Ca2+ release via IP3 receptors shapes the cytosolic Ca2+ transient for hypertrophic signalling in ventricular cardiomyocytes
Hilary Hunt, Agne Tilunaite, Greg Bass, Christian Soeller, H. Llewelyn, Roderick, Vijay Rajagopal, Edmund J. Crampin

TL;DR
This study models how IP3 receptor-mediated Ca2+ release influences the shape of cytosolic Ca2+ transients, revealing its role in hypertrophic signalling in heart cells and linking Ca2+ duty cycle to NFAT activation.
Contribution
The paper introduces a novel model of IP3R and RyR interaction, showing how IP3R activation modulates Ca2+ transient shape and hypertrophic signalling in cardiomyocytes.
Findings
IP3R activation prolongs Ca2+ transient duration.
IP3 concentration affects Ca2+ transient amplitude.
Increased Ca2+ duty cycle correlates with NFAT activation.
Abstract
Calcium (Ca2+) plays a central role in mediating both contractile function and hypertrophic signalling in ventricular cardiomyocytes. L-type Ca2+ channels trigger release of Ca2+ from ryanodine receptors (RyRs) for cellular contraction, while signalling downstream of Gq coupled receptors stimulates Ca2+ release via inositol 1,4,5-trisphosphate receptors (IP3Rs), engaging hypertrophic signalling pathways. Modulation of the amplitude, duration, and duty cycle of the cytosolic Ca2+ contraction signal, and spatial localisation, have all been proposed to encode this hypertrophic signal. Given current knowledge of IP3Rs, we develop a model describing the effect of functional interaction (cross-talk) between RyR and IP3R channels on the Ca2+ transient, and examine the sensitivity of the Ca2+ transient shape to properties of IP3R activation. A key result of our study is that IP3R activation…
Click any figure to enlarge with its caption.
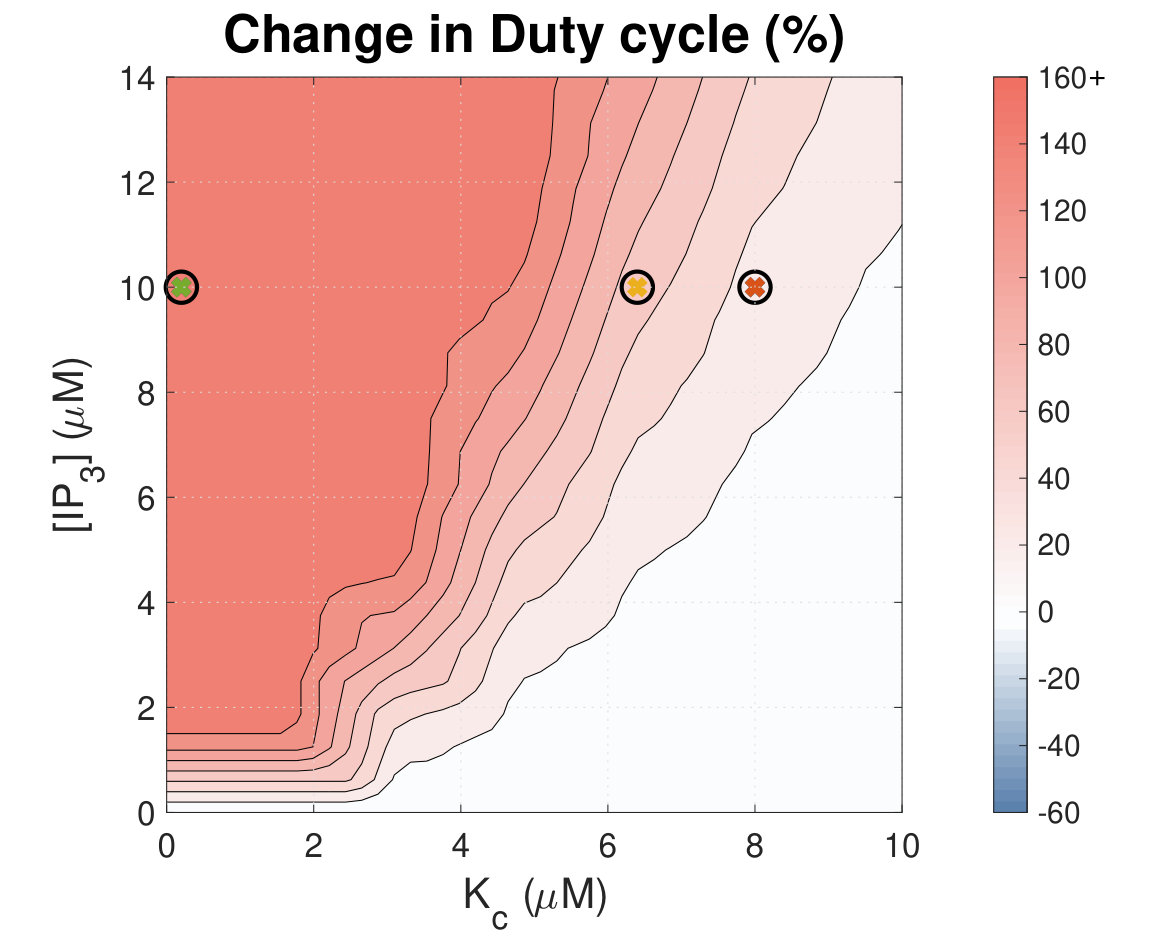 Figure 1
Figure 1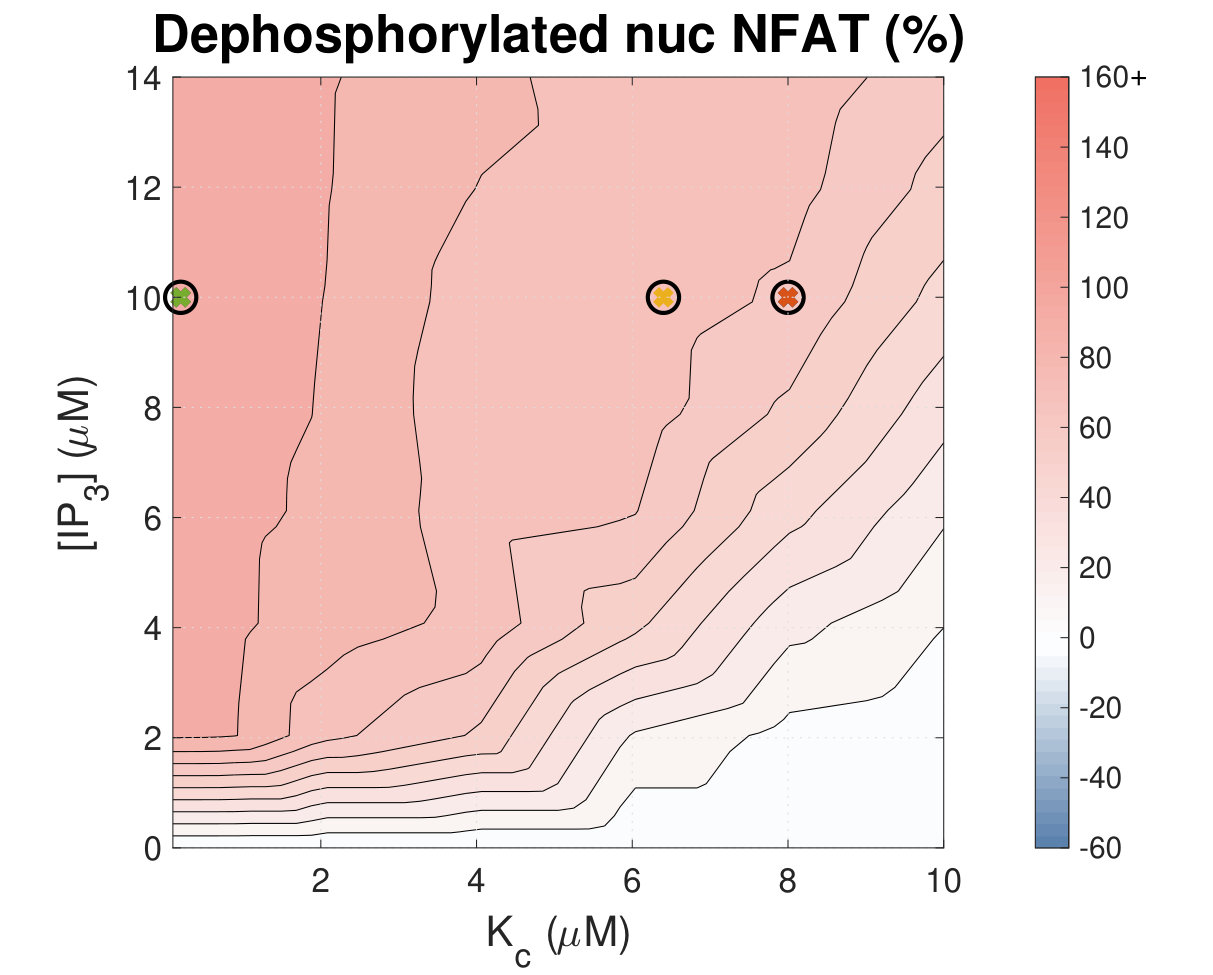 Figure 9
Figure 9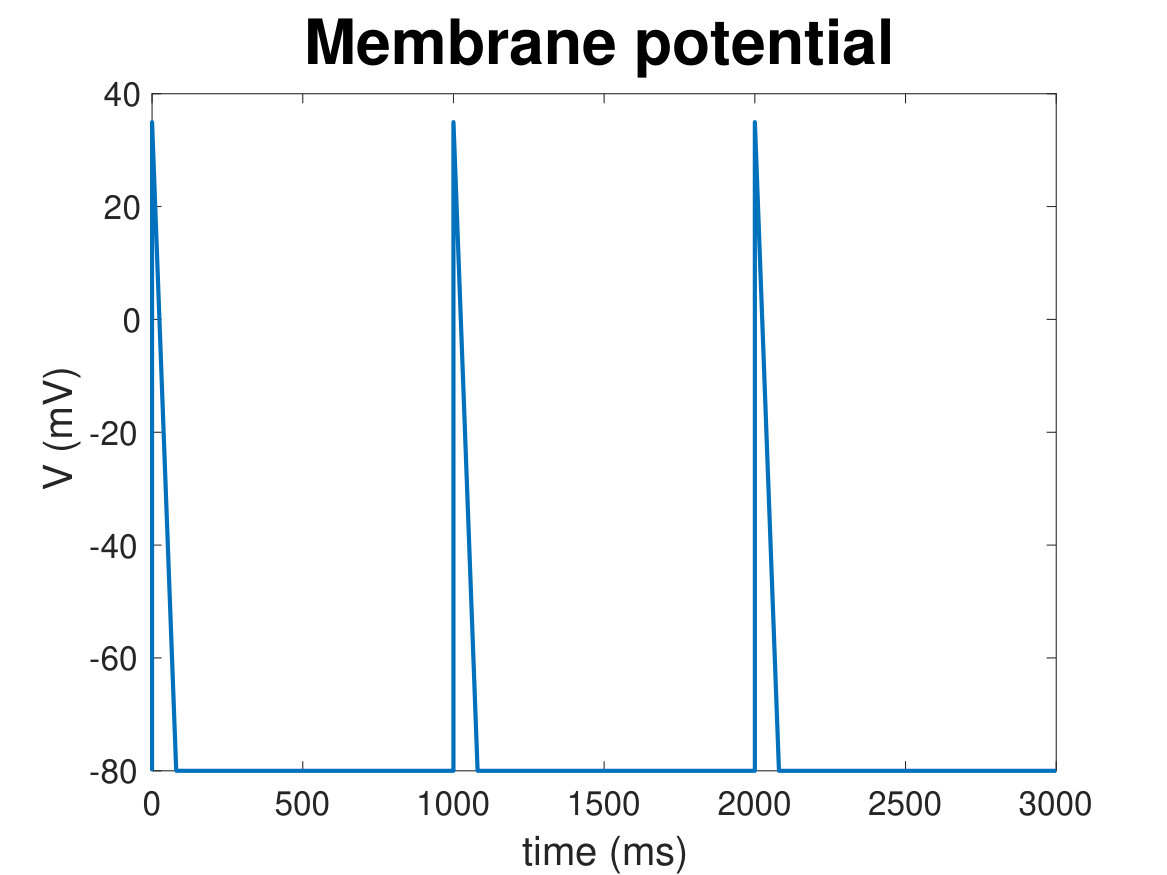 Figure 3
Figure 3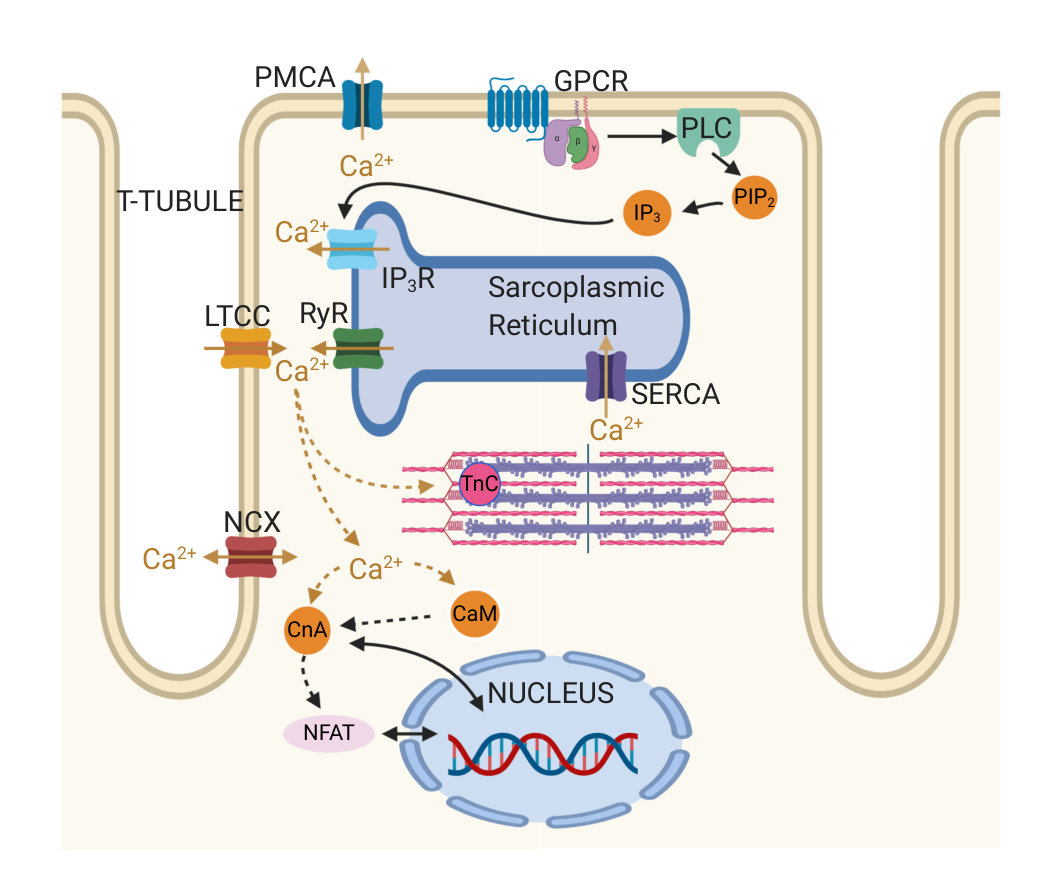 Figure 4
Figure 4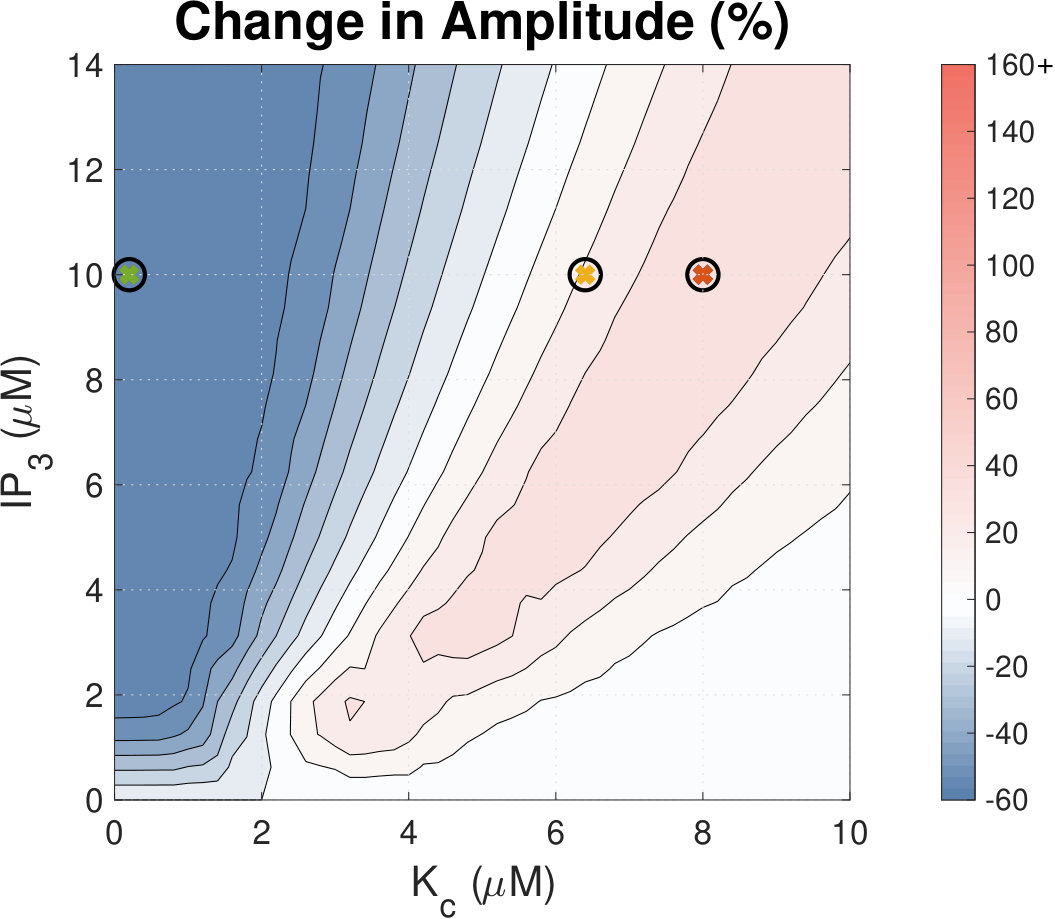 Figure 5
Figure 5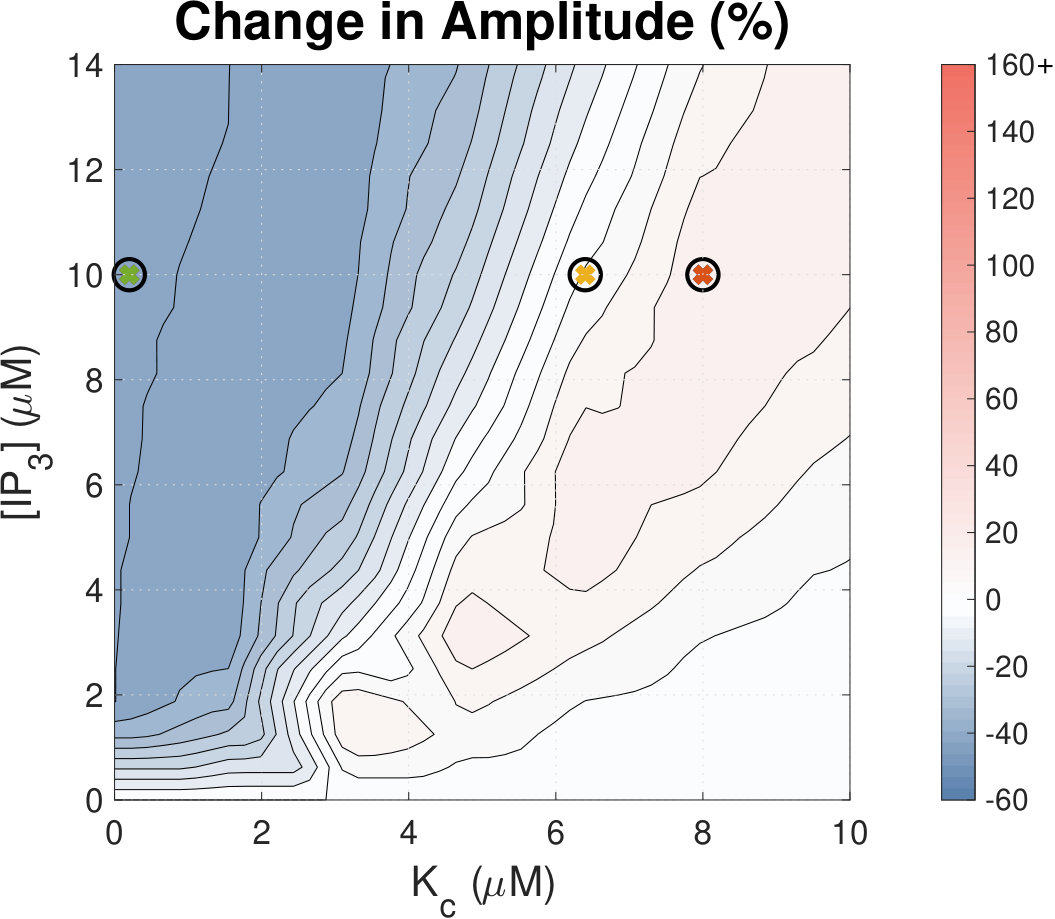 Figure 6
Figure 6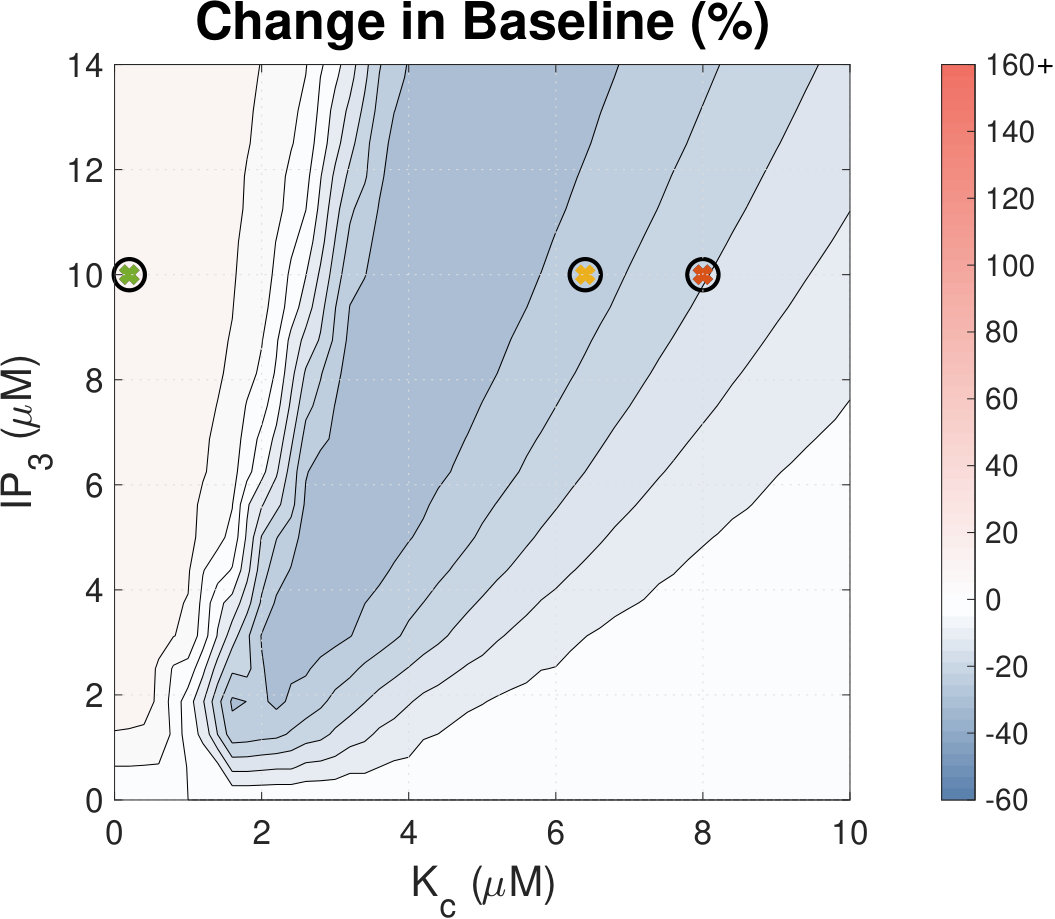 Figure 7
Figure 7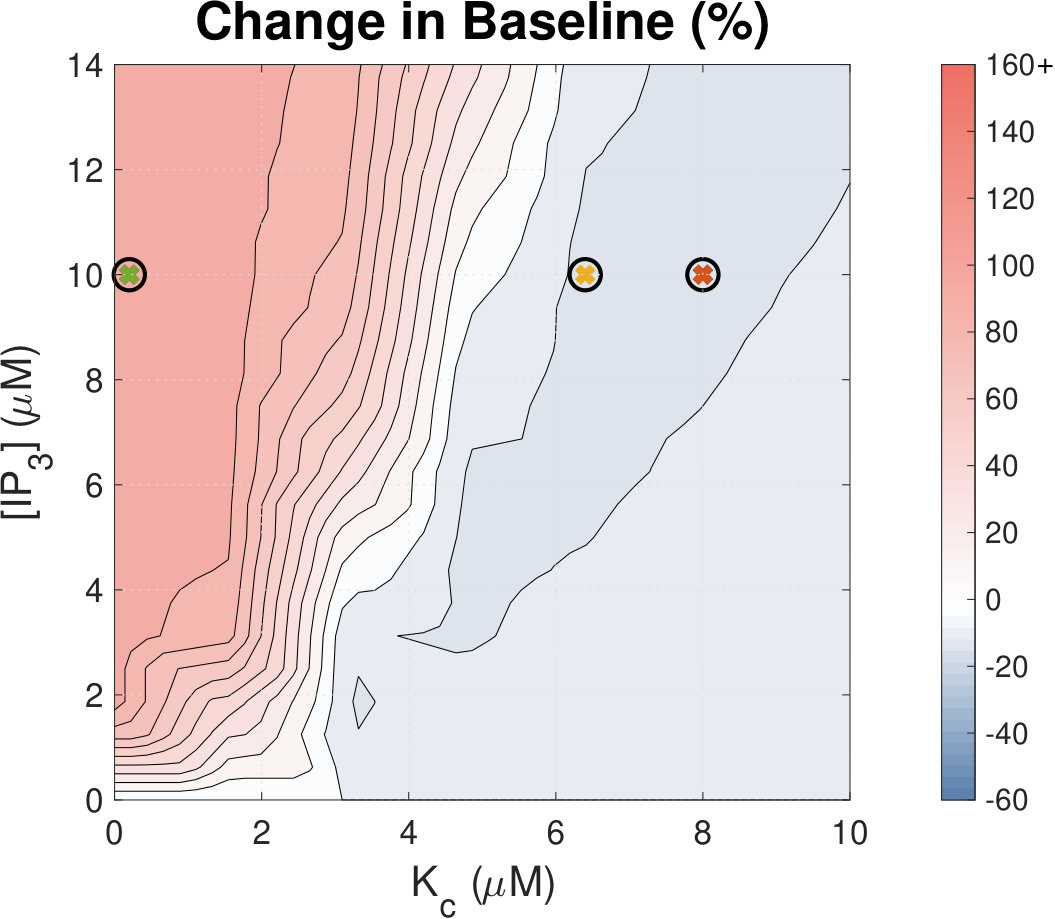 Figure 8
Figure 8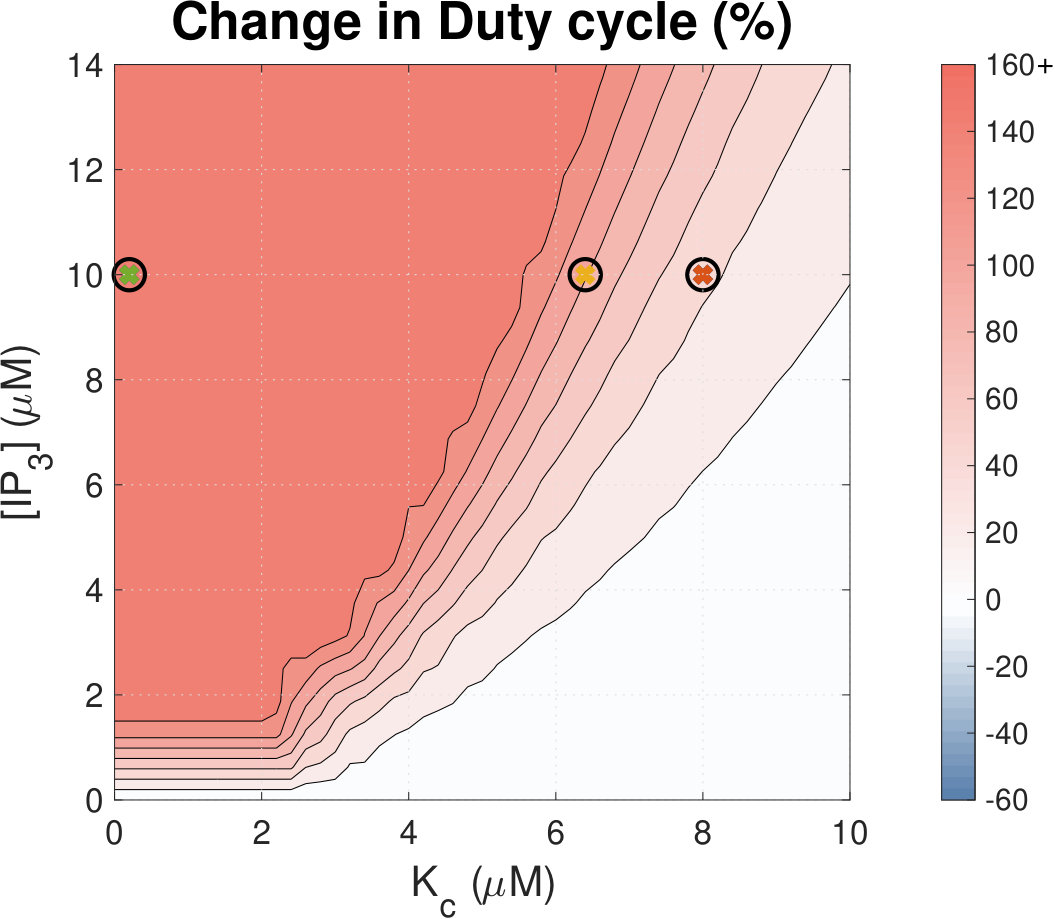 Figure 9
Figure 9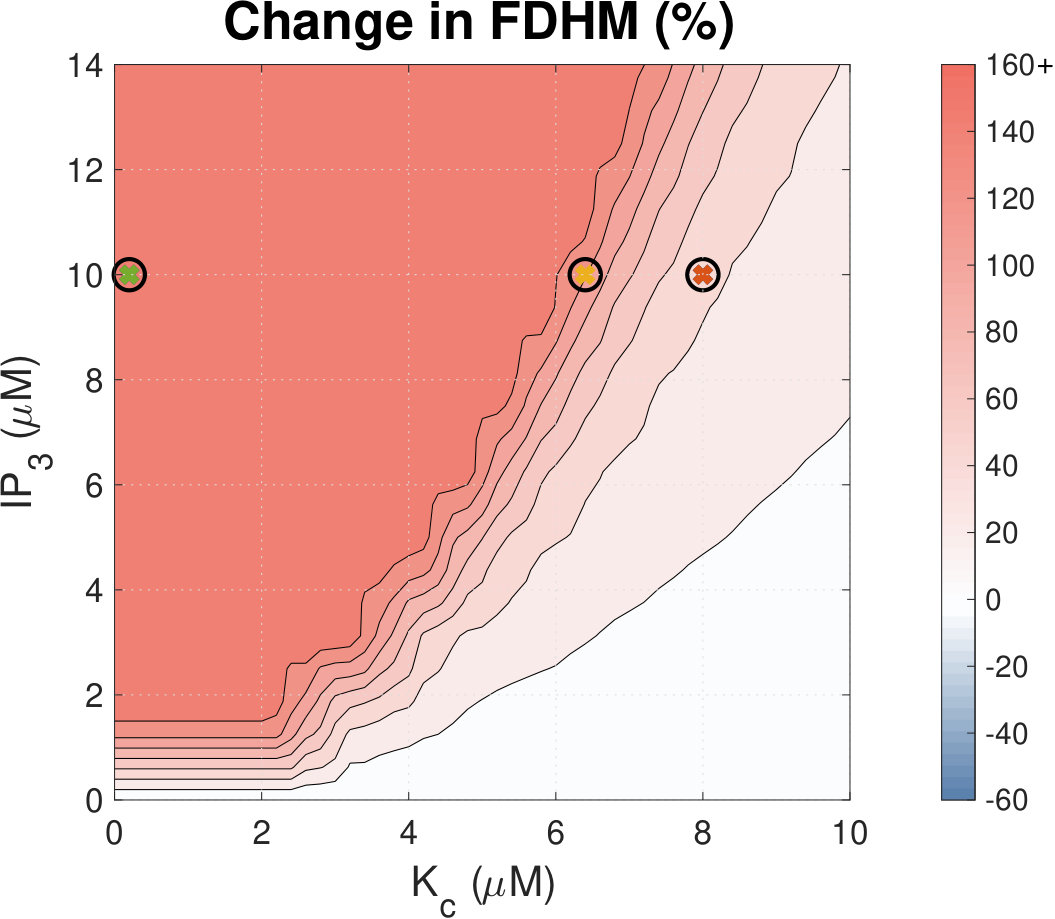 Figure 10
Figure 10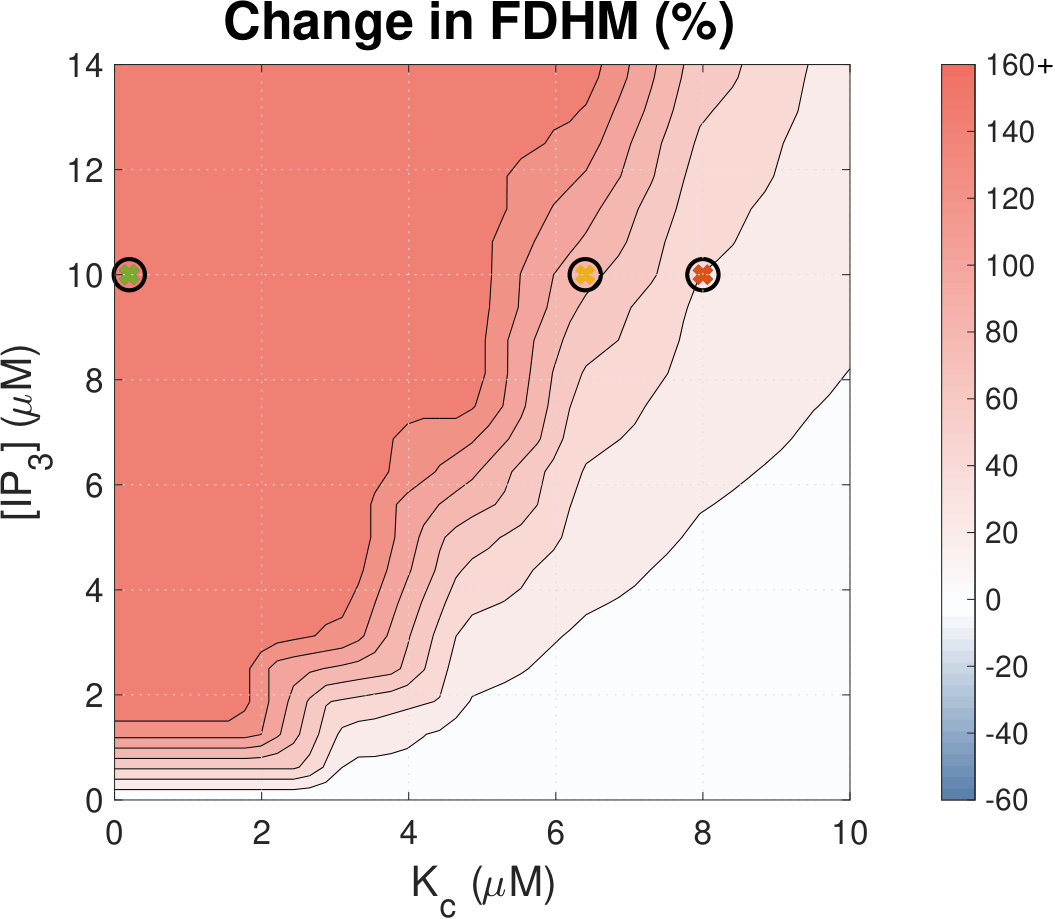 Figure 11
Figure 11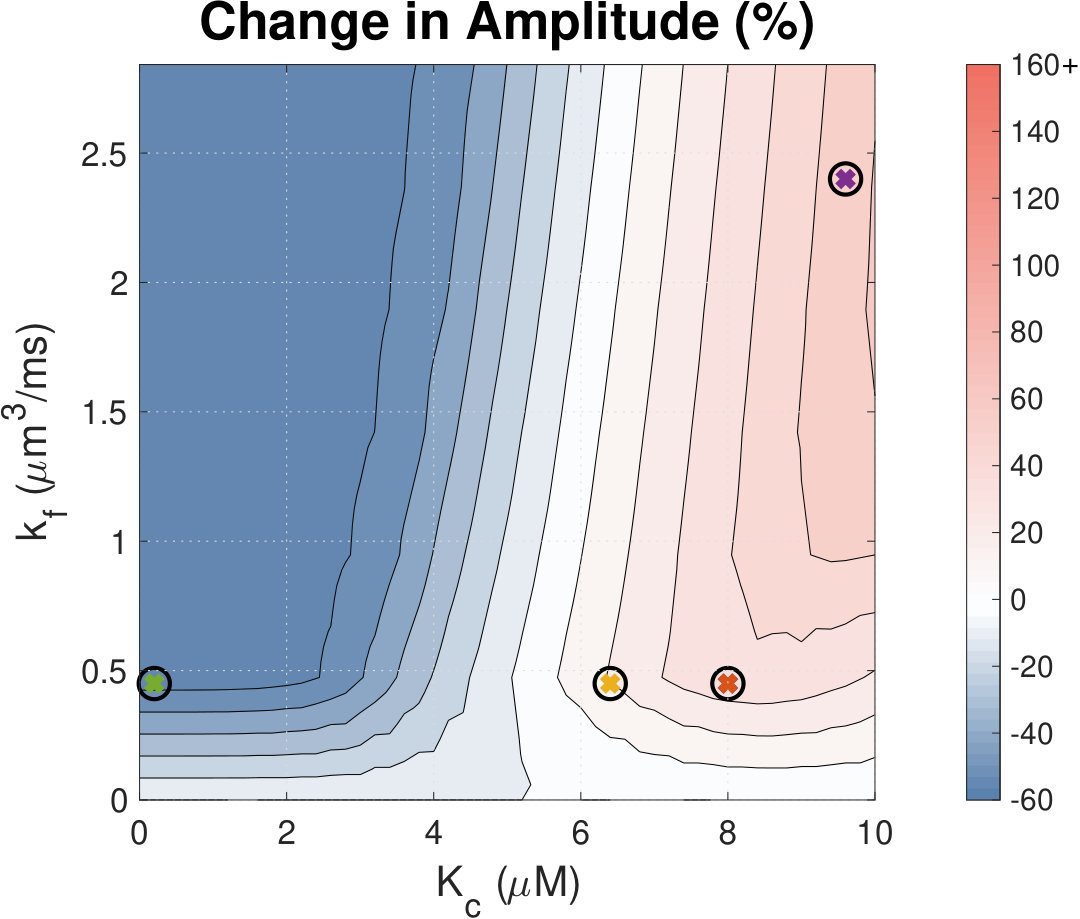 Figure 12
Figure 12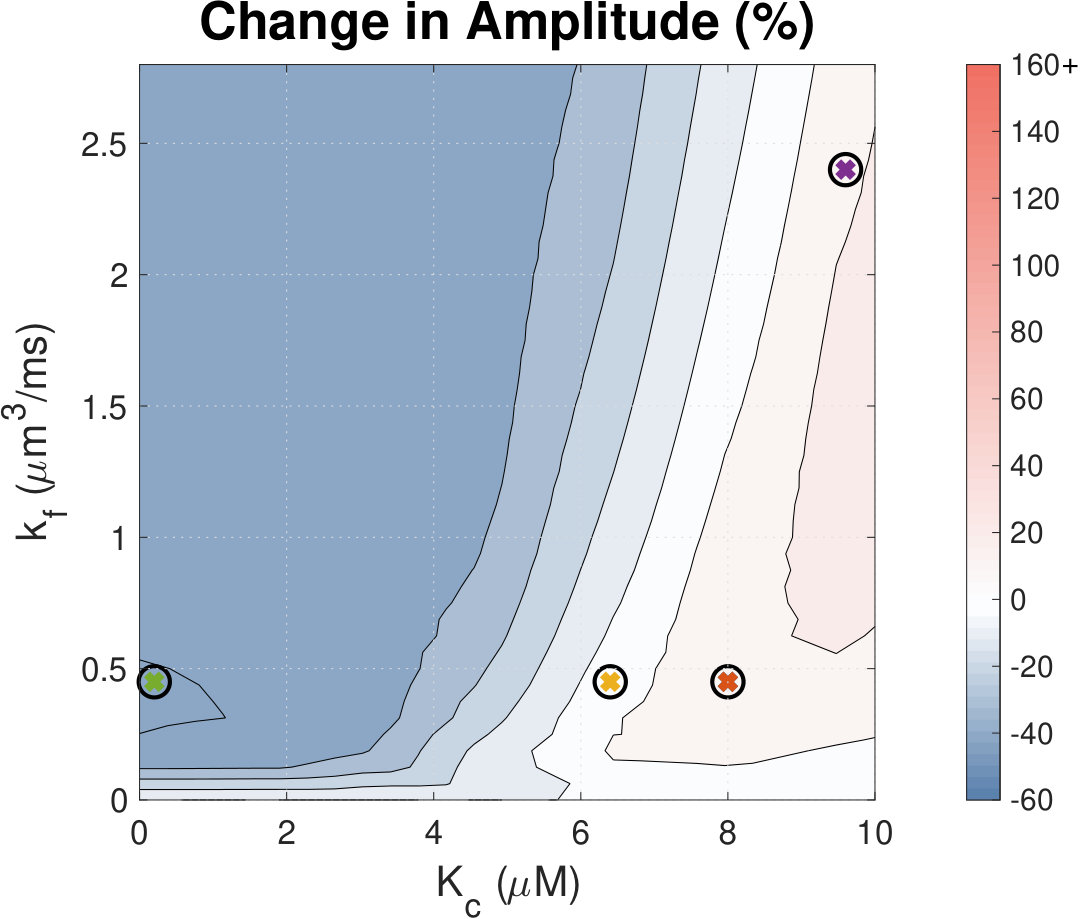 Figure 13
Figure 13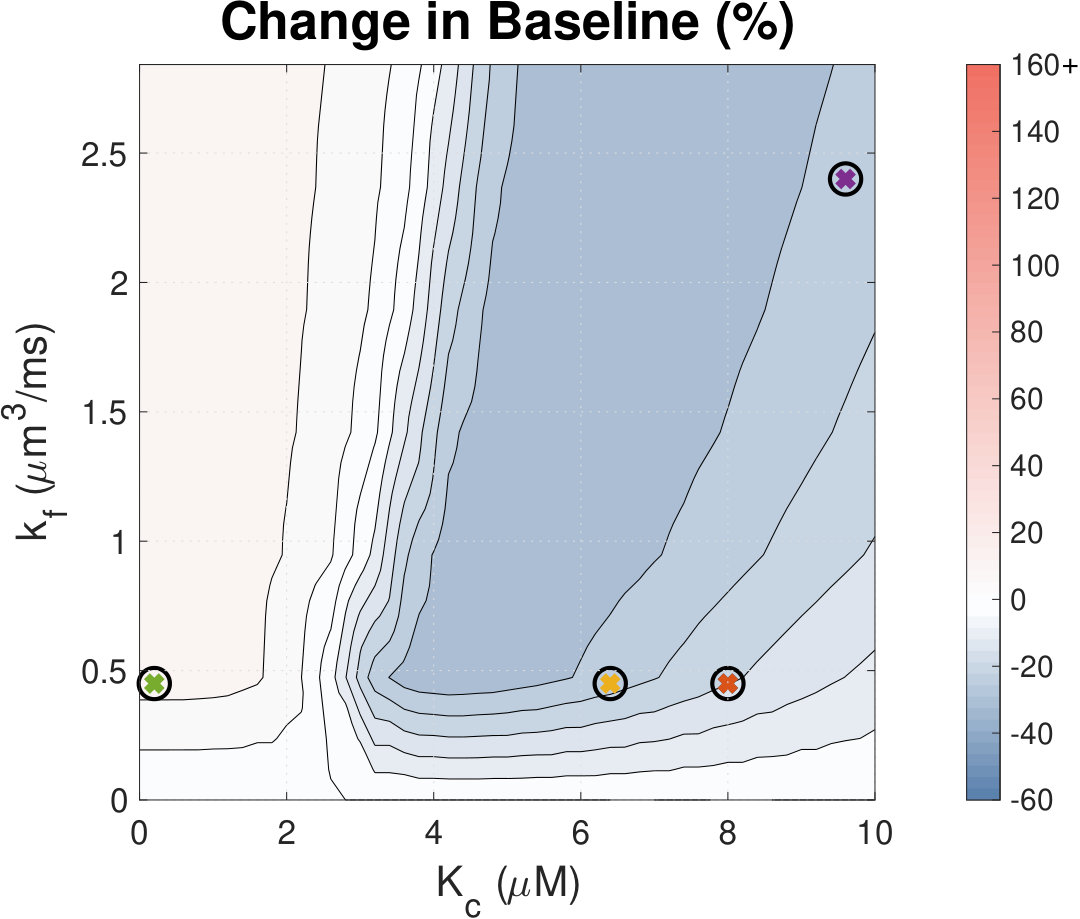 Figure 14
Figure 14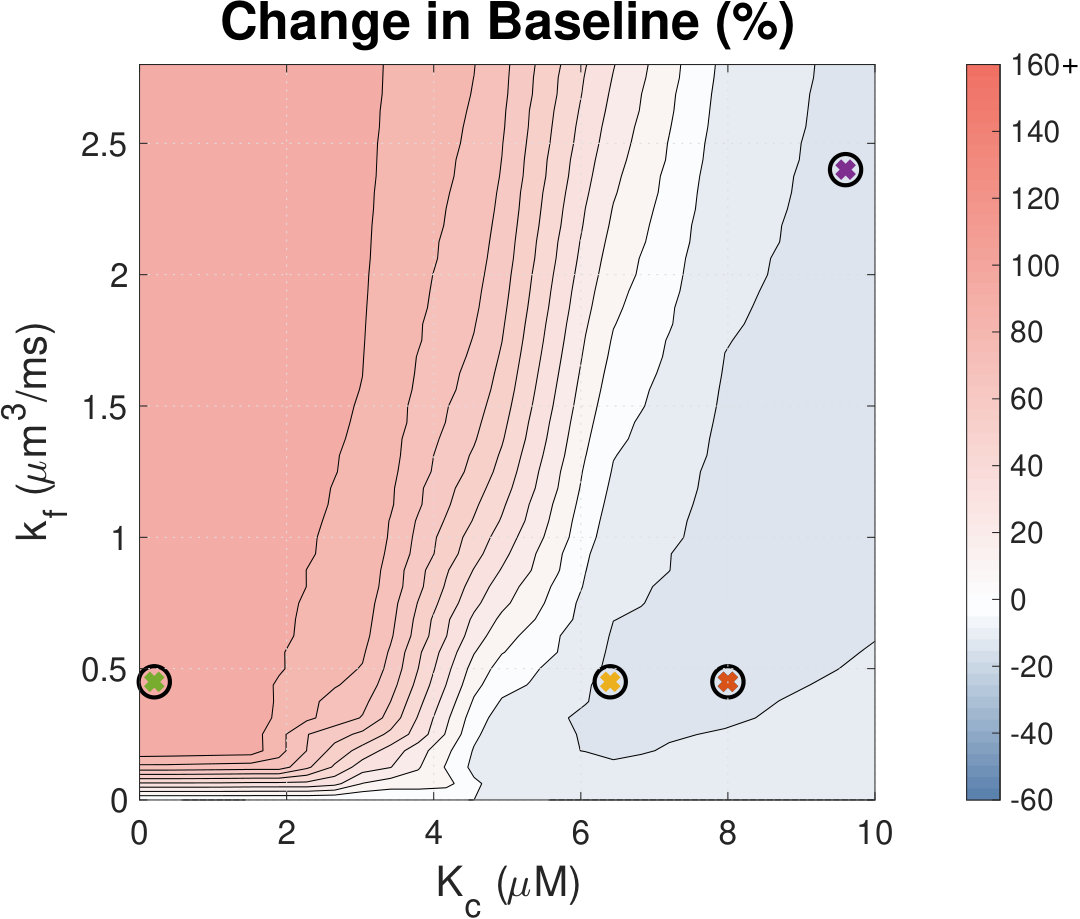 Figure 15
Figure 15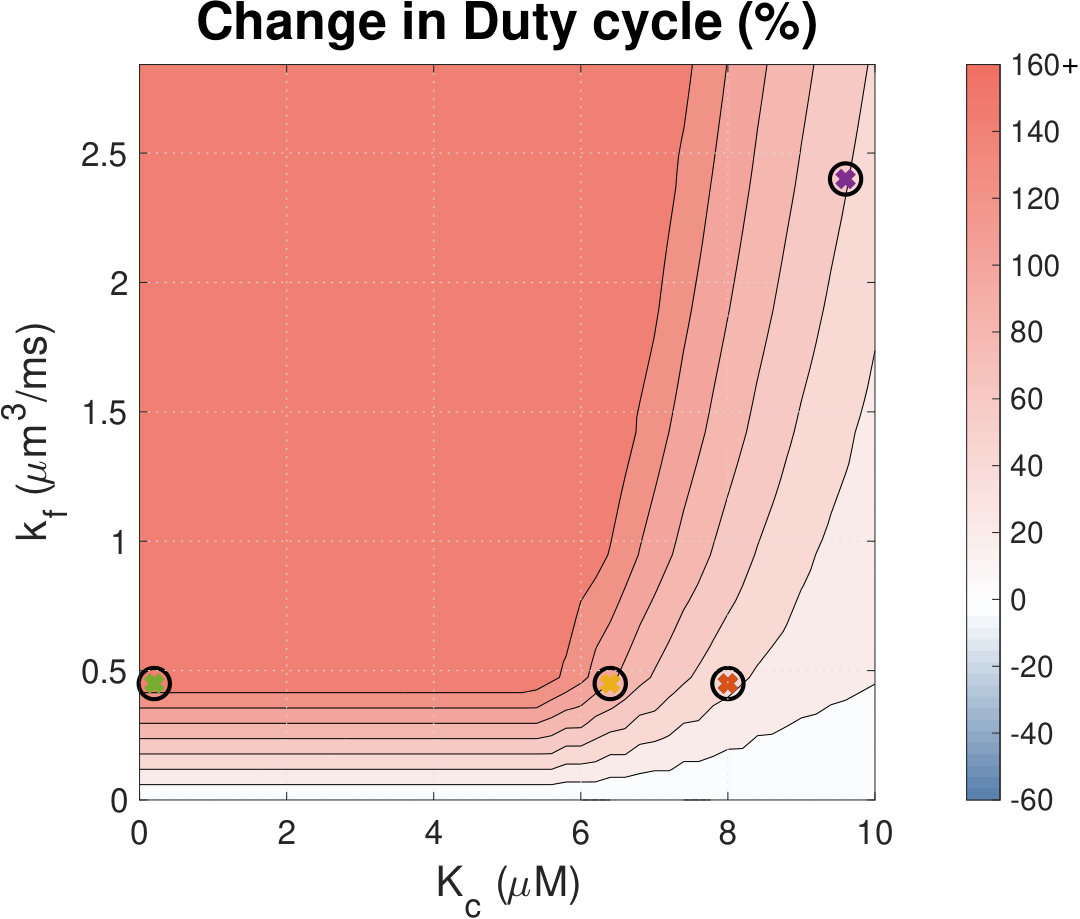 Figure 16
Figure 16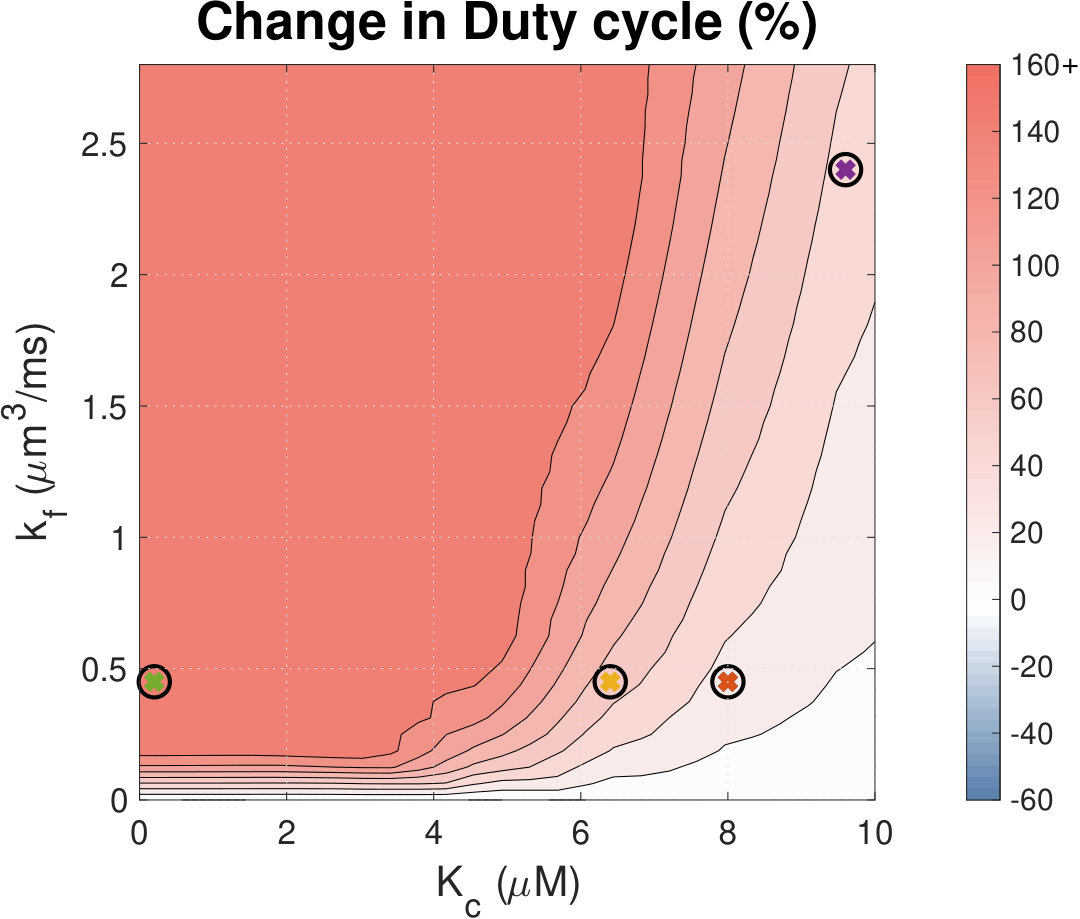 Figure 17
Figure 17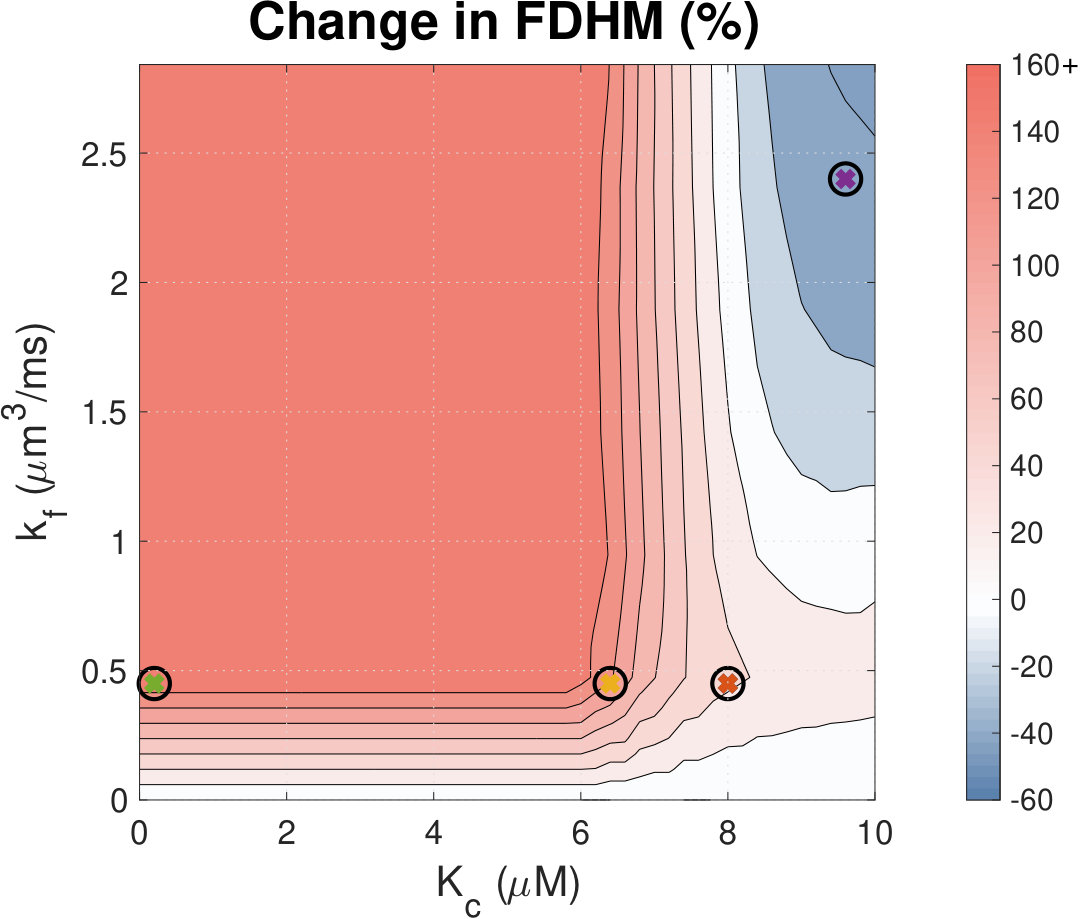 Figure 18
Figure 18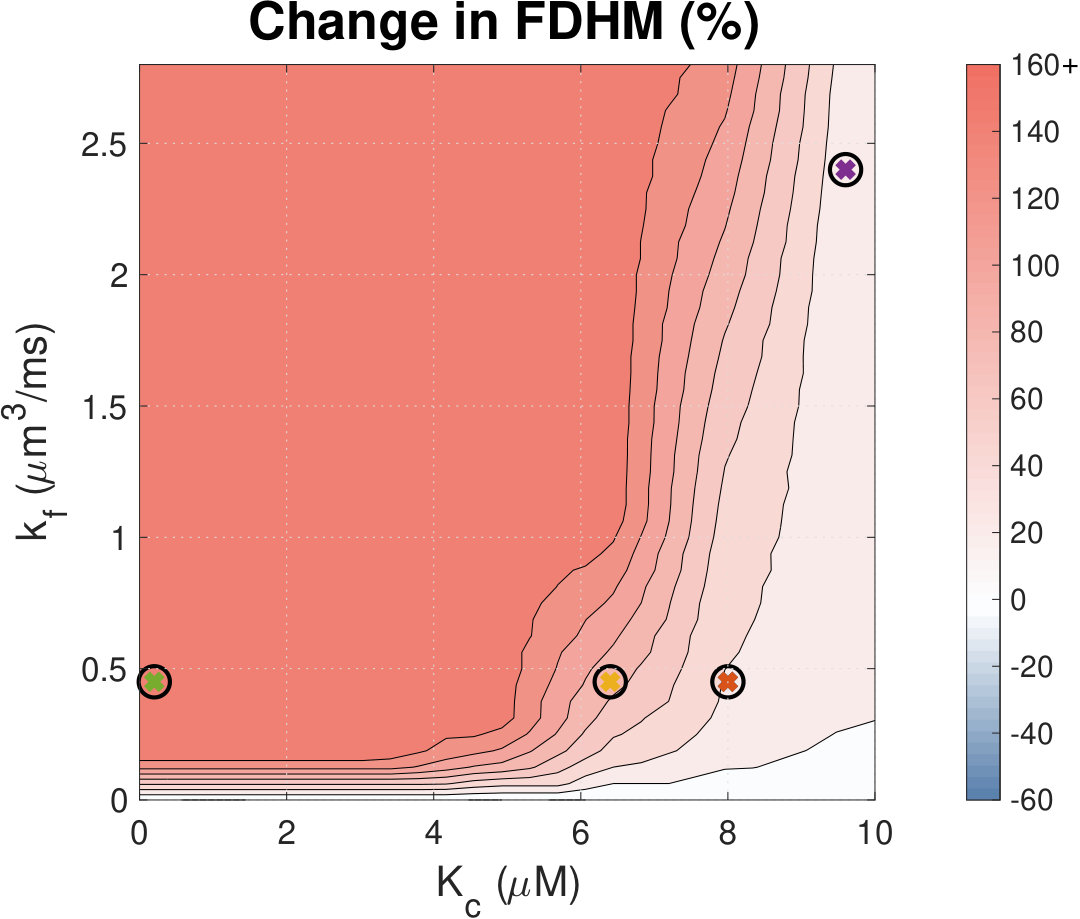 Figure 19
Figure 19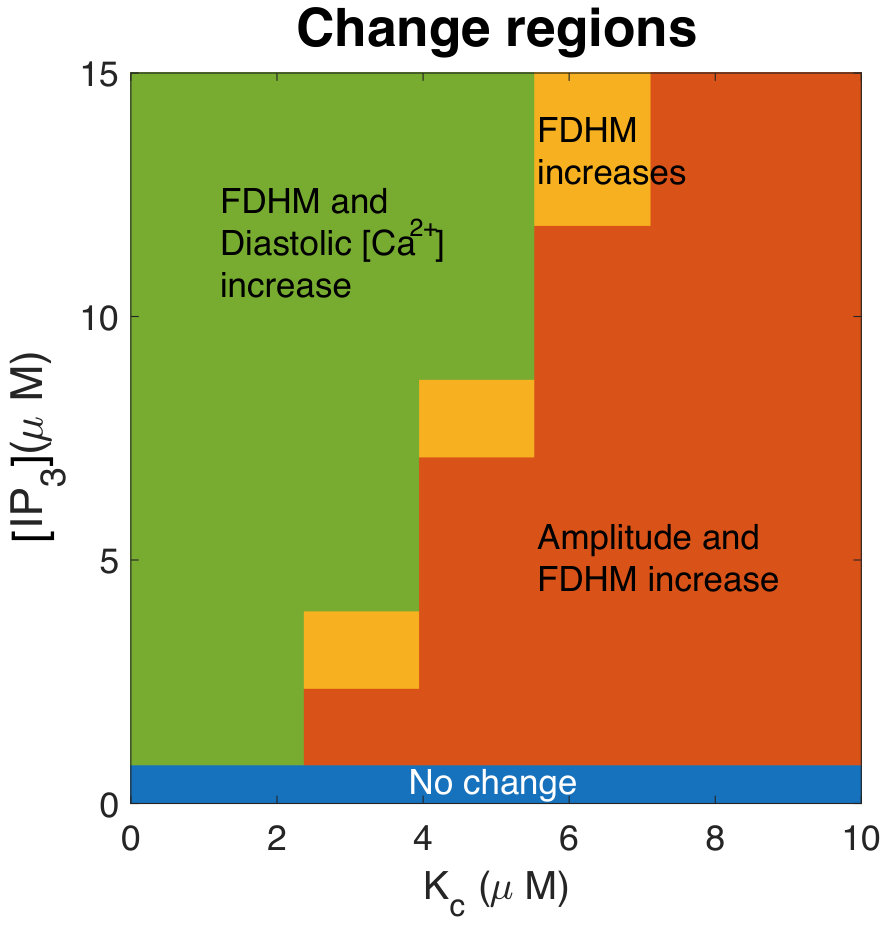 Figure 20
Figure 20 Figure 21
Figure 21 Figure 22
Figure 22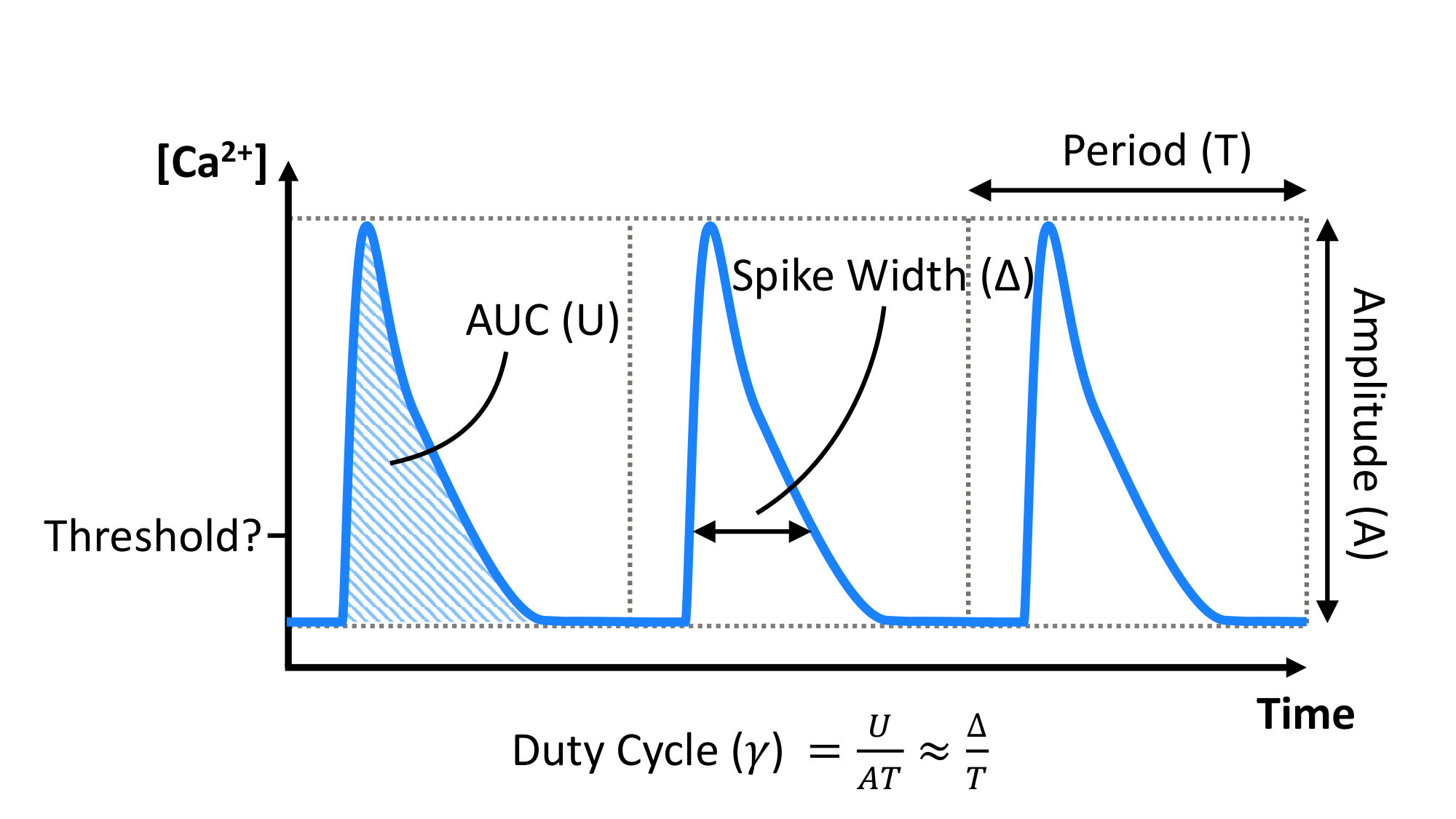 Figure 23
Figure 23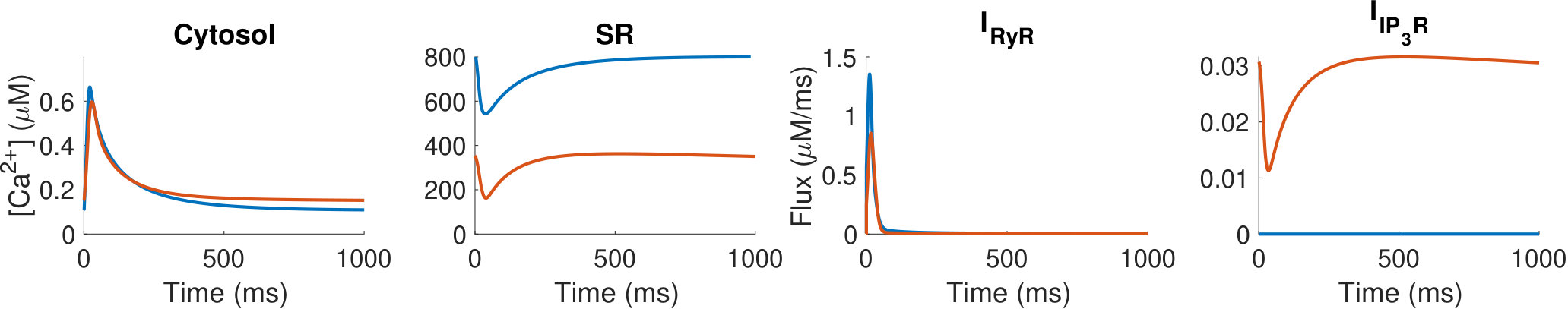 Figure 24
Figure 24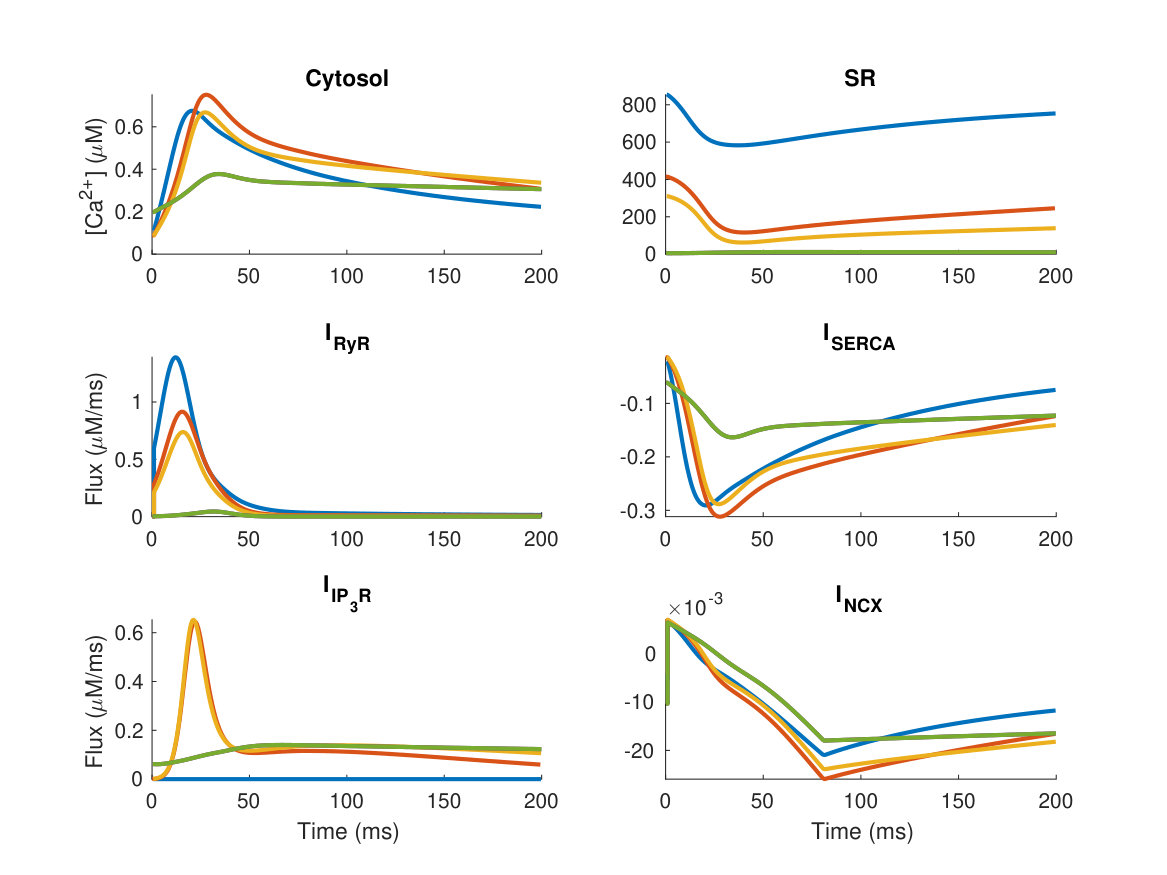 Figure 25
Figure 25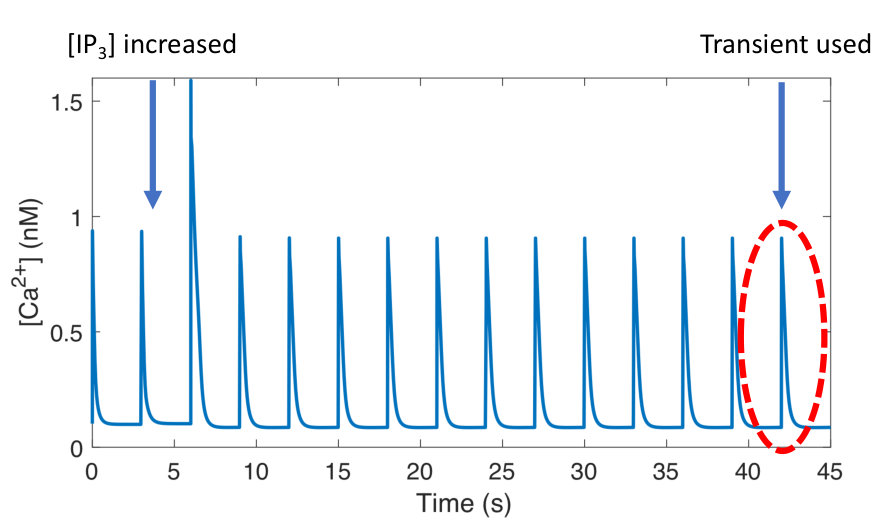 Figure 26
Figure 26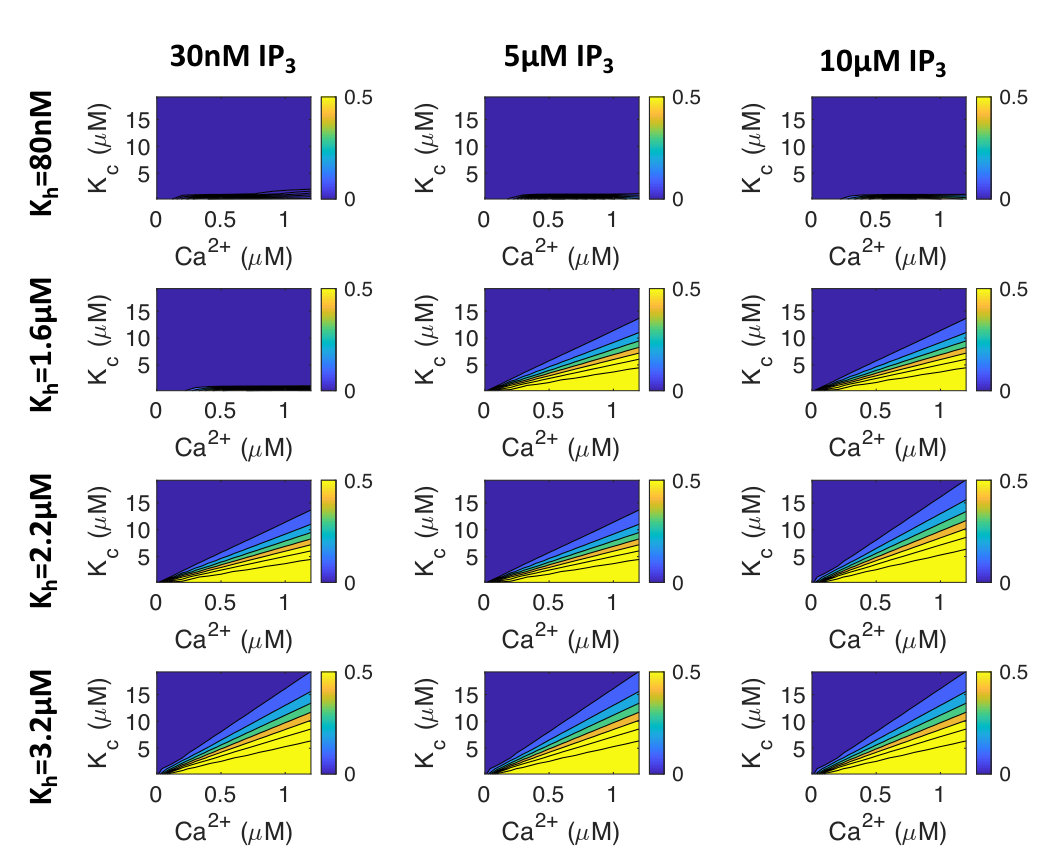 Figure 27
Figure 27| Cell State | IP3 | ET-1 | |
| Rat | \pbox2.5cmAmplitude: | ||
| Duration: | |||
| Basal Ca2+: | |||
| SCTs: | \pbox2.5cm r▲(proven_inositol_2006) r◆(harzheim_increased_2009) | ||
| – | |||
| r◆(harzheim_increased_2009) | |||
| r▲(proven_inositol_2006) r▲(harzheim_increased_2009) | \pbox3.8cm r▲(proven_inositol_2006) r◆(higazi_endothelin-1-stimulated_2009) r▲(harzheim_increased_2009) | ||
| – | |||
| r◆(harzheim_increased_2009) | |||
| r▲(proven_inositol_2006) r▲(harzheim_increased_2009) | |||
| Other species | \pbox2.5cmAmplitude: | ||
| Duration: | |||
| Basal Ca2+: | |||
| SCTs: | \pbox2.5cm m▲(signore_inositol_2013) m◆(escobar_role_2012) | ||
| – | |||
| m▲(escobar_role_2012) | |||
| – | \pbox2.5cm h▲(signore_inositol_2013) m▲(signore_inositol_2013) | ||
| – | |||
| m▲(signore_inositol_2013) b▲(domeier_ip3_2008) | |||
| h▲(signore_inositol_2013) m▲(signore_inositol_2013) | |||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Ca2+ release via IP3 receptors shapes the cytosolic Ca2+ transient for hypertrophic signalling in ventricular cardiomyocytes
Hilary Hunt
Systems Biology Laboratory, School of Mathematics and Statistics and Melbourne School of Engineering, University of Melbourne, Australia
Agnė Tilūnaitė
Systems Biology Laboratory, School of Mathematics and Statistics and Melbourne School of Engineering, University of Melbourne, Australia
Greg Bass
Systems Biology Laboratory, School of Mathematics and Statistics and Melbourne School of Engineering, University of Melbourne, Australia
Christian Soeller
Living Systems Institute, University of Exeter, UK
H. Llewelyn Roderick
Laboratory of Experimental Cardiology, Department of Cardiovascular Sciences, KU Leuven, Belgium
Vijay Rajagopal*222These authors contributed equally to the supervision of this work.
Cell Structure and Mechanobiology Group, Department of Biomedical Engineering, Melbourne School of Engineering, University of Melbourne, Australia
Edmund J. Crampin*
Systems Biology Laboratory, School of Mathematics and Statistics and Melbourne School of Engineering, University of Melbourne, Australia
ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, School of Chemical and Biomedical Engineering, University of Melbourne, Australia
Abstract
Calcium (Ca2+) plays a central role in mediating both contractile function and hypertrophic signalling in ventricular cardiomyocytes. L-type Ca2+ channels trigger release of Ca2+ from ryanodine receptors (RyRs) for cellular contraction, while signalling downstream of Gq coupled receptors stimulates Ca2+ release via inositol 1,4,5-trisphosphate receptors (IP3Rs), engaging hypertrophic signalling pathways. Modulation of the amplitude, duration, and duty cycle of the cytosolic Ca2+ contraction signal, and spatial localisation, have all been proposed to encode this hypertrophic signal. Given current knowledge of IP3Rs, we develop a model describing the effect of functional interaction (cross-talk) between RyR and IP3R channels on the Ca2+ transient, and examine the sensitivity of the Ca2+ transient shape to properties of IP3R activation. A key result of our study is that IP3R activation increases Ca2+ transient duration for a broad range of IP3R properties, but the effect of IP3R activation on Ca2+ transient amplitude is dependent on IP3 concentration. Furthermore we demonstrate that IP3-mediated Ca2+ release in the cytosol increases the duty cycle of the Ca2+ transient, the fraction of the cycle for which [Ca2+] is elevated, across a broad range of parameter values and IP3 concentrations. When coupled to a model of downstream transcription factor (NFAT) activation, we demonstrate that there is a high correspondence between the Ca2+ transient duty cycle and the proportion of activated NFAT in the nucleus. These findings suggest increased cytosolic Ca2+ duty cycle as a plausible mechanism for IP3-dependent hypertrophic signalling via Ca2+-sensitive transcription factors such as NFAT in ventricular cardiomyocytes.
\papertype
Article
\corrauthor[*][email protected]; [email protected]
{sigstatement}
Many studies have identified a role for IP3R-mediated Ca2+ signalling in cardiac hypertrophy, however the mechanism by which this signal is communicated within the cardiomyocyte remains unclear. We present a mathematical model of functional interactions between RyR and IP3R channels. We show that IP3-mediated Ca2+ release is capable of providing a modest increase to the duty cycle of the calcium signal, which has been shown experimentally to lead to NFAT activation, and hence hypertrophic signalling. Through a parameter sensitivity analysis we demonstrate that the duty cycle is increased with IP3 over a broad parameter regime, indicating that this mechanism is robust, and we show that an increase in Ca2+ duty cycle increases nuclear NFAT activation. These findings suggest a plausible mechanism for IP3R-dependent hypertrophic signalling in cardiomyocytes.
Introduction
Calcium is a universal second messenger that plays a role in controlling many cellular processes across a wide variety of cell types; ranging from fertilisation, cell contraction, and cell growth, to cell death (berridge_calcium_2003, clapham_calcium_2007). Precisely how Ca2+ fulfills each of these roles while also ensuring signal specificity remains unclear in many cases. Ca2+ can be used to transmit signals in a variety of ways. Signal localisation, and amplitude and frequency modulation have been widely explored (berridge_am_1997, berridge_calcium_2006, bootman_update_2009), however, mechanisms for information encoding in the cumulative signal (i.e. area under the curve (AUC), proportional-integral-derivative (PID) controller, or duty cycle (DC)) have also been proposed (purvis_encoding_2013, uzhachenko_computational_2016, hannanta-anan_optogenetic_2016). Determining which method of information encoding is relevant to a specific signalling pathway requires determining what type of signal encoding the system is capable of, and whether the downstream effector of the signal is capable of temporal signal integration, high or low pass filtering, or threshold filtering.
In cardiac myocytes, discrete encoding of multiple Ca2+-mediated signals is particularly pertinent because of the essential and continuous role Ca2+ plays in excitation-contraction coupling (ECC). Of particular significance is the involvement of Ca2+ in hypertrophic growth signaling. How Ca2+ can communicate a signal in the hypertrophic signalling pathway concurrent with the cytosolic Ca2+ fluxes that drive cardiac muscle contraction is still largely unresolved (roderick_calcium_2007, hohendanner_cytosolic_2015). Understanding this mechanism is important as pathological hypertrophic remodelling is a precursor of heart failure and a common final pathway of cardiovascular diseases including hypertension and coronary disease (jefferies_mechanisms_2018, tham_pathophysiology_2015, gilbert_calcium_2019).
During each heartbeat, on depolarisation of the membrane Ca2+ enters the cell via L-type Ca2+ channels (LTCC), triggering larger Ca2+ release from the sarcoplasmic reticulum (SR) via ryanodine receptors (RyRs), which then induces contraction. The activation of Ca2+ release via RyRs by the Ca2+ arising via LTCCs is known as calcium-induced calcium release (CICR), and results in a 10-fold increase in cytosolic Ca2+ concentration (relative to resting Ca2+ concentration of 100 nM). Sarco-endoplasmic reticulum Ca2+ pumps (SERCA) and other Ca2+ sequestration mechanisms subsequently withdraw the released Ca2+ back into the SR and out of the cytosol (hinch_simplified_2004, vierheller_multiscale_2015) reverting the cell to its relaxed state. Ca2+ also plays a central role in hypertrophic signalling. Hypertrophic stimuli such as endothelin-1 (ET-1) bind to G-protein-coupled receptors at the cell membrane to stimulate generation of the intracellular signalling molecule inositol 1,4,5-trisphosphate (IP3). After IP3 binds to and activates its cognate receptor, inositol 1,4,5-trisphosphate receptors (IP3R), on the SR and nuclear envelope, Ca2+ is released into the cytosol and nucleus respectively (higazi_endothelin-1-stimulated_2009, harzheim_increased_2009) (see Figure 1). This Ca2+ signal arising from IP3Rs has been shown in multiple mammalian species to produce a distinct Ca2+ signal that, through activation of pro-hypertrophic pathways including those involving NFAT, induces hypertrophy within cardiomyocytes. (higazi_endothelin-1-stimulated_2009, nakayama_ip3_2010, rinne_isoform-_2010).
In healthy adult rat ventricular myocytes (ARVMs), various effects of IP3 on global Ca2+ transients associated with ECC have been described, summarised in Table Introduction. While application of GPCR agonists that stimulate IP3 generation produces robust effects on ECC associated IP3 transients and contraction, the direct contribution of IP3 to these actions varies between studies (signore_inositol_2013, proven_inositol_2006, harzheim_increased_2009, domeier_ip3_2008, ljubojevic_early_2014, olivares-florez_nuclear_2018). For example, in rabbit the effect of ET-1 on Ca2+ transient amplitude is sensitive to the IP3R inhibitor 2-APB (domeier_ip3_2008), whereas in healthy rats IP3R inhibition with 2-APB was without effect (smyrnias_contractile_2018). In mice 2-APB abrogated an increase in ECC associated Ca2+ transients brought about by AngII (olivares-florez_nuclear_2018). Responses have also been variable when IP3 was directly applied to cardiac myocytes. In healthy rat, IP3 produced no or a modest effect on Ca2+ transient amplitude (proven_inositol_2006, harzheim_increased_2009), whereas in rabbit (domeier_ip3_2008) a more substantial effect was observed. These differences in the effect of IP3 have been ascribed in part to the greater dependence of rat myocytes on SR Ca2+ release to the Ca2+ transient than rabbit myocytes(domeier_ip3_2008). Notably, both ET-1 and IP3 elicit arrhythmogenic effects whereby they promote the generation of spontaneous calcium transients, manifest as a prolonged Ca2+ transient with additional peaks, and they increase the frequency of Ca2+ sparks (proven_inositol_2006, harzheim_increased_2009, domeier_ip3_2008, nakayama_ip3_2010). A more profound role for IP3 signalling is observed in hypertrophic ventricular myocytes, with ECC-associated Ca2+ transients of greater amplitude reported. Underlying these effects, IP3R expression is elevated in hypertrophy (harzheim_elevated_2010). Hence, a question remains as to what independent effect IP3R activation has on the cytosolic Ca2+ transient in healthy ventricular cardiac myocytes.