Phenotypic Profiling of High Throughput Imaging Screens with Generic Deep Convolutional Features
Philip T. Jackson, Yinhai Wang, Sinead Knight, Hongming Chen, Thierry, Dorval, Martin Brown, Claus Bendtsen, Boguslaw Obara

TL;DR
This paper demonstrates how pre-trained deep convolutional features can be used to analyze high throughput cellular images, enabling phenotypic clustering that aids drug discovery and compound triage.
Contribution
It introduces a method to reduce image dimensionality using pre-trained CNN features and applies unsupervised clustering to identify phenotypic groups in HTI screens.
Findings
Embedding space groups similar cellular phenotypes effectively.
Clustering reveals distinct phenotypic clusters for further analysis.
Workflow improves robustness of HTI screening and compound triage.
Abstract
While deep learning has seen many recent applications to drug discovery, most have focused on predicting activity or toxicity directly from chemical structure. Phenotypic changes exhibited in cellular images are also indications of the mechanism of action (MoA) of chemical compounds. In this paper, we show how pre-trained convolutional image features can be used to assist scientists in discovering interesting chemical clusters for further investigation. Our method reduces the dimensionality of raw fluorescent stained images from a high throughput imaging (HTI) screen, producing an embedding space that groups together images with similar cellular phenotypes. Running standard unsupervised clustering on this embedding space yields a set of distinct phenotypic clusters. This allows scientists to further select and focus on interesting clusters for downstream analyses. We validate the…
Click any figure to enlarge with its caption.
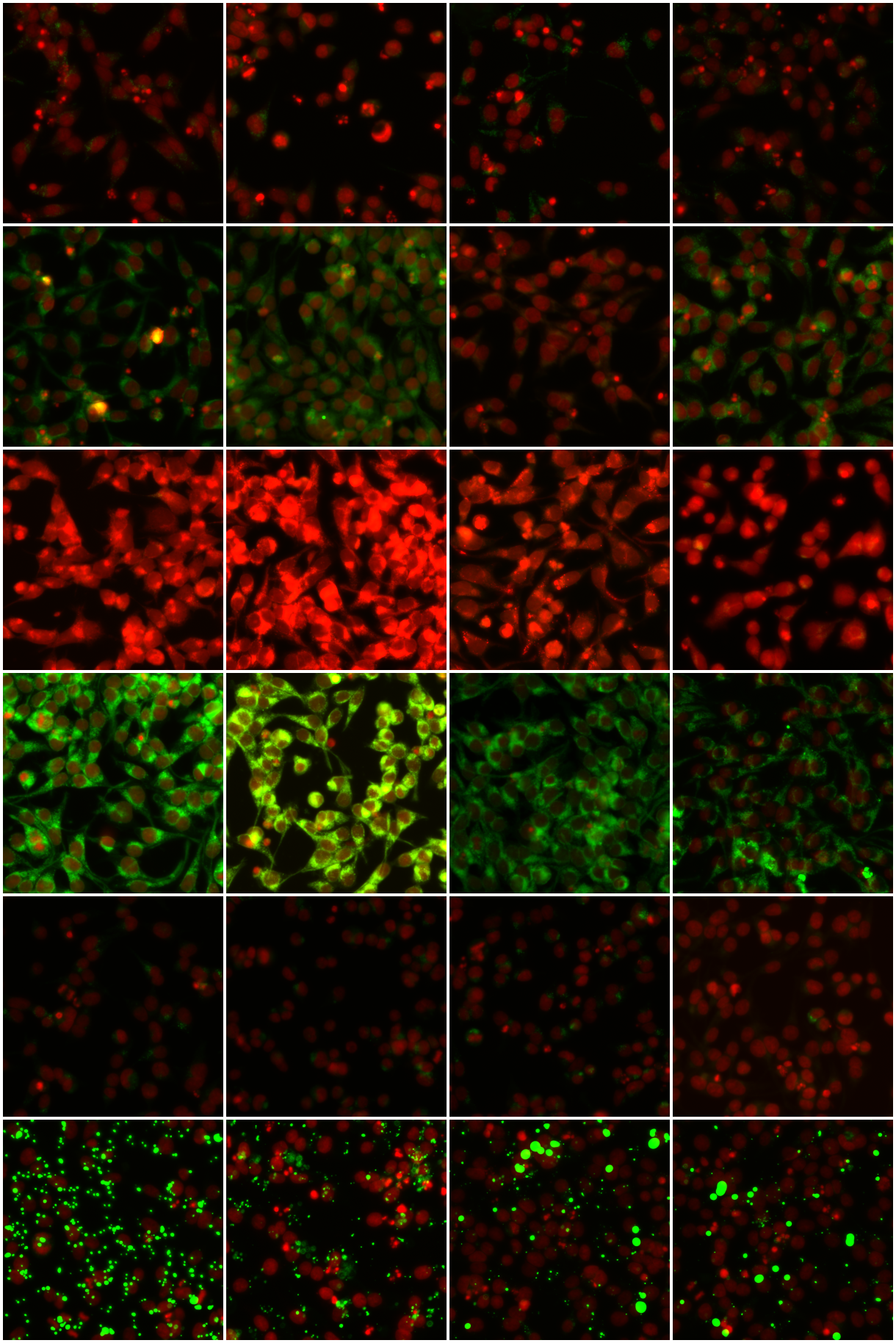 Figure 1
Figure 1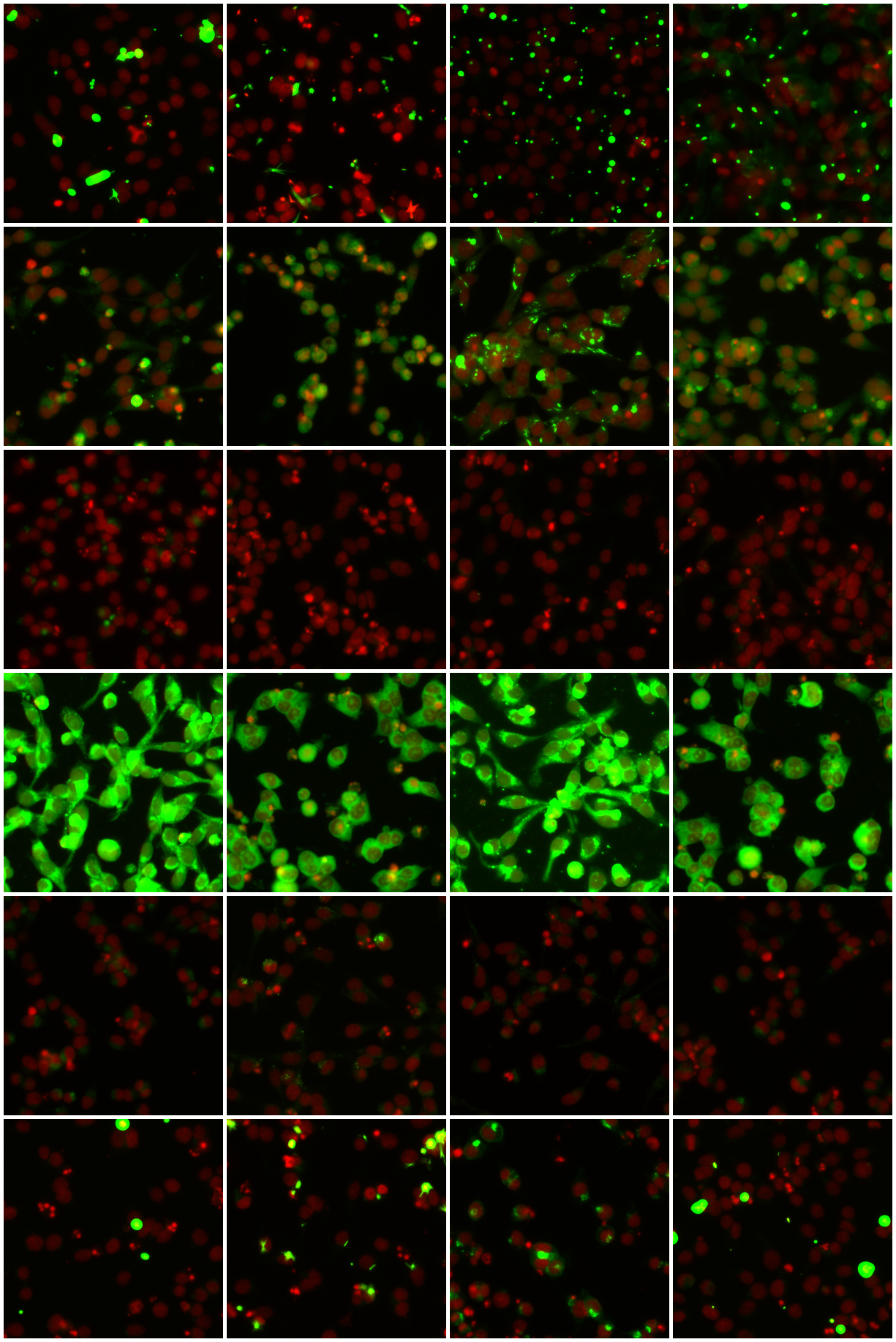 Figure 2
Figure 2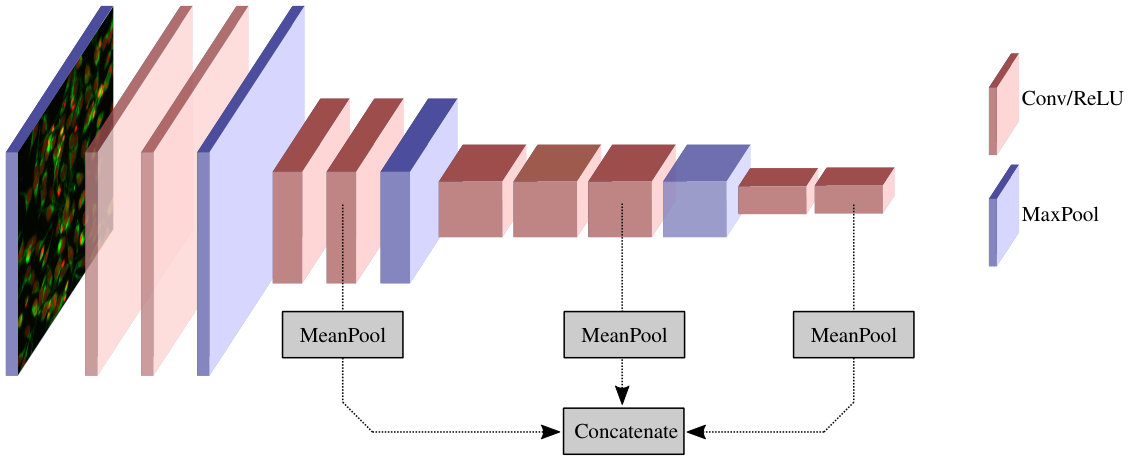 Figure 3
Figure 3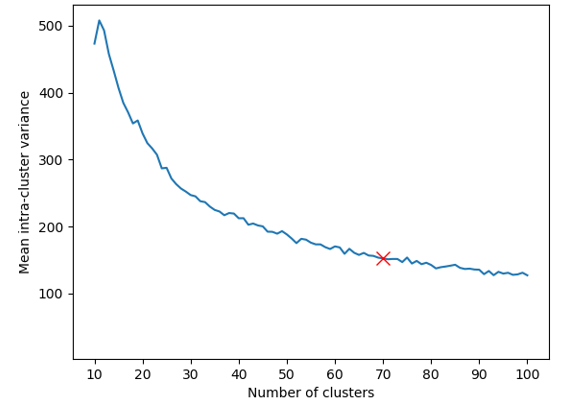 Figure 4
Figure 4 Figure 5
Figure 5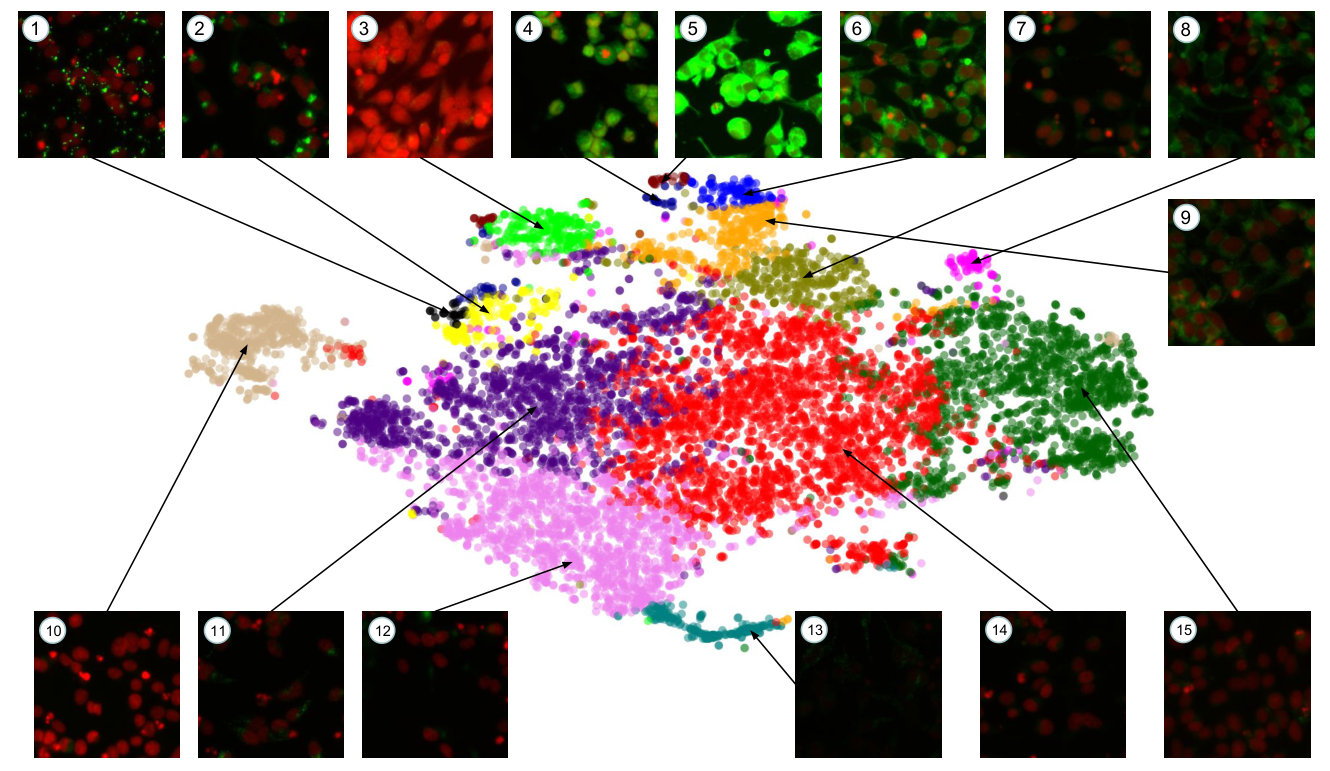 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCell Image Analysis Techniques · Computational Drug Discovery Methods · Image Processing Techniques and Applications
\finalcopy
Phenotypic Profiling of High Throughput Imaging Screens with Generic Deep Convolutional Features
Philip T. Jackson1
Yinhai Wang2
Sinead Knight2
Hongming Chen2
Thierry Dorval2
Martin Brown2
Claus Bendtsen2
Boguslaw Obara1
1Department of Computer Science
Durham University
2IMED Biotech Unit
AstraZeneca
[email protected], [email protected]
Abstract
*While deep learning has seen many recent applications to drug discovery, most have focused on predicting activity or toxicity directly from chemical structure. Phenotypic changes exhibited in cellular images are also indications of the mechanism of action (MoA) of chemical compounds. In this paper, we show how pre-trained convolutional image features can be used to assist scientists in discovering interesting chemical clusters for further investigation. Our method reduces the dimensionality of raw fluorescent stained images from a high throughput imaging (HTI) screen, producing an embedding space that groups together images with similar cellular phenotypes. Running standard unsupervised clustering on this embedding space yields a set of distinct phenotypic clusters. This allows scientists to further select and focus on interesting clusters for downstream analyses. We validate the consistency of our embedding space qualitatively with t-sne visualizations, and quantitatively by measuring embedding variance among images that are known to be similar. Results suggested the usefulness of our proposed workflow using deep learning and clustering and it can lead to robust HTI screening and compound triage. *
1 Introduction
Modern drug discovery is a rapidly evolving field with the use of modern AI technology. Nonetheless, it remains expensive and time consuming for the introduction of any new medicine (almost $3 billion, >10 years, [3]). Rather than hand-designing and testing novel drugs individually, the modern pharmaceutical approach is to use a library of compounds (e.g. 2 million compounds), and filter them with a sequence of complex imaging and biochemical tests. Typically for a high throughput screen, a single dose experiment is designed to remove vast majority of irrelevant compounds, e.g. by 100 folds.
Following the spectacular rise of deep learning techniques in computer vision, natural language processing and numerous scientific applications in recent years, deep learning has increasingly been applied to the field of chemoinformatics [2]. However, most recent work (e.g. [1]) has focused on predicting the biological effects of chemical compounds directly from chemical structure representations such as SMILES strings [9]. Morphological profiling is a complementary approach that can be used to predict a broad range of biological effects [4]. In this approach, candidate drugs are applied to cell cultures and imaged with high throughput fluorescence microscopy; depending on the bioactivity of the drugs, this can cause a variety of morphological changes to occur, yielding clues as to what effect a compound has on the cells. Indeed, [4] shows that a single network can be transferred to predict the outcomes of many other targeted assays.
Despite the rich information provided by morphological profiling, it is sometimes unknown which exact morphological phenotypes one should screen for. An example of this scenario is a high throughput screen conducted by AstraZeneca, in which novel compounds are screened for inhibition of IDOL - Inducible Degrader of the Low density lipoprotein receptor (LDLR) [5]. In this screen, HEK293S human embryonic kidney cells were engineered to express both Low Density Lipoprotein Receptor - Green Fluorescence Protein (LDLR-GFP) and IDOL. Since IDOL degrades LDLR, the presence of green fluorescence is an indicator of IDOL inhibition. However, due to the complex and poorly understood interactions between novel compounds and human cells, presence of the GFP signal is a necessary but not sufficient condition to infer IDOL inhibition - the actual phenotypic appearance of genuine hits is not known at the screening stage. In situations like this, the expert knowledge and intuition of a biologist is required to identify phenotypes that are indicative of genuine hits. Due to the high volume of images in a HTI screen, this cannot be done manually for each individual image, and due to the unknown nature of the target phenotype, supervised learning is not applicable.
In this paper, we propose a novel procedure for computing feature vectors for cellular images using a pre-trained convolutional neural network (CNN). The resulting feature vector space can then be partitioned by unsupervised clustering, allowing us to decompose a HTI screen into a small set of visually distinct phenotypes. The expert judgement of biologists can then be applied to whole phenotype clusters rather than individual images, allowing a HTI screen to be filtered rapidly for interesting compounds. The feature vectors can also be embedded in 2D space for visualization using dimensionality reduction techniques such as t-sne [6], providing a visual summary of the phenotypic distribution.
In Section 2 we describe our feature extraction pipeline. Section 3 discusses clustering and in Section 4 we demonstrate and evaluate our approach on a high throughput imaging dataset (IDOL).
2 Feature Extraction
CNNs trained on large datasets such as ImageNet have been found to learn a hierarchy of features, with early layers learning general, task-agnostic features pertaining to texture and shape primitives, and later layers learning more task specific features [10]. Despite the obvious differences between ImageNet images (which are generally photographs) and fluorescence micrographs, the early convolutional layers of a CNN trained on ImageNet are general enough to respond to the differences in shape, colour and texture in fluorescent labeled cellular images.
Our feature extraction begins by computing feature maps for a cellular image, by feeding it through a pre-trained CNN (Figure 1). The CNN architecture we use is VGG16 [8], pre-trained on ImageNet. Rather than taking final fully-connected layer activations as our feature vector, we extract our features by mean pooling the (pre-activation) feature maps of three early convolutional layers, and concatenating the means to produce a feature vector of length (one component for each feature map in the chosen layers). Extracting features from early convolutional layers rather than final fully connected layers has a number of advantages for large images in which the objects of interest are relatively small and homogeneously distributed.
Firstly, high level representations learned by CNNs contain information that is immediately relevant for identifying the classes they are trained to recognize. In the case of ImageNet, these are everyday objects such vehicles and animals. High level features are unlikely to be very descriptive for cellular images, which differ substantially from the ImageNet images and training classes, but lower level features are still general enough to capture information about shape, texture and colour.
Secondly, because of the fixed weight matrix connecting the first fully connected layer to the last convolutional layer, fully connected layers require the input image to be of a fixed size. By using only the convolutional layers of the network, we avoid the need to downsample our images to (for the VGG network), which would discard valuable high frequency information.
Thirdly, different layers capture information at different scales. Higher level layers have larger receptive fields, and describe patterns of greater size and complexity, but successively discard the higher frequency information captured by lower layers. By extracting features from the fourth, seventh and ninth convolutional layers, we obtain a multi-scale representation (see Figure 2).
3 Clustering
To discover distinct phenotypes in the dataset, we perform k-means clustering in feature space. The optimal choice for the number of clusters is a trade-off between making the clusters as homogeneous as possible, and keeping their number low. Figure 3 shows the mean intra-cluster embedding variance as a function of ; since we observe diminishing returns past , we choose as the optimal number of clusters.
4 Results
The most crucial aspect to validate is that our feature extraction does indeed embed similar images at similar points in the feature space. We can evaluate this quantitatively, by measuring the variance (concretely, mean squared distance to centroid) of different groups of image embeddings. The compounds tested in this dataset are chemically clustered by AstraZeneca in Extended-Connectivity Fingerprint embedding space [7], resulting in chemical clusters. We would expect images corresponding to compounds from the same chemical cluster to have more tightly clustered feature vectors than the dataset as a whole, because similar compounds may lead to similar morphological changes in images. As expected, the mean intra-cluster feature variance is that of the dataset as a whole. Furthermore, we would expect different images captured from the same well on an assay plate to be clustered more tightly still, because these images all correspond to the same compound. We observe the mean intra-well variance (4 images were captured per well) to be that of the full dataset.
To validate the quality of our clusters, we display samples from six phenotypic clusters in Figure 4. We see that k-means has identified relatively homogeneous clusters, further validating the quality of our embeddings.
Figure 5 shows a t-sne visualization of our entire dataset, with phenotypic clusters detected by k-means labelled by colour and annotated with representative samples from that cluster. To speed up the t-sne process, we used Principal Component Analysis (PCA) to reduce the dimensionality of our features from to . These principal components explain of the variance in the embedding space. Clusters and in Figure 5 show genuine GFP expression, while the others are judged as uninteresting by the biologists, and can be discarded, resulting in a 47-fold reduction. Using the full clusters would result in more precise filtering and greater reduction still.
5 Conclusion
We have developed a novel workflow for high throughput screening. In this workflow, images were represented as deep learning feature vectors from a pre-trained convolutional neural network. This was followed by the clustering of images with similar image phenotypes. This facilitates scientists to select interesting clusters for downstream screening in an attempt to find hit compounds. Because it uses generic convolutional features extracted from a pre-trained convolutional neural network, our method requires no training and can be applied to any cellular screen dataset without hyperparameter tuning - a significant saving in time. Our visualizations allow scientists to quickly assess the distribution of cellular morphologies in a high throughput imaging screen, or within a smaller subset of compounds, such as a chemical cluster. Meanwhile, our proposed workflow allows scientists to select interesting/promising phenotypes and quickly retrieve chemical clusters that show a high prevalence of said phenotypes.
Future work could gain further insight into the biological processes at work by investigating the relationship between our morphological embeddings and the ECFP embeddings of the chemicals that produced them, perhaps by predicting ECFP embeddings from morphological embeddings using supervised learning. Our techniques could also be applied to other imaging modalities, such as tissue pathology and mass spectrometry imaging, with minimal modification needed.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1[1] Esben Jannik Bjerrum. Smiles enumeration as data augmentation for neural network modeling of molecules. ar Xiv preprint ar Xiv:1703.07076 , 2017.
- 2[2] Hongming Chen, Ola Engkvist, Yinhai Wang, Marcus Olivecrona, and Thomas Blaschke. The rise of deep learning in drug discovery. Drug Discovery Today , 23(6):1241–1250, June 2018.
- 3[3] Joseph A Di Masi, Henry G Grabowski, and Ronald W Hansen. Innovation in the pharmaceutical industry: new estimates of r&d costs. Journal of health economics , 2016.
- 4[4] Jaak Simm et al. Repurposing High-Throughput Image Assays Enables Biological Activity Prediction for Drug Discovery. Cell Chemical Biology , pages 611–618.e 3, May 2018.
- 5[5] Sinead Knight, Helen Plant, Lisa Mc Williams, David Murray, Rebecca Dixon-Steele, Anet Varghese, Paul Harper, Anna Ramne, Paula Mc Ardle, Susanna Engberg, et al. Enabling 1536-well high-throughput cell-based screening through the application of novel centrifugal plate washing. SLAS DISCOVERY: Advancing Life Sciences R&D , pages 732–742, 2017.
- 6[6] Laurens van der Maaten and Geoffrey Hinton. Visualizing data using t-sne. Journal of machine learning research , 9(Nov):2579–2605, 2008.
- 7[7] David Rogers and Mathew Hahn. Extended-connectivity fingerprints. pages 742–754, 2010.
- 8[8] Karen Simonyan and Andrew Zisserman. Very deep convolutional networks for large-scale image recognition, 2014.