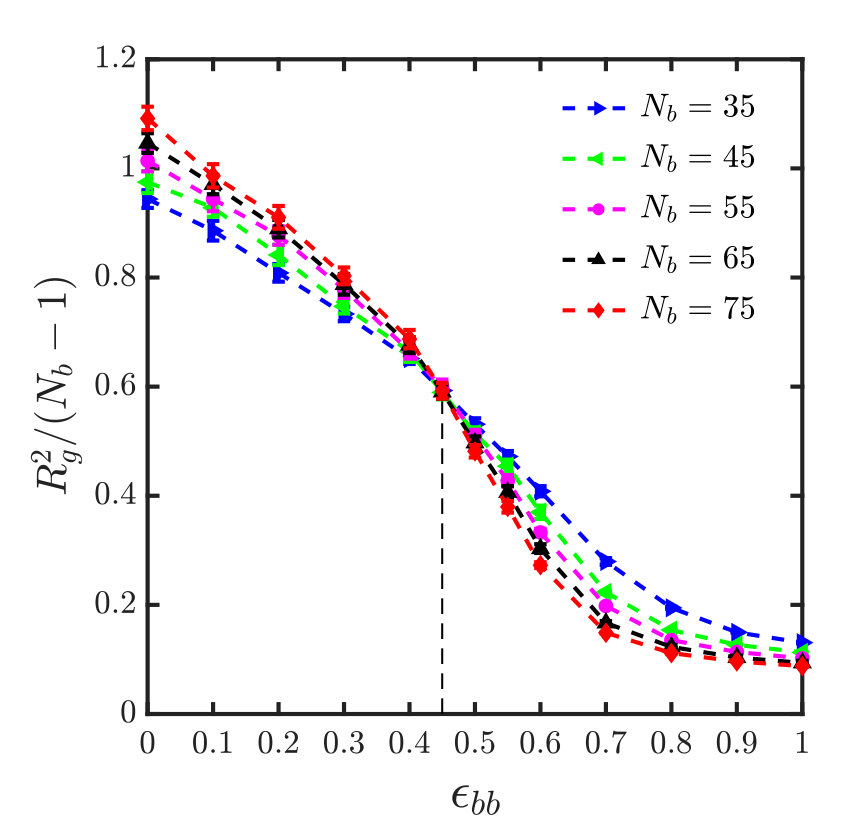 Figure 9
Figure 9| (from scaling) | (from ) | ||
|---|---|---|---|
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Universality of the collapse transition of sticky polymers
Aritra Santra
Department of Chemical Engineering, Monash University, Melbourne, VIC 3800, Australia
Kiran Kumari
IITB-Monash Research Academy, Indian Institute of Technology Bombay, Mumbai, Maharashtra - 400076, India
Department of Biosciences and Bioengineering, Indian Institute of Technology Bombay, Mumbai, Maharashtra - 400076, India
Department of Chemical Engineering, Monash University, Melbourne, VIC 3800, Australia
R. Padinhateeri
Department of Biosciences and Bioengineering, Indian Institute of Technology Bombay, Mumbai, Maharashtra - 400076, India
B. Dünweg
Max Plank Institute of Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
Department of Chemical Engineering, Monash University, Melbourne, VIC 3800, Australia
J. Ravi Prakash
Department of Chemical Engineering, Monash University, Melbourne, VIC 3800, Australia
Abstract
The universality of the swelling of the radius of gyration of a homopolymer relative to its value in the state, independent of polymer-solvent chemistry, in the crossover regime between and athermal solvent conditions, is well known. Here we study, by Brownian dynamics, a polymer model where a subset of monomers is labelled as “stickers”. The mutual interaction of the stickers is more attractive than those of the other (“backbone”) monomers, and has the additional important characteristic of “functionality” , i.e., the maximum number of stickers that can locally bind to a given sticker. A saturated bond formed in this manner remains bound until it breaks due to thermal fluctuations, a requirement which can be viewed as an additional Boolean degree of freedom that describes the bonding. This, in turn, makes the question of the order of the collapse transition a non-trivial one. Nevertheless, for the parameters that we have studied (in particular, ), we find a standard second-order collapse, using a renormalised solvent quality parameter that takes into account the increased average attraction due to the presence of stickers. We examine the swelling of the radius of gyration of such a sticky polymer relative to its value in the altered state, using a novel potential to model the various excluded volume interactions that occur between the monomers on the chain. We find that the swelling of such sticky polymers is identical to the universal swelling of homopolymers in the thermal crossover regime. Additionally, for our model, the Kuhn segment length under conditions is found to be the same for chains with and without stickers.
I Introduction
Solutions of sticky polymers consist of chains with sticky groups that can form reversible physical bonds, which in turn lead to the formation of reversible gels and networks. The ability to tune different microscopic parameters of sticky polymer chains, like the number of stickers per chain, the position of the stickers on the polymer backbone, the strength of associations of the stickers and the solution temperature (or backbone solvent quality) has led to the use of sticky polymer solutions in a number of different applications, such as rheology modifiers, adhesives, biomedical implants, adsorbents and many such moreGlass, Schulz, and Zukoski (1991); Kirchhof, Goepferich, and Brandl (2015). Due to the relative affinity of sticky groups for each other, sticky polymer chains are more collapsed or less swollen at a given temperature compared to the corresponding homopolymer made up of only the backbone or non-sticky monomers of the same molecular weight. For example, at the temperature for the homopolymer, simple linear polymer chains in a dilute solution follow random walk (RW) statistics, whereas, the introduction of sticky groups leads to a decrease in the size of the chain due to relatively poorer solvent quality. Indeed the whole phase diagram for homopolymers Grosberg and Khokhlov (1994); Rubinstein and Colby (2003) is expected to be modified due to the presence of stickers Semenov and Rubinstein (1998); Rubinstein and Semenov (1998); Dobrynin (2004). In many applications like mist control or drag reduction of aviation fuel, it is necessary to have long, swollen, physically associated polymer chains in a single-phase solutionDavid et al. (2009); David, Wei, and Kornfield (2009). For such applications, it is important to estimate the renormalised solvent quality of a sticky polymer solution in order to have some knowledge of chain conformations and the relative location of the system in the phase-space of temperature and concentration. In this paper we address the question of how to compute the solvent quality of dilute sticky polymer solutions and show that the swelling behaviour of sticky polymers in terms of a renormalised solvent quality follows the same universal behaviour as a standard second-order transition. As will be discussed subsequently, we consider the possibility that a first-order collapse might also exist in a certain parameter region for the particular model studied here, making the current observation a non-trivial one.
For homopolymers, it is well-known that in the limit of large molecular weight, static properties of polymer chains in dilute solution, such as the radius of gyration , follow universal power laws in both and athermal solvents. Furthermore, experiments and theoretical studies indicate that in the region between and athermal solvents universal behaviour in terms of crossover scaling is still observed. For such systems the mean size of the polymer is a function of both the temperature () and the molecular weight () which combine to form a single variable, the solvent quality, , where indicates the temperature corresponding to a -solvent and is a chemistry-dependent constant. A plot of the swelling ratio, , which is the ratio of in a good solvent to that in a -solvent, against the solvent quality , for , collapses data on a universal master curve for a wide variety of polymer-solvent systems with an appropriate choice of the constant Hayward and Graessley (1999); Miyaki and Fujita (1981); Vidakovic and Rondelez (1985); Bercea et al. (1999). Within the framework of Brownian dynamics (BD) simulations, this collapse has been demonstrated by Kumar and Prakash Kumar and Prakash (2003).
To the best of our knowledge, there are no studies exploring the universal swelling behaviour of dilute solutions of sticky polymers. In this paper we have used a novel potential, proposed by Soddemann et al.Soddemann, Dünweg, and Kremer (2001) (which we denote as the SDK potential) to investigate the effect of stickers on the solvent quality and the swelling behaviour of sticky polymers in dilute solutions, using the methodology of Kumar and Prakash Kumar and Prakash (2003). The sticky macromolecules are modelled as multi-sticker chains with equispaced stickers positioned along the backbone of each chain (except at the chain ends where there are no stickers) separated by spacer (or backbone) monomers. A sticker is assumed to associate with only one other sticker (i.e. with functionality ). Such systems can be easily designed in experimentsDavid et al. (2009); David, Wei, and Kornfield (2009); Guo et al. (2005). The key idea is to investigate various systems characterised by different parameters, and to check if the typical characteristics of a second-order collapse are observed. Since this indeed appears to be valid in our model, we are able to verify the universality of the swelling of sticky polymer solutions.
The advantage of using the SDK potential is that it can be used to represent both the backbone monomer-monomer interactions, and the sticker monomer-monomer interactions, with a simple choice of the attractive well-depth of the potential. In the former case this is denoted by , while in the latter it is denoted by . We find that the effective solvent quality of a sticky polymer solution can be represented in terms of these variables, along with the clear identification of the -temperature. As a consequence, the swelling of sticky polymer chains can be examined as a function of the various parameters that control their static properties.
The plan of the paper is as follows. Section II discusses the principal governing equations and the details of the various interactions. The description of the universal swelling of homopolymers, which forms the framework within which the universal behaviour of sticky polymer solutions is discussed, is taken up in Section III. The determination of the -temperature of sticky polymer solutions, in terms of the appropriate value of the well depth of the SDK potential, is considered in IV.1, while the universal swelling of sticky polymers is examined in IV.2. The main conclusions are summarised in Section V. In order to make the discussion of sticky polymer solutions the central focus of the paper, the estimation of the well depth of the SDK potential that determines the -point for homopolymer solutions is described in an Appendix, while the optimisation of the cut-off radius of the SDK potential and the considerations that lead to the choice of a particular value, are discussed in the Supplementary Information.
II Basic equations and simulation details
On the mesoscopic scale, polymers are modelled as a sequence of coarse-grained bead-spring chains with beads connected by entropic springsBird et al. (1987). In this study we have simulated a single chain, in an implicit-solvent framework, with the chain configuration specified by the set of position vectors . The evolution of bead positions in BD simulations is governed by the following Ito stochastic differential equation,
[TABLE]
Here the length and time scales are non-dimensionalised with and , respectively where is the absolute temperature, is the Boltzmann constant, is the spring constant, and is the Stokes friction coefficient of a spherical bead of radius where is the solvent viscosity. is a non-dimensional Wiener process with mean zero and variance . is a non-dimensional tensor whose evaluation requires the decomposition of the diffusion tensor , defined as , where is the Kronecker delta, is the unit tensor, and is the hydrodynamic interaction tensor. Defining the matrices and as block matrices consisting of blocks each having dimensions of , with the -th block of containing the components of the diffusion tensor , and the corresponding block of being equal to , the decomposition rule for obtaining can be expressed as . The bonded interactions between the beads are represented by a spring force, , and the non-bonded excluded volume (EV) interactions are denoted by . We use the regularized Rotne-Prager-Yamakawa (RPY) tensor to compute hydrodynamic interactions (HI),
[TABLE]
where
[TABLE]
with
[TABLE]
and
[TABLE]
Here, the hydrodynamic interaction parameter is the dimensionless bead radius in the bead-spring model, defined as . In all the simulations reported here in which hydrodynamic interactions have been implemented, a value of equal to 0.25 has been used. Unless explicitly stated, however, hydrodynamic interactions have been turned off by setting .
We use a finitely extensible nonlinear elastic (FENE) spring potential to represent the interaction between adjacent beads,
[TABLE]
where is the dimensionless maximum stretchable length of a single spring, and is used to non-dimensionalise energy. All the simulations reported in this work use a value of . Note that the notation used here is the same as the more commonly used FENE -parameter. The spring force on a bead resulting from is denoted by .
Note that a large value of , as used here, implies a very soft potential that admits the possibility of self-crossing of the chain. This would pose a severe problem if we were interested in the dynamics of dense systems. However, our present investigation aims at statics in the dilute limit, and future studies on dynamics will be restricted to the dilute limit too. It is well known that for these properties topological constraints do not play a role. Rather on the contrary, self-crossings are expected to speed up the exploration of phase space, and are hence advantageous for our purposes.
The excluded volume interactions between pairs of beads on the chain is modelled by a novel potential, , proposed by Soddemann-Dünweg-Kremer Soddemann, Dünweg, and Kremer (2001),
[TABLE]
The potential has a minimum at , and the value of the non-dimensional distance is taken to be 1 in the present study. The quantity is the attractive well depth of the potential. As can be seen from Eq. (8), the repulsive part of the SDK potential is modelled by a truncated Lennard-Jones (LJ) potential similar to the Weeks-Chandler-Anderson (WCA) potential while the attractive contribution is modelled with a cosine function. Unlike the LJ potential, which has a long attractive tail, the short ranged attractive tail of the SDK potential smoothly approaches zero at a finite distance , which leads to an increase in the simulation efficiencySoddemann, Dünweg, and Kremer (2001). It is worth noting that in the SDK potential corresponds to a purely repulsive WCA potential, and the solvent quality reduces with increasing values of . An advantage of the SDK potential over the LJ potential is that the complete range of solvent qualities, from poor to athermal, can be explored by varying the single parameter, , which can alter the attractive component of the SDK potential without affecting the repulsive force. The constants and are determined by applying the two boundary conditions, namely, at , and at . Based on these two boundary conditions, and are calculated by solving the following set of equations,
[TABLE]
In order to solve the above set of equations, it is required to choose a reasonable value of the cut-off radius, . In the original study by Soddemann et al.Soddemann, Dünweg, and Kremer (2001) the cut-off radius of the potential, , was chosen to be in order to include only the first neighboring shell of interactions, determined from the first minimum of the pair correlation function. For , the values of and are calculated to be and , respectivelySoddemann, Dünweg, and Kremer (2001), and the resultant SDK potential has been used to investigate various equilibrium properties of the solutions of polymer chains using molecular dynamics (MD) and Monte Carlo (MC) simulationsSoddemann, Dünweg, and Kremer (2001); Steinhauser (2005). In the context of the current Brownian dynamics simulations, however, we find that using leads to the prediction of unphysical asymptotic scaling behaviour in the poor solvent limit. We were able to “cure” the problem by using a value of . A detailed discussion of the problems encountered with the original cut-off radius, and the process by which the revised value was arrived at is given in the Supplementary Information. All subsequent results reported here with the SDK potential are for .
At this point, it is appropriate to elaborate on what is meant by the concept of the functionality , and the choice of its value of one in the model. For each pair of monomers and , we introduce a Boolean variable . is zero whenever at least one of the two monomers is a backbone monomer, while for a pair of sticker monomers, is zero if no bond exists between the two stickers, and for a bonded pair of sticker monomers. The attractive strength, of the SDK potential for a pair of monomers and is then given by
[TABLE]
where, as mentioned earlier, backbone monomer-monomer interactions are denoted by , and the sticker monomer-monomer interactions are denoted by . Typically, . The variables can be considered to be additional degrees of freedom whose dynamics are coupled to the dynamics of the monomer coordinates according to the following simple update rules:
Whenever two stickers come within the cutoff radius of the SDK potential, , the value of is changed from zero to one, provided that both stickers are not bonded to other stickers. 2. 2.
As long as these two stickers are within the interaction range , is maintained at the value one, regardless of how closely they might be approached by other stickers. 3. 3.
As soon as the distance between two stickers becomes greater than , is reset to zero, and new bondings may occur.
It is clear that the monomer coordinates, together with the values, provide sufficient information to calculate the interaction energy of the system uniquely. Furthermore, it is also clear that the update rules give rise to a well defined configuration space of the system in the sense of Statistical Mechanics, such that the partition function exists. As long as there are only pairs of stickers and within interaction range, it is clear that the corresponding has to be one, while in a situation, where, for example, three stickers are all within interaction range of each other, there are three possibilities to form the bond, corresponding to the three sides of the triangle formed by the three stickers. Since all three cases are dynamically accessible, each of them must appear in the partition function. In this context, it should be emphasised that our simulation setup aims at modelling reversible sticker bonds.
Note also that other update rules, and/or other values of , may well be conceivable, which would then give rise to a different configuration space, and a correspondingly altered Statistical Mechanics of the system.
The observation that we are dealing with additional degrees of freedom makes the existence of a second-order transition a non-trivial and subtle question. There are various examples in Statistical Mechanics where the coupling to an additional degree of freedom turns a second-order phase transition into a first-order transition. Some of these examples, which are relevant to the present work, are discussed in Section V.
The simulations are carried out for different chain lengths, , ranging from 25 to 90 beads per chain, with an equilibration run of about Rouse relaxation times (estimated analytically as given in Bird et al. (1987)) and a production run of to Rouse relaxation times with a non-dimensional time step size . Data from each independent trajectory in the simulations are collected at an interval of to non-dimensional time steps over the entire production run and time averages are calculated over each of the trajectories. Average equilibrium properties and error of mean estimates are evaluated over an ensemble of such independent time averages consisting of to independent trajectories. In the case of sticky polymers, an additional pre-equilibration run of 2 to 3 Rouse times is carried out with a chain without stickers.
III Universal swelling of homopolymers
In order to discuss the universal swelling of homopolymers, it is necessary to first determine the radius of gyration of a homopolymer under -conditions. This in turn requires the determination of the well depth of the SDK potential that corresponds to the -temperature for homopolymers. This question is taken up in the Appendix, where the value of is estimated by two means, first by determining the value of at which the chain obeys random walk statistics, and second by determining the value of that leads to the second osmotic virial coefficient being zero. As shown in detail in the Appendix, for a SDK potential with cut-off radius , we find that the -point occurs at .
The swelling of homopolymers interacting with an SDK potential as the source of the excluded volume force is investigated in this section. The results are compared with the swelling of experimental polymer-solvent systems and earlier predictions of BD simulations, where the excluded volume interactions are modelled by a narrow Gaussian potential given byÖttinger (1996); Prakash and Öttinger (1999)
[TABLE]
Here, , is a non-dimensional parameter that measures the range of interactions, and is the non-dimensional strength of excluded volume interactions. In the context of the narrow Gaussian potential, the solvent quality is defined by , which takes into account the dependence on both temperature and chain length. Kumar and Prakash performed BD simulations with the narrow Gaussian potential to obtain the universal swelling ratio as a function of solvent quality Kumar and Prakash (2003). Basically, they obtained at a particular value of by carrying out simulations for different chain lengths , where the parameter was evaluated using the expression , for each choice of . The data accumulated for various values of was then extrapolated to the limit of to obtain the asymptotic value, at the chosen value of . The results are plotted in Fig. 1 and fitted with an expression suggested earlier by renormalisation group calculations Schäfer (1999); Freed (1987); des Cloizeaux and Jannink (1990)
[TABLE]
with fit parameters , , and Yamakawa (1971); Schäfer (1999); Kumar and Prakash (2003). The fitted curve is the universal thermal crossover swelling curve predicted by BD, and acts as a reference for collapsing swelling data for a range of polymer-solvent systems as discussed below.
Kumar and Prakash Kumar and Prakash (2003) showed that experimental data acquired previously Miyaki and Fujita (1981) for versus , in a variety of different polymer-solvent systems, could also be described by the same universal curve. This is done by defining the experimental solvent quality by , where and adjusting , which is a chemistry dependent constant, in order to achieve data collapse Kumar and Prakash (2003). Swelling data Miyaki and Fujita (1981) for polystyrene in cyclohexane at C, and in benzene at C and C, obtained in this manner are shown in Fig. 1. We have adopted a similar approach to check whether polymer chains with the SDK potential follow the same universal swelling behaviour. The solvent quality is defined here in terms of the potential well depth as
[TABLE]
where is a constant dependent on the interaction potential and is equivalent to the temperature dependent term, , defined earlier. Note that the factor is defined in such a way that in the limit of a -solvent its value is zero, while in the good solvent limit (), . The value of is obtained by the following procedure. Simulations are carried out for different values of chain length and well depths , and the mean-squared radius of gyration is calculated in each case. The swelling, , relative to the size of the chain under -solvent conditions, (obtained from a simulation with ), is calculated in each case, and the corresponding values of are determined from the universal swelling curve given by Eq. (13). Finally, values of obtained in this manner are plotted as a function of , and the resultant curve is fitted with a straight line as shown in Fig. 2. From the slope one finds .
The swelling of polymer chains with and , obtained with the SDK potential for a set of values of obtained in this manner, is compared in Fig. 1 with earlier results from BD simulations and experimental measurements of the swelling of polystyrene in cyclohexane and benzene. It is clear that the SDK potential reproduces the universal swelling behaviour in the thermal crossover regime obtained previously with the narrow Gaussian potential. It is worth noting here, however, that in the case of the SDK potential, we have not extrapolated finite chain data to the long chain limit, as was done for the narrow Gaussian potential. We found that this was unnecessary since the results for and were already lying on the universal curve.
IV -point and swelling of sticky polymers
As mentioned earlier, the inclusion of sticky groups decreases the effective solvent quality. The purpose of this section is to present results which establish, within the studied parameter range, that the collapse of the sticky polymer chain is a standard second order transition. We show this by demonstrating that all the methods that have been applied to homopolymers, both for the localisation of the transition (as discussed in the Appendix), and for the study of the universal scaling for the swelling (as discussed in the previous section) can be carried over.
IV.1 -point for sticky polymer solutions
IV.1.1 Scaling of the radius of gyration
For the sticky chain, we keep and fixed, and study as a function of , which we use as the control parameter that drives the transition. Following the procedure described in Appendix A for homopolymers, intersection plots for versus are presented in Fig. 3, which allows us to find the -point for various choices of and , . The error in is estimated by linear interpolation between the data points adjacent to the intersection, combined with standard error propagation. We will show shortly that for our model, can be determined by an alternative simpler and perhaps more accurate procedure.
It is interesting to note that the ratio at the -point, denoted by , assumes a constant value, as shown in Fig. 4, for monomers interacting via the SDK potential, irrespective of the spacer length and backbone solvent quality. It is also evident that the value of the ratio is the same for both homopolymers and sticky polymers, suggesting that the Kuhn step length is the same in both cases, and is independent of the presence of sticky groups. This implies that one does not need to do simulations for each and every system in order to estimate the -point and calculate the corresponding . This, of course, simplifies matters substantially. It is worth emphasising, however, that this is most probably a special feature of our model, and probably will no longer be true for chemically more realistic models.
IV.1.2 Calculation of the second virial coefficient
For sticky polymers, the estimation of the -point from the second virial coefficient was carried out as was done for homopolymers (see Appendix B), with a set of about configurations of chain pairs. Since the Boolean variables (see Section II) were not stored in the course of the simulations, it was necessary in situations where there were three stickers within interaction distance of each other, to define by some rule. For simplicity, we picked the two stickers with the lowest monomer indices to be bonded. For the rare case of four or more stickers, we proceeded in an analogous fashion.
Figures 5 shows the effective interaction potentials and second virial coefficients for systems with spacer lengths , and . One sees that even though the method is hampered by similar problems as in the homopolymer case, it is nevertheless possible to locate the -point with reasonable accuracy, which is actually significantly better than that obtained from the scaling of . The results are summarised in Table 1. With increase in spacer length () and sticker strength, the sampling gets poorer and less efficient, as explained in Appendix B, such that it was not possible to determine the -point by the virial coefficient method satisfactorily.
IV.1.3 The -surface for sticky polymers
Since the location of the -point, , depends on both and , the full phase diagram of the system is three-dimensional, with a two-dimensional surface of -points separating the good and poor solvent regions. Figure 6 is a schematic representation of such a surface, where we confine attention to the physically interesting case . This implies that under conditions for the chain as a whole, the backbone is in a relatively good solvent (), meaning that the conformations are the result of a competition between backbone-backbone repulsion and sticker-sticker attraction.
IV.2 Universal swelling of sticky polymers
In order to study the universal swelling for the sticky chain, a suitable definition of the solvent quality parameter is required. Based upon the observation that we have chosen as the independent control parameter that drives the transition, it is logical to generalise Eq. (14) as
[TABLE]
where is a material dependent function of spacer length () and backbone monomer interaction strength (). In the limit of , the effective solvent quality becomes the same as that of the corresponding homopolymer, and the sticky chain becomes indistinguishable from it (see Eq.(11)). By definition, for the overall solvent quality .
For obtaining the swelling curve for sticky polymers, one can exploit the simplification that comes from the constancy of the ratio , independent of all other parameters, which renders the need for extra simulations to calculate superfluous. Simulations are carried out for different values of , , , and , and the swelling ratio, , is then calculated for each set of these parameters. For values of and for which is known, can be estimated using the same technique as was used to find in the case of homopolymers, discussed in Section III. The results are tabulated in Table 2. Plotting versus , as displayed in Fig. 7, shows that the swelling of sticky polymers relative to its state follows the same universal curve already presented in Fig. 1 for homopolymer systems.
The existence of the universality of the swelling of sticky polymers, which has been established above, can be used to determine the -point , along with , for any pair of values of and , without the complicated analysis of subsection IV.1. One only needs data for two different values, while , , and are being kept fixed. The swelling curve allows us to transform the values to corresponding solvent qualities . Therefore, inserting all known parameters into Eq. (15) gives rise to two equations with two unknowns which, after solution, provide the desired values and . A sample calculation to demonstrate the above method is as follows. The parameters chosen are , , and the two sticker strengths are and . Considering , the values of obtained from the simulations for the given set of parameters are equal to and for and , respectively. The corresponding values of are estimated from Eq. (13) to be and for and , respectively. Substituting the values of in Eq. (15) and simultaneously solving the two linear equations for the unknowns, gives and , which is, within error bars, consistent with the previously estimated values of and (see Fig. 3 (a) and Table 2).
A comparison of the values of obtained from the semi-analytical estimation procedure discussed above with that from full blown simulations as carried out in the context of Fig. 3, for different spacer monomers and backbone solvent qualities , is shown in Fig. 8. The plot shows a satisfactory agreement between the two methods and implies that the calculation of using Eq. (15) provides a reasonable estimate of the -point for the sticky polymer systems. Fig. 8 also suggests, as expected intuitively, that the sticker strength at the -point, , increases monotonically with spacer length, , for a given value of . With an increase in spacer monomers, the sticker density along the polymer backbone decreases, and it requires a much higher attractive strength for the stickers to make the chain follow RW statistics. In all our simulations, the value of the sticker strength is taken to be greater than , while . The corresponding increase in solvent quality for the backbone is thus compensated by an increased attraction between the stickers. At , the backbone is in a -solvent condition, and under such circumstances the sticker strength at the -point, , is equal to , which is the limiting value of , indicated by the constant straight line in Fig. 8 and discussed further in the context of Eq. (15).
V Discussion and conclusions
Using the Soddemann-Dünweg-Kremer potential to model excluded volume interactions, and defining a renormalised solvent quality for sticky polymer solutions, the swelling of the radius of gyration has been shown to be identical to the universal swelling of homopolymers in the thermal crossover regime. Additionally, the Kuhn segment length under conditions, for our model, is found to be the same for chains with and without stickers. This allows, in combination with the known universal swelling curve, a fairly easy determination of the two-dimensional -surface embedded in the three-dimensional phase diagram.
The collapse transition observed here is a standard second-order transition and as a consequence, all scaling laws and universal properties are faithfully reproduced. In the parameter range that we have studied, a strong first-order transition can be ruled out. In hindsight this is perhaps not too surprising, since the only theoretical possibility for the existence of first-order behaviour arises from a strong coupling of the conformational degrees of freedom to the Boolean degrees of freedom that describe functionality. As seen from Eq. (11), the strength of the coupling is directly proportional to . In the parameter range where we did the simulations, this difference was never very large. For this reason, the possibility of a first-order transition in the opposite limit cannot be ruled out within the framework of the current investigation.
One might then consider a situation where the backbone is under very good solvent conditions, , while is so large that nevertheless a collapse would occur. Note that in such a situation the transition would be entropy-driven rather than energy-driven. This is so because the condition for the bonds would lead to a complete saturation, with no residual attraction being left. The reason for a collapsed conformation would then be merely entropic because such a state allows for many more possibilities to form bonds than a swollen chain, which would only allow association of stickers that are near each other on the chain. Note, however, that such a situation would essentially be impossible to simulate with standard Brownian dynamics, simply because the breaking of a once-formed bond would be extremely rare. It might be possible to investigate such a situation with advanced Monte Carlo algorithms like parallel tempering Earl and Deem (2005), but this is beyond the scope of the present investigation.
There are a number of previous studies, in the context of models for both synthetic and biological polymer solutions, where a first-order rather than a second-order transition has been observed. For instance, in the model proposed by Jeppesen and Kremer Jeppesen and Kremer (1996) for the phase-behaviour of polyethylenoxide in water, each monomer has a Boolean degree of freedom that enters the interaction energy. Depending on the strength of the coupling parameter, they find a second or first-order transition, with the first order transition being entropy driven for reasons similar to those discussed above. More recently, Scolari and Lagomarsino (2015) have developed a model for the folding of chromosomes due to self-attraction, and the formation of loops due to bridging proteins. Their model is similar to that used here for sticky polymers, with the bridging interactions (which are distributed uniformly along the backbone of the polymer) playing the role of stickers. However, their model differs from the one used here in certain key aspects. Firstly, the interaction energy in their system can be completely determined from the position coordinates of the beads, and secondly their functionality is not restricted to one. As a result, there are no additional Boolean degrees of freedom, and complex micellar structures are formed with multiple stickers forming clusters. Within the framework of such a model, for certain parameter values, they observe a first order collapse transition which is driven by competition between the energy gained from forming a core of bridging monomers versus the entropy lost by looping backbone monomers. Another relevant recent work where a first-order transition has been observed is by Michieletto, Orlandini, and Marenduzzo (2016), who have examined the 3D dynamics of chromatin folding coupled to 1D dynamics of epigenetic spreading, with a semiflexible bead-spring chain as a model for chromatin fiber. In this model, each bead can have two possible colours, with the colour denoting the epigenetic state and like colours attracting each other. The addition of the colour variable to position coordinates, leads to additional Boolean degrees of freedom, with the interaction energy not being calculable by chain conformation alone. Beads are recoloured periodically with a standard Metropolis acceptance criteria based on the energy difference between beads that are spatially proximate. Unlike in the present model, where the number of stickers is fixed, the number of strongly attracting like-coloured monomers is not constant but calculated dynamically based on proximity and energy of neighbouring monomers. It is observed that a critical value of attraction between like colours exists that separates the chain conformations into a swollen state, with the colours distributed homogeneously along the chain, and a collapsed globular state, with one colour dominant. It is argued that the first-order transition arises because of the coupling between 3D folding dynamics of the polymer and the 1D epigenetic spreading. Interestingly, in contrast to the present model, a second-order transition is never observed for the parameter values that have been examined.
Though the universal swelling of the radius of gyration in dilute sticky polymer solutions in the thermal crossover regime has been demonstrated here with the help of the Soddemann-Dünweg-Kremer potential, which has many desirable properties, we expect this behaviour to be independent of the specific choice of the excluded volume potential. We hope that this intriguing behaviour predicted by simulations will be tested and validated with careful experiments in the future.
Conflicts of interest
There are no conflicts to declare.
Appendix: The -point for homopolymers
A. The radius of gyration
For a linear polymer chain, the mean-squared radius of gyration follows the universal scaling law , where the value of the Flory exponent, , depends on the solvent quality. At the -temperature, linear polymer chains follow RW statistics, with , leading to the ratio to be independent of the chain length, . Whereas, in the case of good and poor solvents, takes the values and , respectivelyRubinstein and Colby (2003). For polymer chains with the SDK potential representing the excluded volume force, the temperature dependence can be captured with the potential well-depth, . As mentioned earlier, (which is equivalent to a WCA potential), represents the athermal limit, where the chain is fully swollen. With increasing values of , a unique value is reached, where the repulsive and attractive interactions between pairs of beads are precisely balanced, leading to -like conditions. The value of at the -point can be estimated by plotting the ratio versus for different chain lengths, , and finding the point of intersection at which curves for different values of intersect, as shown in Fig. 9 Steinhauser (2005); Huissmann, Blaak, and Likos (2009). Following this procedure, the -point for a homopolymer chain with beads connected by FENE springs having a maximum stretchable length of , and as the cut-off radius of the SDK potential, is found to occur at . As discussed in the Supplementary Information, a value of for the cut-off radius leads to the -point occurring for a well depth , which leads to unphysical asymptotic scaling in the poor solvent limit.
B. The second virial coefficient
In addition to the determination of the -point from the scaling of radius of gyration, there is an alternative method based on the second osmotic virial coefficient (), which involves the determination of the potential of mean force, , between a pair of polymer chains with their centres of mass separated by a distance Dautenhahn and Hall (1994); Withers et al. (2003); Narros, Moreno, and Likos (2013). Following the procedure discussed by Dautenhahn et al.Dautenhahn and Hall (1994), is calculated as follows. Two independent chain configurations are chosen from two sets of equilibrated single chain conformations (each having 2000 conformations) and are randomly oriented with respect to each other, with their centres of mass separated by a distance . For a set of values of ranging from 0 to 5 times the radius of gyration, all such possible two chain configurations (about configuration pairs) are considered. For each configuration pair , the interaction energy, , between the two chains is evaluated by computing the pair-wise potential for all pairs of beads, one taken from each chain such that
[TABLE]
Here is computed using the SDK potential to account for the pair-wise interaction of the beads and the indices and corresponds to chain 1 and 2, respectively. Finally the effective potential is evaluated from from the expression
[TABLE]
where the term on the right hand side of Eq. (17) is an ensemble average over all configuration pairs. The second virial coefficient is then easily calculated by evaluating the integral Rubinstein and Colby (2003)
[TABLE]
Positive values of indicate that the polymer solution lies in the good solvent regime, while negative values indicate that it is under poor solvent conditions. At the -point, .
Figure 10 (a) shows the effective potential, , as a function of the distance, , between the centres of mass. For short chains, the potential of mean force depends on chain length, but for sufficiently long chains it saturates within error bars Krakoviack, Hansen, and Louis (2003); Withers et al. (2003); Narros, Moreno, and Likos (2013). These are typically fairly small but non-negligible for deep well depths, small distances, and long chains. At small distances sampling is difficult, since for most random pairs the Boltzmann factor is very small due to chain overlap and the strong repulsion of the SDK potential. Therefore, the average is strongly dominated by those few configurations where this is not the case, and this gives rise to an effectively very small sample size Krakoviack, Hansen, and Louis (2003); Dautenhahn and Hall (1994); Narros, Moreno, and Likos (2013). This problem is aggravated for increasing chain lengths and well depths.
Figure 10 (b) is a plot of as a function of the potential well depth, . vanishes at , independently of chain length. This estimate of the -point is in good agreement, within error bars, with the value calculated previously from the scaling of (). Since both methods lead to approximately the same estimate, the value has been used for all further calculations.
Acknowledgements
This research was supported under the Australian Research Council’s Discovery Projects funding scheme (project number DP190101825).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Glass, Schulz, and Zukoski (1991) J. E. Glass, D. N. Schulz, and C. F. Zukoski, ACS Symposium Series 462 , 2 (1991).
- 2Kirchhof, Goepferich, and Brandl (2015) S. Kirchhof, A. M. Goepferich, and F. P. Brandl, Euro. J. Pharmaceutics and Biopharmaceutics 95 , 227 (2015).
- 3Grosberg and Khokhlov (1994) A. Y. Grosberg and A. R. Khokhlov, Statistical physics of macromolecules (AIP Press, New York, 1994).
- 4Rubinstein and Colby (2003) M. Rubinstein and R. H. Colby, Polymer Physics (Oxford University Press, 2003).
- 5Semenov and Rubinstein (1998) A. N. Semenov and M. Rubinstein, Macromolecules 31 , 1373 (1998).
- 6Rubinstein and Semenov (1998) M. Rubinstein and A. N. Semenov, Macromolecules 31 , 1386 (1998).
- 7Dobrynin (2004) A. V. Dobrynin, Macromolecules 37 , 3881 (2004).
- 8David et al. (2009) R. L. A. David, M. Wei, D. Liu, B. F. Bathel, J. P. Plog, A. Ratner, and J. A. Kornfield, Macromolecules 42 , 1380 (2009).