Electric dipole polarizability of group-IIIA ions using PRCC: Large correlation effects from nonlinear terms
Ravi Kumar, S. Chattopadhyay, B. K. Mani, D. Angom

TL;DR
This study calculates the electric dipole polarizability of group-IIIA ions using advanced relativistic coupled-cluster methods, highlighting the importance of nonlinear terms and various quantum electrodynamics corrections for accurate results.
Contribution
The paper introduces the use of PRCC with nonlinear terms and QED corrections to accurately compute polarizabilities of group-IIIA ions, showing significant effects of nonlinear contributions.
Findings
Nonlinear terms in PRCC significantly affect polarizability values.
Breit interaction correction is largest for Al+ and decreases for heavier ions.
Vacuum polarization and self-energy corrections increase with ion mass.
Abstract
We compute the ground-state electric dipole polarizability of group-IIIA ions using the perturbed relativistic coupled-cluster (PRCC) theory. To account for the relativistic effects and QED corrections, we use the Dirac-Coulomb-Breit Hamiltonian with the corrections from the Uehling potential and the self-energy. The effects of triple excitations are considered perturbatively in the PRCC. Our PRCC results for are good in agreement with the previous theoretical results for all the ions. From our computations we find that the nonlinear terms in PRCC have significant contributions and must be included to obtain the accurate value of for group-IIIA ions. For the correction from the Breit interaction, we find that it is largest for Al and decreases as we go towards the heavier ions. The corrections from the vacuum polarization and the self-energy increase from lighter…
Click any figure to enlarge with its caption.
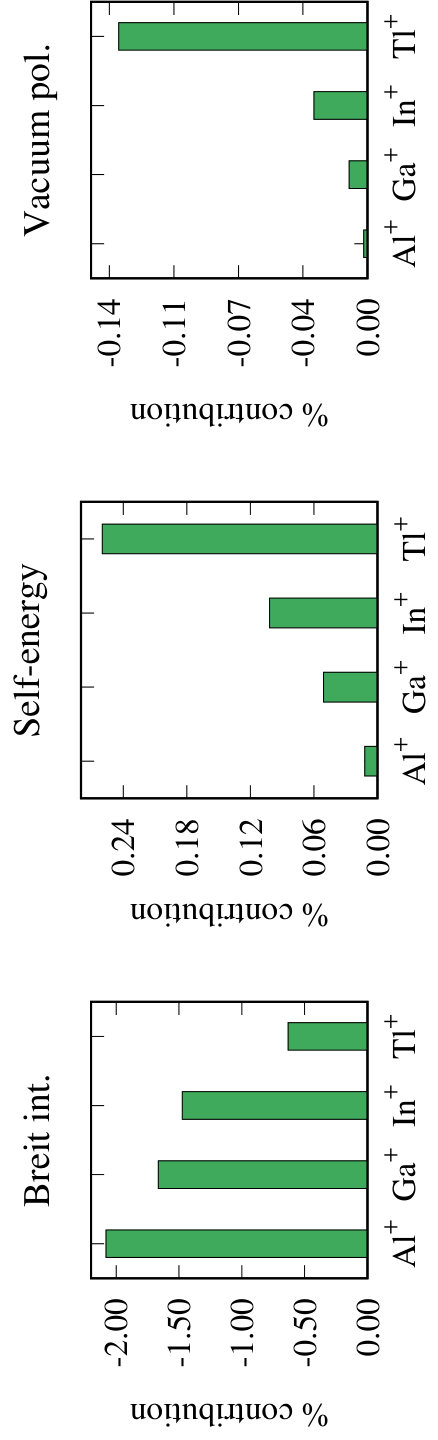 Figure 1
Figure 1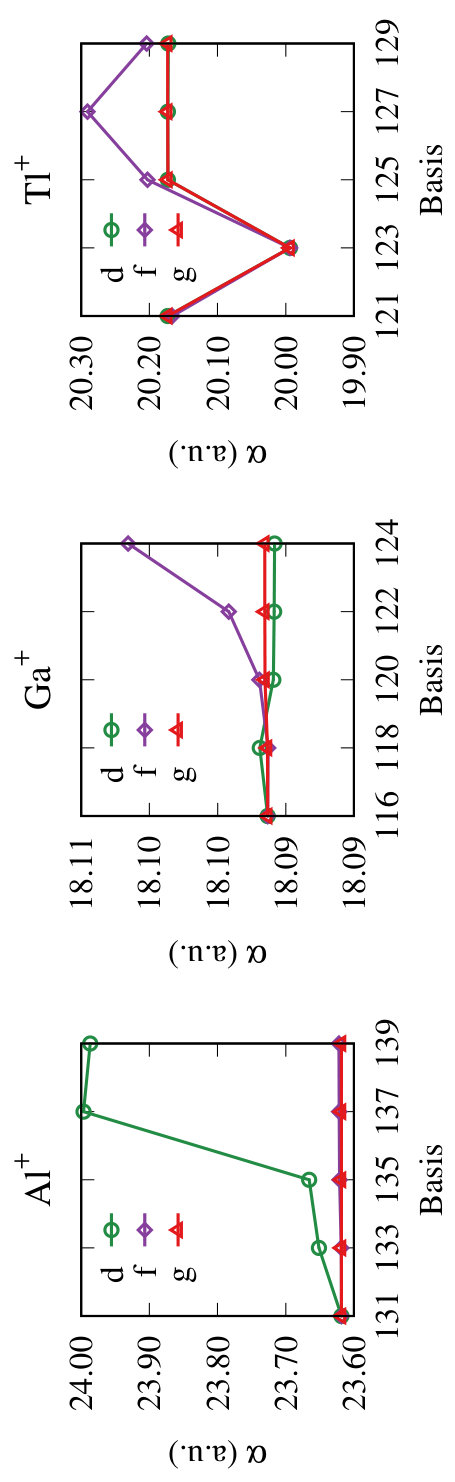 Figure 2
Figure 2| Ion | ||||||
|---|---|---|---|---|---|---|
| 0.0046 | 2.258 | |||||
| 0.0020 | 2.038 | 0.0020 | 2.105 | |||
| 0.0046 | 2.258 | 0.0048 | 2.215 | 0.0045 | 2.120 | |
| 0.0053 | 1.862 | 0.0052 | 1.870 | 0.0058 | 1.880 | |
| 0.0570 | 1.895 | 0.0498 | 1.820 | 0.0615 | 1.955 | |
| No. of orbitals | Basis size | |
| B+ | ||
| 107 | 9.292 | |
| 129 | 9.346 | |
| 151 | 9.358 | |
| 168 | 9.413 | |
| 173 | 9.413 | |
| Al+ | ||
| 131 | 23.618 | |
| 148 | 23.652 | |
| 159 | 23.789 | |
| 166 | 23.999 | |
| 169 | 23.999 | |
| Ga+ | ||
| 116 | 18.050 | |
| 132 | 18.050 | |
| 152 | 18.053 | |
| 172 | 18.056 | |
| 177 | 18.056 | |
| In+ | ||
| 123 | 24.748 | |
| 139 | 24.746 | |
| 150 | 24.744 | |
| 162 | 24.744 | |
| Tl+ | ||
| 134 | 20.026 | |
| 147 | 20.173 | |
| 156 | 20.129 | |
| 161 | 20.215 | |
| 171 | 20.216 | |
| Method | ||||
|---|---|---|---|---|
| PRCC(DC) | 23.9989 | 18.0556 | 24.7449 | 20.2159 |
| Breit int. | -0.4994 | -0.3006 | -0.3647 | -0.1283 |
| Vacuum pol. | -0.0004 | -0.0018 | -0.0070 | -0.0274 |
| Self-energy | 0.0028 | 0.0090 | 0.0249 | 0.0526 |
| Total | 23.5019 | 17.7621 | 24.3981 | 20.1128 |
| Ion | Present | Method | Previous | Method |
|---|---|---|---|---|
| work | works | |||
| 12.809 | LPRCC | 9.62 Safronova et al. (2011) | CI+all-order | |
| 9.413 | PRCC | 9.57 Archibong and Thakkar (1990) | CCD+ST(CCD) | |
| 9.415 | PRCC(T) | 9.64(3) Cheng and Mitroy (2012) | CICP | |
| 28.624 | LPRCC | 24.05 Safronova et al. (2011) | CI+all-order | |
| 23.502 | PRCC | 24.14(8) Kállay et al. (2011) | RCC | |
| 23.516 | PRCC(T) | 23.78(15)111 Finite-field using energies from RCI calculations. Yu et al. (2013) | Finite-field | |
| 24.07(41)222 Finite-field using energies from RCC calculations. Yu et al. (2013) | Finite-field | |||
| 24.12 Hamonou and Hibbert (2008) | CI | |||
| 24.14(12) Mitroy, J. et al. (2009) | CICP | |||
| 24.20(75) Reshetnikov et al. (2008) | Sum-rule | |||
| 24.2 Archibong and Thakkar (1991) | MBPT | |||
| 21.722 | LPRCC | 17.95 Cheng and Mitroy (2013) | CICP | |
| 17.762 | PRCC | 18.14(44) Reshetnikov et al. (2008) | Sum-rule | |
| 17.814 | PRCC(T) | |||
| 30.167 | LPRCC | 24.01 Safronova et al. (2011) | CI+all-order | |
| 24.398 | PRCC | 24.16(3)333 Finite-field using energies from RCI calculations. Yu et al. (2015) | Finite-field | |
| 24.467 | PRCC(T) | 24.33(15)444 Finite-field using energies from RCC calculations. Yu et al. (2015) | Finite-field | |
| 18.8(13) Reshetnikov et al. (2008) | Sum-rule | |||
| 22.834 | LPRCC | 19.60 Zuhrianda et al. (2012) | CI+all-order | |
| 20.113 | PRCC | 12.7(12) Reshetnikov et al. (2008) | Sum-rule | |
| 20.129 | PRCC(T) |
| Terms + h.c. | |||||
|---|---|---|---|---|---|
| 9.9782 | 25.2392 | 19.7942 | 27.0396 | 22.3619 | |
| -0.3162 | -0.7448 | -0.6858 | -1.0204 | -1.1296 | |
| 0.2740 | 0.6074 | 0.5088 | 0.6818 | 0.5700 | |
| -0.1804 | -0.3342 | -0.3222 | -0.4134 | -0.1352 | |
| 0.0106 | 0.0206 | 0.0170 | 0.0212 | -0.0078 | |
| Normalization | 1.0367 | 1.0329 | 1.0696 | 1.0632 | 1.0714 |
| Total | 9.4207 | 23.9989 | 18.0556 | 24.7449 | 20.2159 |
| Ion | ||||||
|---|---|---|---|---|---|---|
| B-spline | ||||||
| 0.00005 | ||||||
| -0.00178 | ||||||
| -0.05990 | ||||||
| -0.40518 | ||||||
| -4.23753 | ||||||
| Orbital | GTO | GRASP2K | SE | |||
| B+ | ||||||
| Al+ | ||||||
| 1.350[-2] | ||||||
| 9.440[-4] | ||||||
| -2.500[-5] | ||||||
| 2.000[-5] | ||||||
| 5.900[-5] | ||||||
| Ga+ | ||||||
| 2.759[-1] | ||||||
| 2.908[-2] | ||||||
| -7.880[-4] | ||||||
| 1.499[-3] | ||||||
| 4.324[-3] | ||||||
| -6.900[-5] | ||||||
| 2.000[-4] | ||||||
| -1.000[-5] | ||||||
| 1.100[-5] | ||||||
| 2.700[-4] | ||||||
| In+ | ||||||
| 1.312 | ||||||
| 1.606[-1] | ||||||
| -9.080[-4] | ||||||
| 1.316[-2] | ||||||
| 3.156[-2] | ||||||
| 2.770[-4] | ||||||
| 2.527[-3] | ||||||
| -2.080[-4] | ||||||
| 2.580[-4] | ||||||
| 5.837[-3] | ||||||
| 5.400[-5] | ||||||
| 4.160[-4] | ||||||
| -2.600[-5] | ||||||
| 3.200[-5] | ||||||
| 5.100[-4] | ||||||
| Orbital | GTO | GRASP2K | SE | |||
|---|---|---|---|---|---|---|
| 7.801 | ||||||
| 1.190 | ||||||
| 1.012[-1] | ||||||
| 1.468[-1] | ||||||
| 2.766[-1] | ||||||
| 3.027[-2] | ||||||
| 3.545[-2] | ||||||
| -2.150[-3] | ||||||
| 4.477[-3] | ||||||
| 7.001[-2] | ||||||
| 7.596[-3] | ||||||
| 8.733[-3] | ||||||
| -5.640[-4] | ||||||
| 1.169[-3] | ||||||
| 0.000 | ||||||
| 0.000 | ||||||
| 1.377[-2] | ||||||
| 1.330[-3] | ||||||
| 1.459[-3] | ||||||
| -6.200[-5] | ||||||
| 1.230[-4] | ||||||
| 1.519[-3] |
| Orbital | GTO | GRASP2K | |
| Al+ | |||
| 4.859[-9] | |||
| 4.851[-9] | |||
| 5.076[-7] | |||
| 5.076[-7] | |||
| 4.973[-6] | |||
| 4.973[-6] | |||
| 2.706[-5] | |||
| 2.706[-5] | |||
| 9.621[-5] | |||
| 9.620[-5] | |||
| 1.975[-4] | |||
| 1.975[-4] | |||
| Ga+ | |||
| -3.936[-4] | |||
| -3.947[-4] | |||
| -5.518[-4] | |||
| 2.304[-4] | |||
| 9.610[-4] | |||
| 1.798[-4] | |||
| -1.695[-4] | |||
| -1.672[-4] | |||
| 3.140[-4] | |||
| -3.164[-4] | |||
| -3.982[-4] | |||
| -3.856[-4] | |||
| 2.148[-4] | |||
| 2.227[-4] | |||
| 3.231[-4] | |||
| 3.366[-4] | |||
| -9.222[-3] | |||
| -9.236[-3] | |||
| Orbital | GTO | GRASP2K | |
| In+ | |||
| -5.307[-8] | |||
| -6.643[-8] | |||
| -1.720[-4] | |||
| -2.334[-4] | |||
| 1.736[-4] | |||
| 2.348[-4] | |||
| 3.585[-3] | |||
| -1.211[-4] | |||
| 1.436[-4] | |||
| 1.621[-4] | |||
| -8.482[-4] | |||
| -6.567[-4] | |||
| 1.045[-3] | |||
| 8.538[-4] | |||
| Tl+ | |||
| -1.125[-2] | |||
| -1.125[-2] | |||
| -6.116[-3] | |||
| -6.115[-3] | |||
| -3.686[-3] | |||
| -3.686[-3] | |||
| -2.382[-3] | |||
| -2.382[-3] | |||
| -1.579[-3] | |||
| -1.579[-3] | |||
| -9.461[-4] | |||
| -9.461[-4] | |||
| -3.565[-4] | |||
| -3.565[-4] | |||
| 1.168[-4] | |||
| 1.165[-4] | |||
| -1.145[-5] | |||
| -1.203[-5] | |||
| 1.991[-2] | |||
| 1.990[-2] | |||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Electric dipole polarizability of group-IIIA ions using PRCC: Large
correlation effects from nonlinear terms
Ravi Kumar
Department of Physics, Indian Institute of Technology, Hauz Khas, New Delhi 110016, India
S. Chattopadhyay
Department of Physics and Astronomy, Aarhus University, DK-8000, Aarhus C
B. K. Mani
Department of Physics, Indian Institute of Technology, Hauz Khas, New Delhi 110016, India
D. Angom
Physical Research Laboratory, Ahmedabad - 380009, Gujarat, India
Abstract
We compute the ground-state electric dipole polarizability of group-IIIA ions using the perturbed relativistic coupled-cluster (PRCC) theory. To account for the relativistic effects and QED corrections, we use the Dirac-Coulomb-Breit Hamiltonian with the corrections from the Uehling potential and the self-energy. The effects of triple excitations are considered perturbatively in the PRCC. Our PRCC results for are good in agreement with the previous theoretical results for all the ions. From our computations we find that the nonlinear terms in PRCC have significant contributions and must be included to obtain the accurate value of for group-IIIA ions. For the correction from the Breit interaction, we find that it is largest for Al+ and decreases as we go towards the heavier ions. The corrections from the vacuum polarization and the self-energy increase from lighter to heavier ions.
pacs:
31.15.bw,31.15.ap,31.15.A-,31.15.ve
I Introduction
The electric dipole polarizability () of atoms or ions is a measure of the interaction with an external electromagnetic field Bonin and Kresin (1997). It is a key parameter, and plays an important role in probing fundamental as well as technologically relevant properties of atoms and ions. Some current and potential implications of in atomic systems include discrete symmetry violations in atomic systems Khriplovich (1991); Griffith et al. (2009), optical atomic clocks Udem et al. (2002); Diddams et al. (2004), condensates of dilute atomic gases Anderson et al. (1995); Bradley et al. (1995); Davis et al. (1995), and the search for the variation in the fundamental constants Karshenboim and Peik (2010).
The recent advances in development of new and improved frequency and time standards in optical domain has elevated the interest in electric dipole polarizability of atoms and ions. One of the important reasons for this is that the is essential to calculate the blackbody radiation (BBR) shift in atomic transition frequency due to ac Stark effect. The BBR shift is one of the dominant environment induced shifts in atomic transition frequency, and contributes to the inaccuracy of atomic clocks. Here, it is to be emphasized that the group-IIIA ions are the promising candidates for accurate optical atomic clocks as they are expected to have low fractional frequency errors Chou et al. (2010); Safronova et al. (2011); Zuhrianda et al. (2012). Despite this important prospect associated with the group-IIIA ions, the ground state polarizability of these ions have not been studied in detail. For example, except for Al+, very little data is available from the previous theoretical calculations. This, perhaps, can be attributed to the complex nature of the correlation effects in these divalent ions.
It can thus be surmised that there is a research gap on the dipole polarizability for the group-IIIA ions. But, considering the experimental developments there are compelling reasons to address this research gap. That is the aim of this work. For this we employ the perturbed relativistic coupled-cluster (PRCC) theory and compute the ground state of the group-IIIA ions and examine the trends in the correlation effects in detail. More precisely, our aim is to: compute accurate value of for B+, Al+, Ga+, In+ and Tl+ ions using the PRCC theory; examine in the detail the contributions from the nonlinear terms in PRCC theory; do a comparative study with the trends observed in the other closed-shell atoms and ions Chattopadhyay et al. (2012a, b, 2013a, 2013b, 2014, 2015); and examine in detail the contributions to from the Breit interaction, the vacuum polarization and the self-energy corrections, and compare with the contributions in other closed-shell atoms and ions.
The PRCC theory is an appropriate many-body theory to account for the correlation effects arising from the external perturbation. It has been used to compute accurate for several atoms and ions in a series of our previous works Chattopadhyay et al. (2012a, b, 2013a, 2013b, 2014, 2015). The essence of PRCC is that it is a relativistic coupled-cluster (RCC) theory Pal et al. (2007); Mani et al. (2009); Nataraj et al. (2011) with an additional set of cluster operators. The latter account for the effects of an internal or external perturbation Hamiltonian. The amplitudes of these cluster operators are obtained by solving a new set of coupled nonlinear equations, this is in addition to the RCC cluster amplitude equations. The added advantage of PRCC is that it does not employ the sum-over-state Safronova et al. (1999); Derevianko et al. (1999) approach to incorporate the effects of a perturbation. The summation over all the possible intermediate states is subsumed in the perturbed cluster operators. In our previous works we have also demonstrated and verified the implementations of Breit interaction Chattopadhyay et al. (2012b), vacuum polarization Chattopadhyay et al. (2013b), and triple excitation in unperturbed Chattopadhyay et al. (2014) and perturbed Chattopadhyay et al. (2015) cluster operators. In the literature, there are other many-body theories which have been used to compute to good accuracy for a variety of atomic systems. A recent review by Mitroy and collaborators Mitroy et al. (2010) provides a detailed overview of these many-body theories and their applications. The remaining part of the paper is organized as follows. In Section II we provide an overview of the RCC and PRCC theories. In Section III we provide the calculational details where we discuss about the basis functions, nuclear potential, etc. used in the present work. The results obtained from our computations are analyzed and discussed in the Section IV. Unless stated otherwise, all the results and equations presented in this paper are in atomic units ( ).
II Theoretical Methods
We use the Dirac-Coulomb-Breit no-virtual-pair Hamiltonian, , to incorporate the relativistic effects in high-Z atoms. It provides a good description of the structure and properties of heavier atoms and ions. For an -electron atom or ion
[TABLE]
where and are the Dirac matrices, is an operator which project out the negative continuum states and is the nuclear potential. Projecting the Hamiltonian with ensures that the Hamiltonian is bounded from below by neglecting the negative-energy continuum states from the calculations. In the present work, this is implemented by selecting only the positive energy states from the finite size basis set. The last two terms, and are the Coulomb and Breit interactions, respectively. The Breit interaction, which represents the inter-electron magnetic interactions, is
[TABLE]
The Hamiltonian satisfies the eigen-value equation
[TABLE]
where, is the exact atomic state and is the corresponding exact energy.
In the presence of external perturbations, the Hamiltonian is modified with the addition of the perturbation interaction terms. For example, the total Hamiltonian in presence of an external electric field is
[TABLE]
where is the interaction Hamiltonian, arising from the interaction between the induced electric dipole moment of the atom and the external electric field . And, is a perturbation parameter. The modified Hamiltonian satisfies the eigen-value equation
[TABLE]
here, and represent the perturbed atomic state and the corresponding perturbed eigen energy, respectively.
To compute and we use RCC Mani et al. (2009) and PRCC Chattopadhyay et al. (2012a, b, 2013a, 2013b, 2014, 2015) theories, respectively. In the RCC theory the ground state atomic wavefunction of a closed-shell atom is
[TABLE]
where is the reference state wave-function and is the cluster operator. The perturbed ground state, based on the PRCC theory, is
[TABLE]
For an -electron closed-shell atom and , where is the order of excitation. An approximation, which captures most of the correlation effects, is the coupled-cluster single and double (CCSD) approximation Purvis and Bartlett (1982). With this approximation
[TABLE]
The cluster operators in second quantized notations are
[TABLE]
where and are the cluster amplitudes, () are single particle creation (annihilation) operators and () represent core (virtual) single-particle states or orbitals. Here, we have used -tensors to represent the perturbed cluster amplitudes to incorporate the rank of in the perturbation Hamiltonian. Besides this modification, are also constrained by the parity and triangular conditions Chattopadhyay et al. (2012b): ; , , and . Where, represents the orbital(total) angular momentum of the single-electron state.
The unperturbed cluster operators used in equation (6) are obtained by solving the coupled nonlinear equations
[TABLE]
The states and are singly- and doubly-excited determinants obtained by replacing and electrons from core orbitals in with virtual electrons, respectively. And, is the normal-ordered Hamiltonian. Similarly, the in equation (7) are solutions of the coupled nonlinear equations
[TABLE]
The above equations are coupled to the equations as these require the values of . We solve these equations using the Jacobi method, and to remedy the slow convergence of this method we employ direct inversion of the iterated subspace (DIIS) Pulay (1980).
In the PRCC theory, the ground state dipole polarizability of close-shell atoms or ions is Chattopadhyay et al. (2015)
[TABLE]
From Eq. (7), using the expression of we can write
[TABLE]
where , and in the denominator is the normalization factor. Considering the computational complexity, we truncate and the normalization to factor to second order in the cluster amplitudes. Based on earlier studies, the contributions from the higher order terms are negligible Chattopadhyay et al. (2014, 2015).
III Calculational Details
The use of basis set with good descriptions of single-electron wavefunctions and energies is critical to get accurate results from RCC and PRCC computations. In this work use the Gaussian-type orbitals (GTOs) Mohanty et al. (1991) as the single-electron basis for RCC and PRCC computations. The GTOs are finite basis set orbitals in which the orbitals are expressed as linear combinations of Gaussian-type functions (GTFs). Specially, the GTFs of the large component of the orbitals have the form
[TABLE]
where , , , , is the GTO index and is the number of GTFs. And, the exponent , where and are two independent parameters optimized separately for each orbital symmetries. This choice of the exponents is referred to as the even-tempered basis set. The small components of orbitals are derived from the large components using the kinetic balance condition Stanton and Havriliak (1984). To incorporate the effects of the finite size of the nucleus we use two-parameter finite size Fermi density distribution
[TABLE]
where, . The parameter is the half charge radius of the nucleus so that , and is the skin thickness.
To generate GTO basis we optimize and parameters so that the orbital energies, both the core and virtual orbitals, match the numerical orbitals obtained from the the GRASP2K code Jönsson et al. (2013). In addition, we also match the the self-consistent field (SCF) energies. It must be mentioned here that the virtual orbitals (for Al+ and Ga+) and (for Ga+, In+, and Tl+) symmetries have significant contributions to the dipole polarizability. Hence, it is essential to optimize the virtual orbitals in and symmetries. The optimized and parameters for all the ions are given in the Table 1. More detailed comparisons of the orbital energies are provided in the Appendix.
To further optimize the orbitals, we incorporate the effects of Breit interaction, vacuum polarization and the self-energy corrections in the orbital basis set. The improved orbitals are then used in RCC and PRCC computations. This leads to, through a change in the cluster amplitudes, a small but important change in the dipole polarizability of all the ions. The detailed comparison of the orbital energy corrections from the afore mentioned effects are given in the Appendix.
IV Results and Discussions
The SCF energies computed from the optimized GTO match very well with the GRASP2K results for all the ions. The largest deviation is of the order hartree and this is observed in the case of In+. For the remaining ions the deviation is much smaller, and lowest is in the case of B+. For which the deviation is of order hartree. A detailed comparison of the SCF and orbital energies is provided in the Appendix. In the Table 2 we show the convergence pattern of with the basis size. As computations with the DCB Hamiltonian are more compute intensive, to determine the converged basis we use the DC Hamiltonian. For example, the computation of for heavy ions like Tl+ with moderate basis set size of 154 orbitals needs a few weeks to complete. As discernible from the table, we start with a moderate basis size orbitals and add orbitals systematically in each symmetry until converges up to atomic units or smaller. For a better description the results of and observed trends of the correlation effects are discussed for each of the ions separately.
IV.1 B+ and Al+
From the results given in Table 2, we find that the change is below in for B+ when the basis set is increased from 168 to 173. So, to minimize the computation time, we consider the basis set with 168 orbitals as optimal, and use it for the further analysis. Similarly, for Al+ we find that a basis set with 166 orbitals is optimal. To analyse the symmetry wise contributions from the virtual orbitals, we plot the values of for each symmetry with respect to the basis size in Fig. 1. From the figure it evident that the virtual orbitals have significant contributions for Al+. We attribute this to the correlation effects arising from the strong mixing of the core electrons with virtual electrons. This indicates that to get high quality results for Al+ it is essential to optimize the virtual orbitals.
The value of computed from the converged basis set is listed in Table 4, for comparison the results reported in previous works are also listed. For B+, as we see from the table, there are very few theoretical results in literature. And, to best of our knowledge, there are no experimental data. From the previous works the average value reported is 9.6. However, our LPRCC result of 12.809, is % larger than the previous theory results and, therefore, indicates missing of important correlation effects. This is in contrast to the trends observed in our previous works Chattopadhyay et al. (2012a, b, 2013a, 2013b, 2014, 2015), where LPRCC provides a reliable result for the ground state polarizability. This could be due to the stronger electron-correlation effects associated with the two-valence nature of the ions. And, which is further enhanced due to the orbital contraction as these are singly charged ions. From our calculations we find that the nonlinear terms in PRCC theory are very important and must be included in the computation of for such a complicated ions. In particular, we find that a nonlinear diagram arising from the PRCC term , has a very large contribution but with opposite sign. We observe this trend for all the ions in the group. Our PRCC and PRCC(T) results, 9.413 and 9.415, respectively, are in good agreement. However, these are slightly lower than the previous results. This difference can be attributed to the effects arising from the inclusion Breit interaction in our computations.
For Al+, among all the ions considered it has the largest number of previous results. However, like in the case of B+ all of these are theoretical results. Except for the RCI based finite-field results Yu et al. (2013), the values of reported in previous works are very close to each other. The average value from the reported values is 24.1. The value 23.78 reported in Ref. Yu et al. (2013) is on the lower side than the others. In terms of the many-body theory method the method used in Kállay et al. Kállay et al. (2011) is closest to ours. They have used the relativistic general-order coupled-cluster theory, where the higher-order cluster excitations are considered by using the many-body diagrammatic techniques based automated programming tools. However, one important difference is that they use DC Hamiltonian, where as we use the DCB Hamiltonian. In fact, this is the main reason for our PRCC and PRCC(T) results of 23.502 and 23.516, respectively, to be lower than that of Ref.Kállay et al. (2011), and others. This is evident from the Table 3 where we list contributions from different effects. Our result of 23.999 based on the DC Hamiltonian is in excellent agreement with the previous values. As we observe from the table, the contribution from the Breit interaction is -0.499, which reduces the DC result of . The contributions from the vacuum polarization and the self-energy correction are -0.0004 and 0.0028, respectively. As shown in the Fig. 2, in terms of percentage these are % and %, respectively. An important point to observe here is that, the contribution from the vacuum polarization is opposite in phase to that of the self-energy correction. Interestingly, we observed the same pattern in our previous work on dipole polarizability of group-IIB elements Chattopadhyay et al. (2015). The inclusion of triples perturbatively improves the result further, and the contribution is 0.014.
In the Table 5 we provide the term wise contributions to . As we observe, for both the ions, the leading order (LO) term is . Quantitatively, it is % and 105% of the total value in the case of B+ and Al+, respectively. This is expected as it subsumes the contributions from the Dirac-Hartree-Fock and the core polarization effects. The next to leading order (NLO) term is , which has a contribution of % of the total value in both the ions. The last two dominant contributions are from and terms, respectively. The contribution from the former is roughly twice than the latter but opposite in phase.
IV.2 Ga+ and In+
From our results we find that for Ga+ both the and virtual orbitals have significant contribution to . This is due to the mixing of these virtual orbitals of and symmetries with the and core orbitals, respectively. Similarly, for In+ there is a mixing between the core electrons and virtual orbitals. In Table 4 we provide the converged value of from our computations and compare with the data available in the literature. As we can observe from the table, there are only two and four previous results in the case of Ga+ and In+, respectively. All of these are theoretical results and to the best of our knowledge, there are no experimental results of for these two ions. For Ga+, the value of , 18.14, from Ref. Reshetnikov et al. (2008) using sum-rule is higher than the CICP value of 17.92 Cheng and Mitroy (2013). Our PRCC (DC) result of 18.056, listed in the Table 3, lies between these two results. Our PRCC result of 17.762 using DCB Hamiltonian is lower than both the previous results. As discussed before, this difference can be attributed to Breit interaction, which has the contribution of -0.3006. The PRCC (T) result of 17.814 shows a marginal increase and the contribution from the perturbative triples is 0.052. The contributions from the vacuum polarization and the self-energy correction are -0.0018 and 0.0090, respectively. Like in Al+, these are of opposite phases, and are larger in magnitude by % and %, respectively than Al+. Interestingly, the Breit contribution to is smaller, by %, than the Al+.
For In+, there is a large variation in the value of reported in the previous works. The lowest value of 18.8 is obtained by using the sum-rule Reshetnikov et al. (2008), while the highest value 24.33 is based on the finite-field method Yu et al. (2015). The value of 24.01 reported based on CI + all-order method Safronova et al. (2011) lies between the two previous results. Our PRCC and PRCC(T) results are 24.398 and 24.467, respectively. These values are higher than the previous results. Quantitatively, our PRCC result is larger by % and % than the values in Refs. Safronova et al. (2011) and Yu et al. (2015), respectively. The contributions from the Breit interaction, vacuum polarization, self-energy correction and the perturbative triples are -0.3647, -0.0070, 0.0249 and 0.069, respectively. In terms of percentage, these are %, %, % and %, respectively of the DC value of . The contributions are larger in magnitude by %, %, % and % than the Ga+ ion. This clearly indicates the importance of incorporating these effects to obtain accurate results.
Examining the term wise contributions, both ions follow the trend of B+ and Al+, where the LO and NLO terms are and , respectively. While the LO contributes %, NLO contributes % of the total value each for both the ions. The next two dominant contributions are from and terms, respectively. The contribution from is % each for Ga+ and In+ ions. And, the contribution from is % each in the case Ga+ and In+.
IV.3 Tl+
The orbital energies obtained from the GTO are in very good agreement with those of numerical orbitals from GRASP2K. The corrections from the self-energy are consistent with the trend observed in the other ions. A detailed comparison and discussions of the orbital energies and various corrections are given in the Appendix. The corrections from the Breit interaction and vacuum polarization requires special attention. The correction from the Breit interaction, , has a different trend than the previous ions. And, that is the negative sign of for , and orbitals. Interestingly, the same pattern was also observed in the case of Hg in our previous work Chattopadhyay et al. (2015) and, as mentioned there, it may be due to the large weight factor associated with the two-electron exchange integrals.
Like in In+ and other ions, is negative for all the orbitals. There is, however, a difference in the phase for orbitals where, unlike the previous ions, is negative for all the orbitals. This trend of negative is consistent with the case of Hg and Ra2+ observed in our previous works Refs. Chattopadhyay et al. (2015) and Chattopadhyay et al. (2013b), respectively. We attribute this to the larger relativistic effects due to the stronger nuclear potential in these ions.
The converged value of is computed by using the optimal basis of 161 orbitals. The computation, like in the previous ions, is incorporating the effects of the Breit interaction, vacuum polarization, and the self-energy corrections. As shown in the Fig. 1 the virtual orbitals of the symmetry have dominant contributions and are important to get the converged value of . This arises from the large mixing between the virtual orbitals and core orbitals.
In the Table 4 we compare our converged result with the previous values. For comparison we found only two theoretical results from the literature. However, there is a large variation in these two results, the value of 19.60 obtained from CI + all-order theory Zuhrianda et al. (2012) is % larger than the value of 12.7 obtained from the sum-rule method Reshetnikov et al. (2008). Our PRCC and PRCC(T) results of 20.113 and 20.129, respectively are in good agreement with the Ref. Zuhrianda et al. (2012). However, our LPRCC result of 22.834 is % larger than the Ref. Zuhrianda et al. (2012). The reason for this is similar to the case of the previous ions–large cancellation due to the contribution from a non-linear diagram arising from the PRCC term . The contribution from the Breit interaction is 0.1209. This has the same phase as in the previous ions but smaller by % than In+. The contributions from the vacuum polarization and the self-energy correction are 0.0258 and 0.0495, respectively. These are also in the same phase as the previous ions but larger by % and 98%, respectively than In+. We also observe that the term wise contributions are also of the same pattern as the previous ions.
To estimate the upper bound on the uncertainty associated with the value of in the present work we have isolated four different sources. While some of these have very small contributions and can be neglected, some are must to be included. The first source of uncertainty is due to the truncation of the basis set in our computations. Since the values of reported in Table 4 are using the converged basis set where the change in is of the order of or less, we neglect this uncertainty also. The second source of uncertainty is due to the truncation of the dressed operator in equation (13) to include the cluster operators up to the second order only. In our previous work Mani and Angom (2010), using an iterative scheme, we have shown that the contribution from the third- and higher-order terms is negligible and therefore we can neglect this uncertainty. The third source of uncertainty is due to the partial inclusion of triples () in the PRCC theory. The maximum contribution from the perturbative triples is % in the case of Ga+. Since the perturbative scheme accounts for the dominant contributions, we can assume % as an upper bound to this uncertainty. The last source of uncertainty in our computation is associated with the frequency-dependent Breit interaction which we do not include in the present work. However, in our previous work Chattopadhyay et al. (2014) using a series of calculations using GRASP2K we estimated an upper bound to be % in the case of Ra. Since Ra has higher than Tl, in this work as well we assume % as an upper bound on the uncertainty due to frequency-dependent Breit interaction. Combining all these sources of uncertainties, the upper bound on the uncertainty in the value of is 0.5%.
V Conclusion
We have computed the ground state electric dipole polarizability group-IIIA ions using the PRCC theory. To account for the relativistic effects and QED corrections, we have used the Dirac-Coulomb-Breit Hamiltonian with the corrections from the Uehling potential and the self-energy. The effects of triple excitations are considered perturbatively. Our results from PRCC and PRCC(T) using the Dirac-Coulomb Hamiltonian are in excellent agreement with the previous results for all the ions. The results using the Dirac-Coulomb-Breit Hamiltonian are, however, lower than most of the previous results except for In+. We attribute this to the effects of the Breit interaction, which is considered in our work but not in the previous works. The other important observation from our computations is that, we need to go beyond the LPRCC to obtain accurate results for the group-IIIA ions. The LPRCC results are on an average % larger than the PRCC results. This could be due to the strong correlation effects arising from the divalent nature of the group-IIIA ions. And, to account for such a large correlation effects the nonlinear terms in PRCC theory must be included.
Based on our analysis of the corrections arising from the Breit interaction we find two trends. First, the contribution for all the ions are negative, and hence reduces the total value of . The same pattern of is also observed in the case of noble-gas atoms Chattopadhyay et al. (2012b), alkaline-earth-metal atoms Chattopadhyay et al. (2014) and the group-IIB elements Chattopadhyay et al. (2015). In the case of singly ionized alkali-metal atoms Chattopadhyay et al. (2013a), however, we get a different trend. Second, in terms of the percentage contribution, we observed the largest contribution of % in the case of Al+. And, as we go towards the heavier ions the contribution decreases, the lowest is %, in the case of Tl+. A similar pattern is also observed in the case of alkaline-earth-metal atoms and the group-IIB elements where heavier atoms Ra and Hg have the smaller contributions, of % and %, respectively, than the lighter ones. In the case of noble-gas atoms, however, we observed an opposite trend where the heaviest atom Rn has the largest contribution of %.
For the Uehling potential and the self-energy corrections, we observed a trend of increasing contributions from the lighter ions to the heavier ions. This is to be expected as the heavier atoms have the larger relativistic effects. The largest contributions are % and % from the Uehling potential and the self-energy corrections, respectively, in the case of Tl+. We observed an opposite trend from the Uehling potential in the case of the group-IIB elements Chattopadhyay et al. (2015), where Zn has larger contribution (%) than the Hg (%). For the self-energy correction, the group-IIB elements show a mix behavior where both Zn and Hg have larger contribution than Cd.
Acknowledgements.
We thank Chandan Kumar Vishwakarma for useful discussions. The results presented in the paper are based on the computations using the HPC facilities at the Indian Institute of Technology Delhi, New Delhi and Physical Research Laboratory, Ahmedabad.
Appendix A Breit interaction, vacuum polarization and self energy corrections
For Breit interaction we use the approach introduced by Grant and Pyper Grant and Pyper (1976) where the Breit interaction operator is expanded as a linear combination of irreducible tensor operators. We employ the expressions given in Ref. Grant and McKenzie (1980) to incorporate the effects of in single-electron basis as well as RCC and PRCC calculations. To analyze the effects of in detail, we compute contributions to SCF energy as well as the single-electron energies for all the ions. The correction to single-electron energy due to Breit interaction is
[TABLE]
where and represent the orbital energies obtained by solving the Dirac-Hartree-Fock and Dirac-Hartree-Fock-Breit orbital equations self-consistently, respectively. Similarly, the correction to the SCF energy is
[TABLE]
where, and are the SCF energies computed using DCB and DC Hamiltonian, respectively. The computed from our implementation on GTO is given in Table 6 where we compare our results with a recently reported code for B-spline basis by Zatsarinny el al. Zatsarinny and Fischer (2016). The contributions to the orbital energies are tabulated in the Tables 7 (for B+, Al+, Ga+, and In+) and 8 (for Tl+) for a quantitative description.
To incorporate the effects of vacuum polarization we consider the expression given by Johnson et al. Johnson et al. (2001), where the Uehling potential Uehling (1935) is generalized to incorporate the effects of the finite size of the nucleus. In our previous work Chattopadhyay et al. (2013b) we had discussed the details of the implementation. To quantify the effects in the present work, we compute the corrections to orbital energies as well as the SCF energy for all the ions. The correction to orbital energy is
[TABLE]
where and are the energies with and without Uehling potential, respectively. Similarly, the correction to SCF energy is
[TABLE]
where, and are the SCF energies computed using DC plus Uehling potential and DC Hamiltonian, respectively. The from our computations are tabulated and compared with the results from the B-spline code Zatsarinny and Fischer (2016) in the Table 6. And, are given in the Tables 7 and 8.
The effects of the self-energy (SE) correction to orbitals are considered through the model Lamb-shift operator introduced by Shabaev et al. Shabaev et al. (2013). For this we use the code QEDMOD Shabaev et al. (2015), developed by the same authors, to compute the corrections to the orbital energies. These corrections to energies are applied and used in the RCC and PRCC computations. A similar analysis was reported for the group-IIB elements in our previous work Chattopadhyay et al. (2015). The data on corrections to orbital energies, computed using QEDMOD code, are listed in the Tables 7 and 8.
Appendix B SCF and orbital energies
In Table 6 we compare the SCF energy from GTO with GRASP2K Jönsson et al. (2013). Similarly, the orbital energies of GTO are tabulated and compared with energies of the numerical orbitals obtained from GRASP2K in the Tables 7 and 8. Considering the Breit correction, the sign of is positive for all the ions and matches with the B-spline results. The positive sign of indicates an increase in the SCF energy, which we attribute to the spatial contraction of the orbitals. Interestingly, we reported the observation of the same trend of in the case of the noble-gas Chattopadhyay et al. (2012b) and group-IIB elements Chattopadhyay et al. (2015). Examining the values listed in the table, we find that our GTO results are in excellent agreement with the B-spline results. The largest difference is of the order of hartree, which occurs in the case Tl+. The last two columns of Table 6 show the comparison of from GTO with the B-spline data. Unlike , from GTO has negative value for all the ions, indicating a decrease in the SCF energy. This decrease in SCF energy implies the relaxation of the orbitals. There is a sign mismatch in the results of B+, though, the contribution is very small. For the remaining ions, both sign as well as magnitude of GTO results are in good agreement with the B-spline data. For better comparison, the results from each ions are discussed separately.
B.1 B+ and Al+
The orbital energies from GTO for core orbitals, given in Table 7, are in excellent agreement with the numerical data for both the ions. The largest difference between the two results is hartree, in the case of orbital of Al+. For remaining orbitals, of both the ions, the difference is even smaller. For virtual orbitals in Al+, provided in Table 9, the difference is of the order of hartree or lower for the orbitals with principal quantum number . For orbitals with the difference increases but still very small, the largest difference is in the case of .
Incorporating the Breit interaction, the change in the orbital energies, is positive for all the orbitals in both the ions. This indicates relaxation of orbitals and a similar trend was observed in our work on group-IIB elements Chattopadhyay et al. (2015). As to be expected the inner core core orbitals, which are closer to the nucleus, have larger corrections. Quantitatively, , hartree, for orbital is two orders of magnitude higher than that of for the orbital in B+. Similarly, in Al+, , hartree, for is three orders of magnitude larger than that of hartree for .
The correction due to the Uehling potential, , is on average two orders of magnitude smaller than the Breit interaction for all the orbitals in both the ions. The other important difference from Breit is that, except for the orbital in B+, all the orbitals tend to contract. The remaining orbitals, and , tend to relax after the inclusion of the Uehling potential. In terms of actual contribution, similar to the trend of Breit interaction, , hartree, is two orders of magnitude larger than , hartree in B+. Similarly, in Al+ is two orders of magnitude larger than . This trend is to be expected, as the vacuum polarization is attractive and short-range potential, it has large effects on the orbitals with finite probability density within the nucleus.
For the self-energy correction, it is negligibly small in the case of B+. So, we provide data for Al+ only. As we observe from the table, like Breit interaction and vacuum polarization corrections, the self-energy correction also is largest for the orbital, and decreases with increasing principal quantum number. Quantitatively, for is times larger than that for . This is to be expected, as the inner core orbitals, which are closer to the nucleus have larger relativistic effects than the others. The other important observation is that, except the orbital, is positive for all the orbitals.
B.2 Ga+ and In+
Like in the case of B+ and Al+ the energies of both the core and virtual orbitals from GTO are in excellent agreement with the GRASP2K data. Quantitatively, for Ga+, the energy of core and virtual orbitals agree up to and hartree, respectively. For In+, the largest difference is of the order of hartree, for the orbital.
For the corrections from the Breit interaction and vacuum polarization to the orbital energies, they follow the same trend as in B+ and Al+. That is, is positive for all the orbitals and is negative for orbitals only. The magnitude of the corrections are, however, orders of magnitudes larger than that of Al+. This trend is to be expected as Ga+ and In+ have stronger nuclear potentials than Al+. In addition, like B+ and Al+, the inner core orbitals have larger corrections than the outer core orbitals.
The self-energy corrections, like in Al+, is positive for all and orbitals and negative for all orbitals for both the ions. Among the remaining orbitals, all orbitals have negative and all orbitals have positive contributions. In terms of the magnitude of the corrections, it is one and two orders larger in Ga+ and In+, respectively than Al+.
B.3 Tl+
Like in the other ions, the orbital energies are in excellent agreement with the GRASP2K data. The largest difference of hartree is observed in the case of orbital. For the virtual orbitals, the difference is of the order of hartree for (), and smaller for the orbitals with . The correction from the Breit interaction , as to be expected, is much more larger than the previous ions. Comparing with In+, it is , , , and times larger in magnitude for , , , and orbitals, respectively. Roughly the same trend of contribution is also observed for all and orbitals. For and () orbitals is and times larger than In+. Among the other orbitals, and has the contributions of and hartrees, respectively.
Like the Breit interaction, corrections from the Uehling potential are also larger than the In+ for all the orbitals. For instance, is , , , and times larger in magnitude for , , , and orbitals, respectively. It is , and times for , and orbitals, respectively. For orbitals, the contributions are slightly smaller, , and times for , and orbitals, respectively. Looking into the corrections to and , they both have the same trend of contributions where is and times larger than In+ for and (), respectively. Among the other orbitals, have the same order of contributions as orbitals.
Considering the correction from the self-energy, there is an important difference in the sign of in comparison to the previous ions. The is positive for all the orbitals, which is in opposite phase to the previous ions. Comparing with In+, in terms of magnitude, among the orbitals, and has the highest and lowest contributions of and times, respectively. Similarly, among the () orbitals, the largest and smallest change from In+ are () and () times for () and () orbitals, respectively. For in the case of and , there is a large change for than . Quantitatively, it is and times, respectively for and in comparison to and times, respectively for and orbitals.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bonin and Kresin (1997) K. Bonin and V. Kresin, Electric-Dipole Polarizabilities of Atoms, Molecules and Clusters (World Scientific Publ., Singapore, 1997).
- 2Khriplovich (1991) I. Khriplovich, Parity Nonconservation in Atomic Phenomena (Gordon and Breach Science Publishers, Philadelphia, 1991).
- 3Griffith et al. (2009) W. C. Griffith, M. D. Swallows, T. H. Loftus, M. V. Romalis, B. R. Heckel, and E. N. Fortson, Phys. Rev. Lett. 102 , 101601 (2009) . · doi ↗
- 4Udem et al. (2002) T. Udem, R. Holzwarth, and T. W. Hansch, Nature 416 , 233 (2002) . · doi ↗
- 5Diddams et al. (2004) S. A. Diddams, J. C. Bergquist, S. R. Jefferts, and C. W. Oates, Science 306 , 1318 (2004) . · doi ↗
- 6Anderson et al. (1995) M. H. Anderson, J. R. Ensher, M. R. Matthews, C. E. Wieman, and E. A. Cornell, Science 269 , 198 (1995) . · doi ↗
- 7Bradley et al. (1995) C. C. Bradley, C. A. Sackett, J. J. Tollett, and R. G. Hulet, Phys. Rev. Lett. 75 , 1687 (1995) . · doi ↗
- 8Davis et al. (1995) K. B. Davis, M. O. Mewes, M. R. Andrews, N. J. van Druten, D. S. Durfee, D. M. Kurn, and W. Ketterle, Phys. Rev. Lett. 75 , 3969 (1995) . · doi ↗