First-principles anharmonic vibrational study of the structure of calcium silicate perovskite under lower mantle conditions
Joseph C. A. Prentice, Ryo Maezono, R. J. Needs

TL;DR
This study uses first-principles anharmonic vibrational calculations to investigate the stability of calcium silicate perovskite's crystal structure under lower mantle conditions, confirming the cubic form's stability.
Contribution
It provides the first comprehensive anharmonic vibrational analysis of CaSiO₃ structures under mantle conditions, supporting the stability of the cubic phase.
Findings
Cubic CaSiO₃ is most stable in the lower mantle.
Anharmonic effects do not alter the stability of the cubic structure.
Thermal expansion does not destabilize the cubic phase.
Abstract
Calcium silicate perovskite (CaSiO) is one of the major mineral components of the lower mantle, but has been the subject of relatively little work compared to the more abundant Mg-based materials. One of the major problems related to CaSiO that is still the subject of research is its crystal structure under lower mantle conditions - a cubic Pmm structure is accepted in general, but some have suggested that lower-symmetry structures may be relevant. In this paper, we use a fully first-principles vibrational self-consistent field method to perform high accuracy anharmonic vibrational calculations on several candidate structures at a variety of points along the geotherm near the base of the lower mantle to investigate the stability of the cubic structure and related distorted structures. Our results show that the cubic structure is the most stable throughout the lower…
Click any figure to enlarge with its caption.
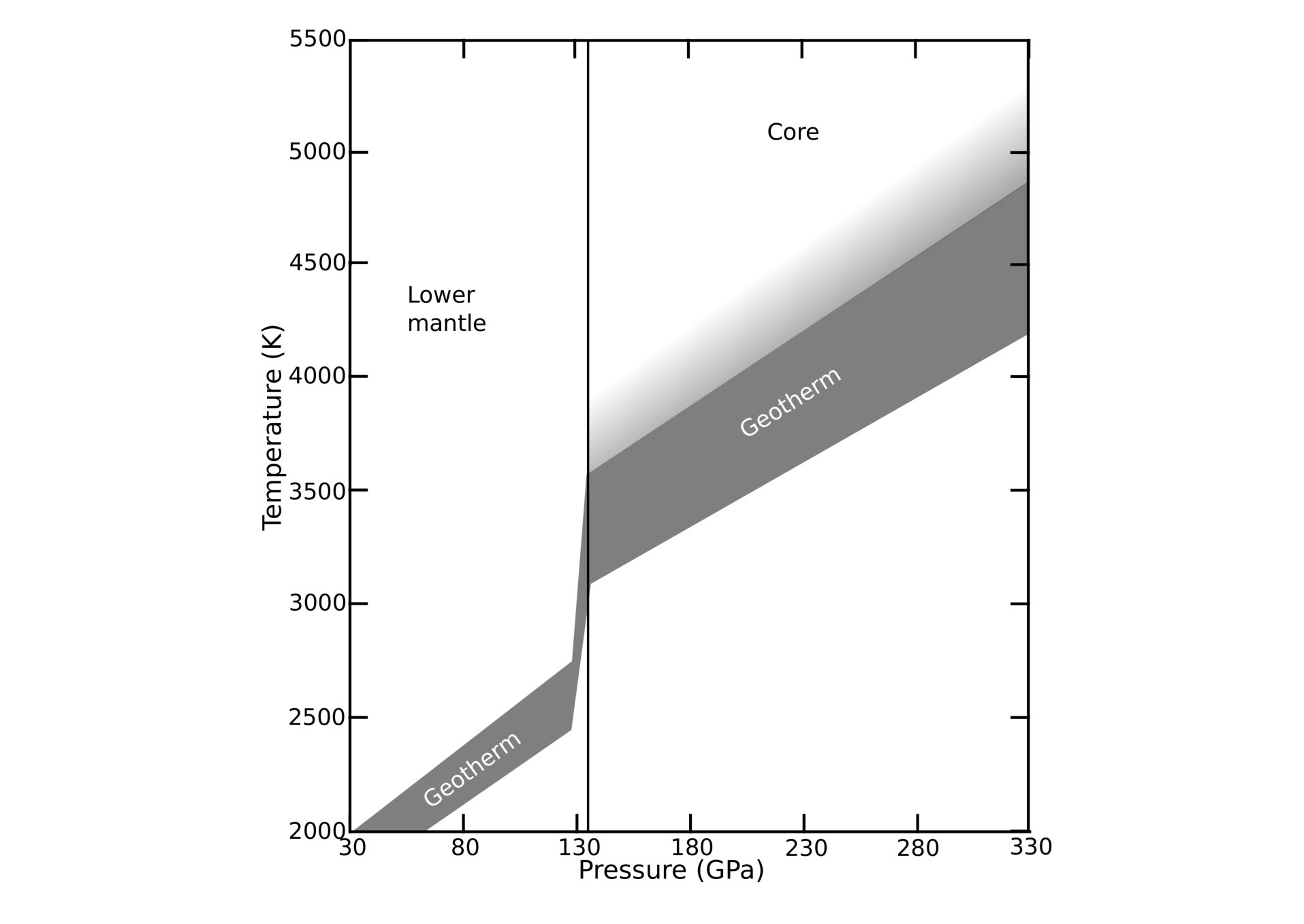 Figure 1
Figure 1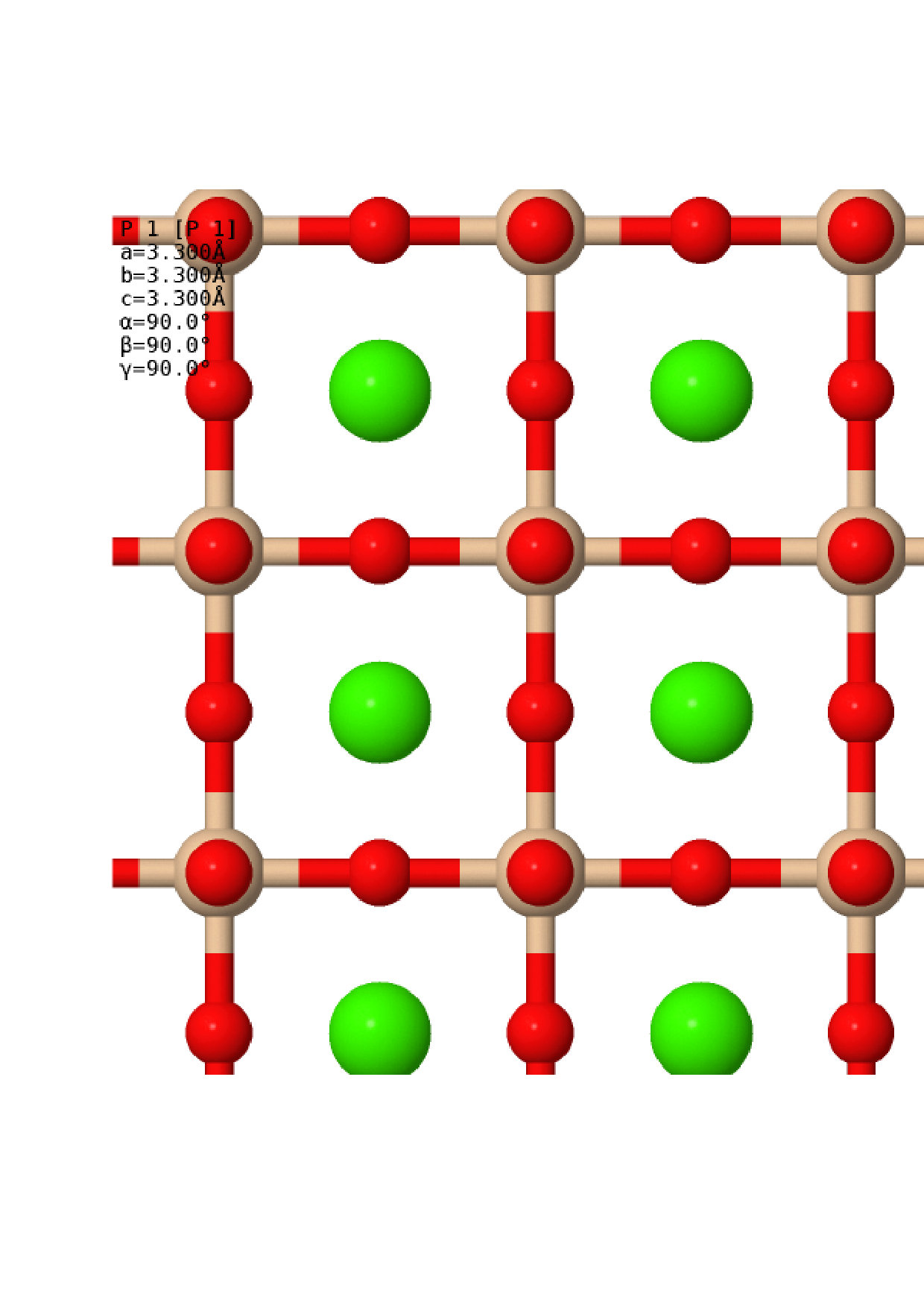 Figure 2
Figure 2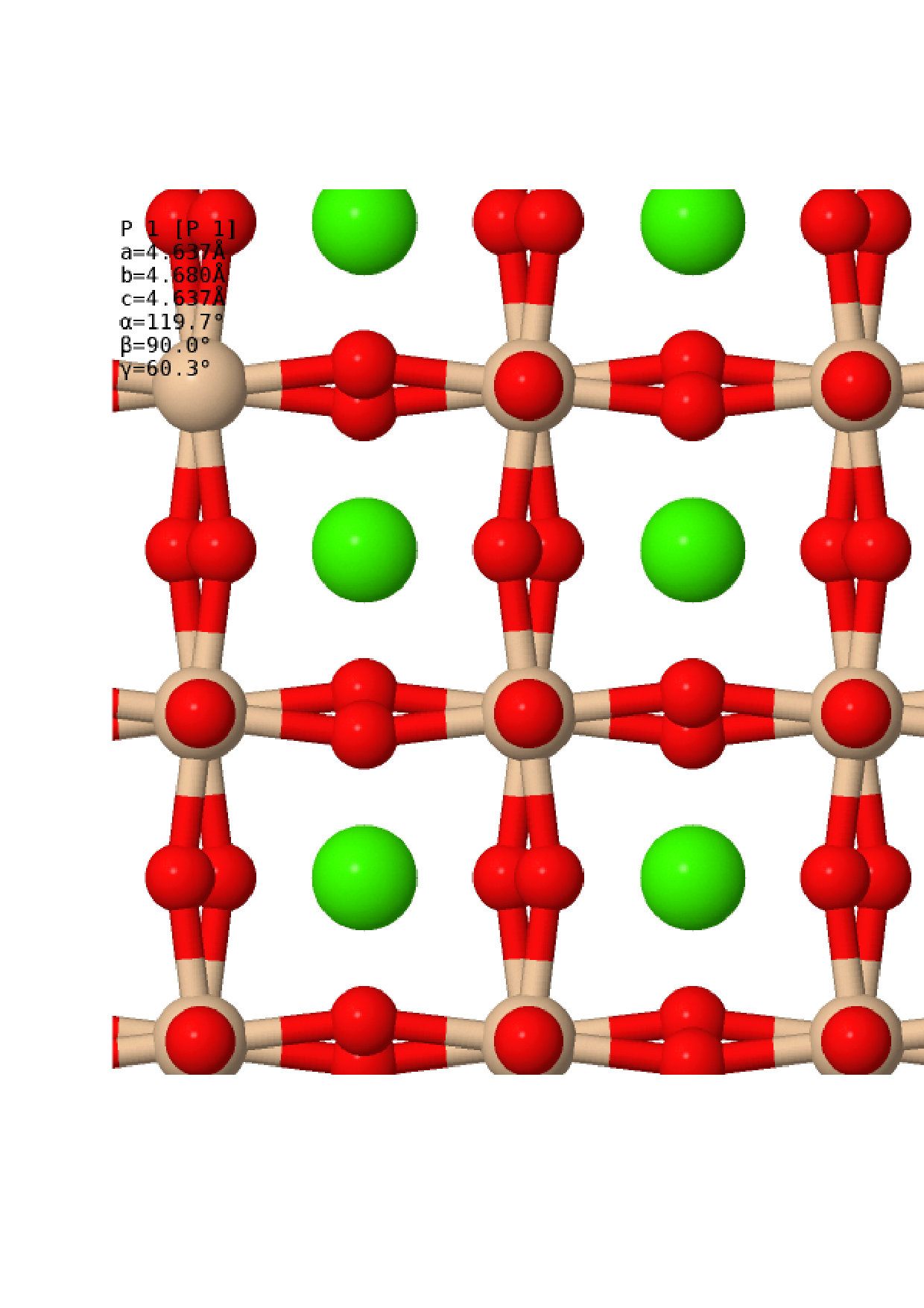 Figure 2
Figure 2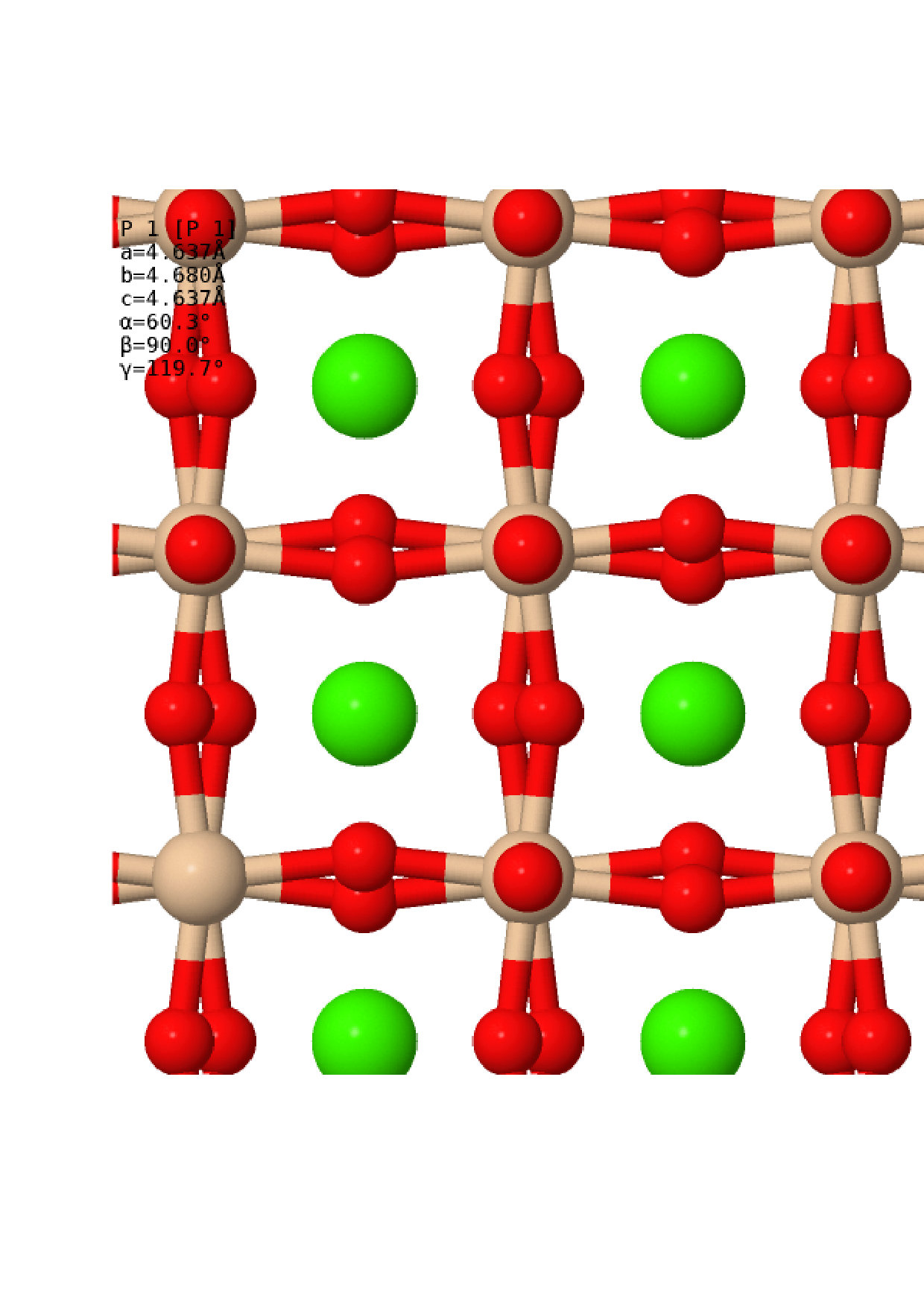 Figure 2
Figure 2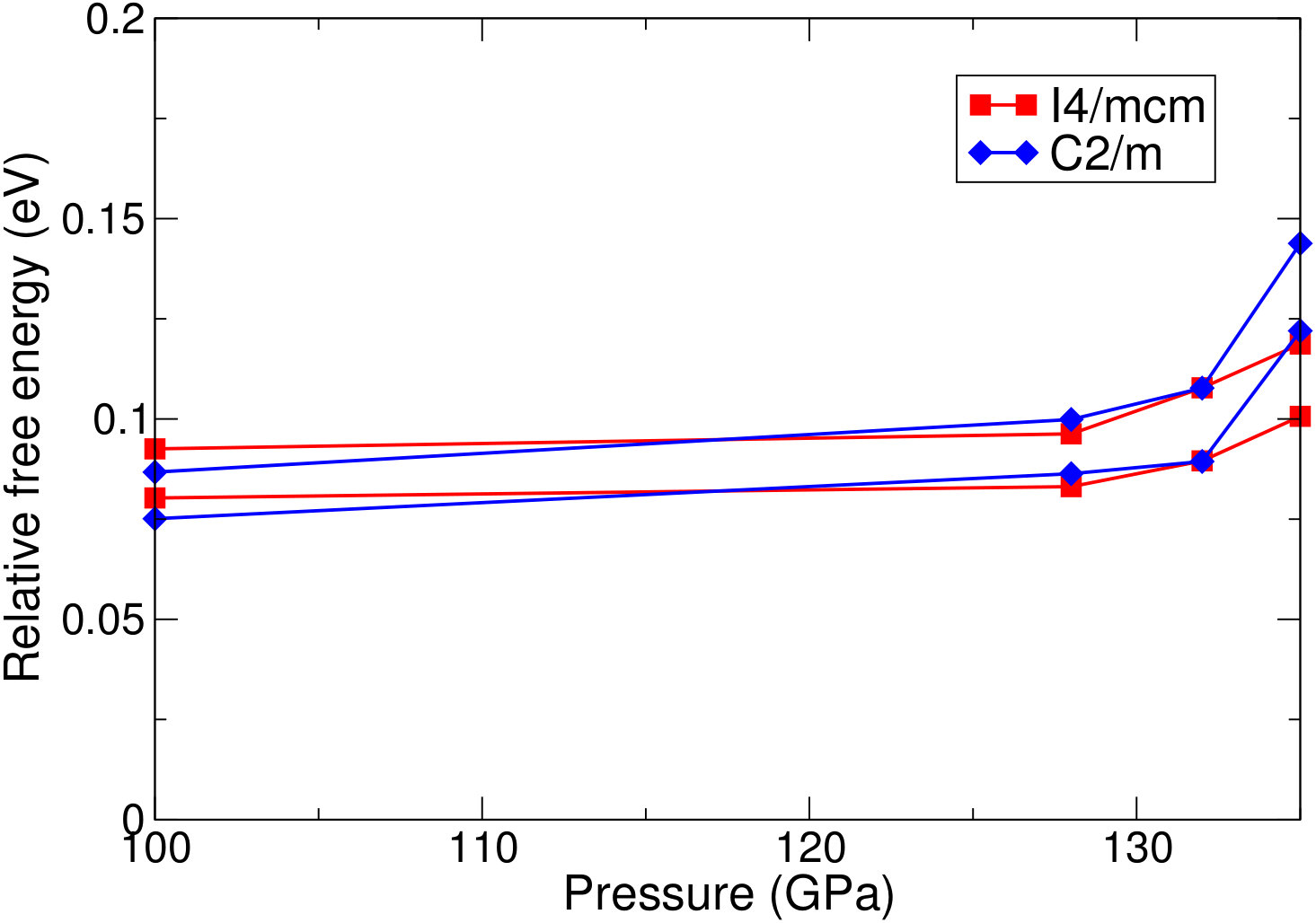 Figure 3
Figure 3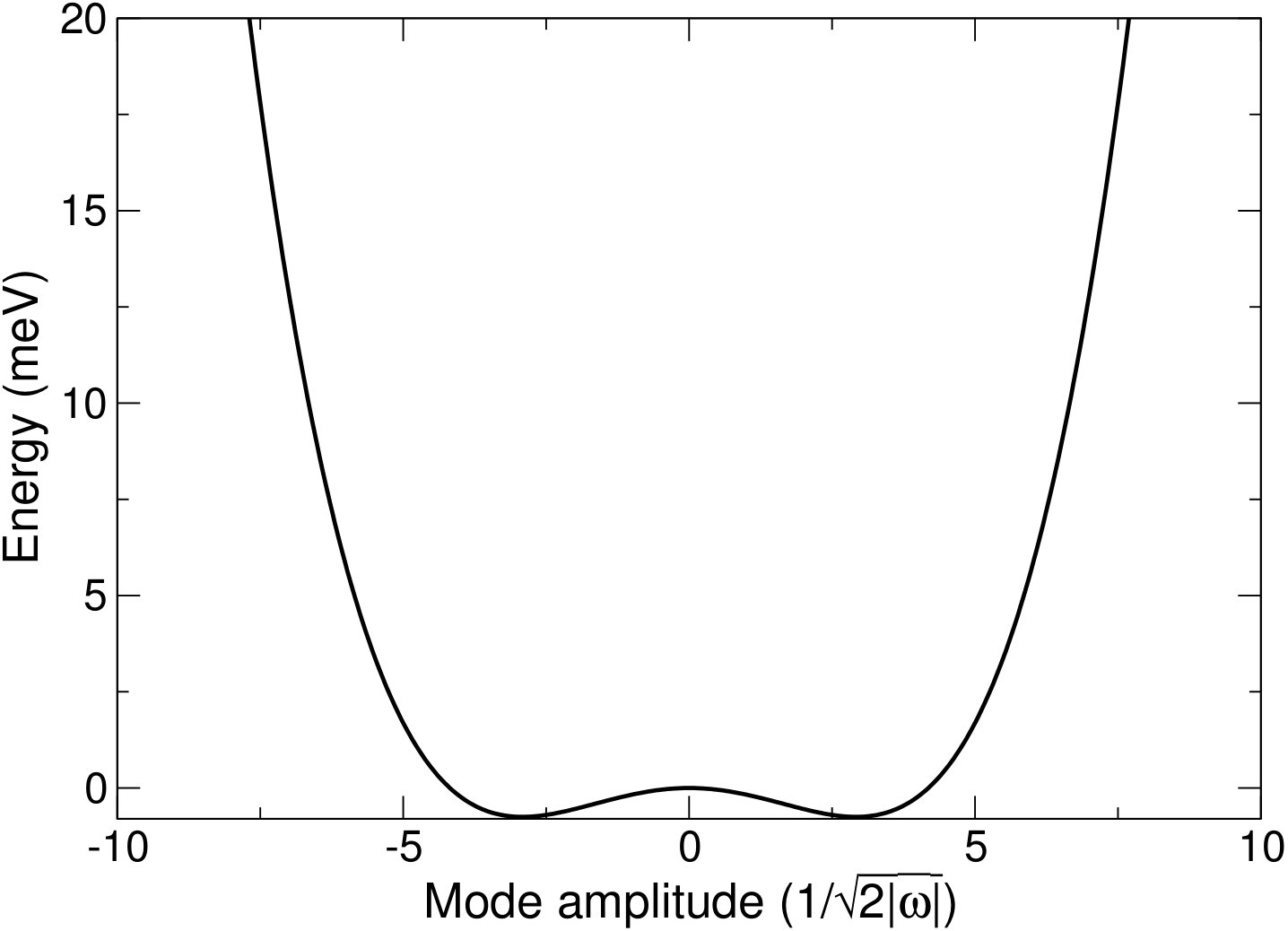 Figure 4
Figure 4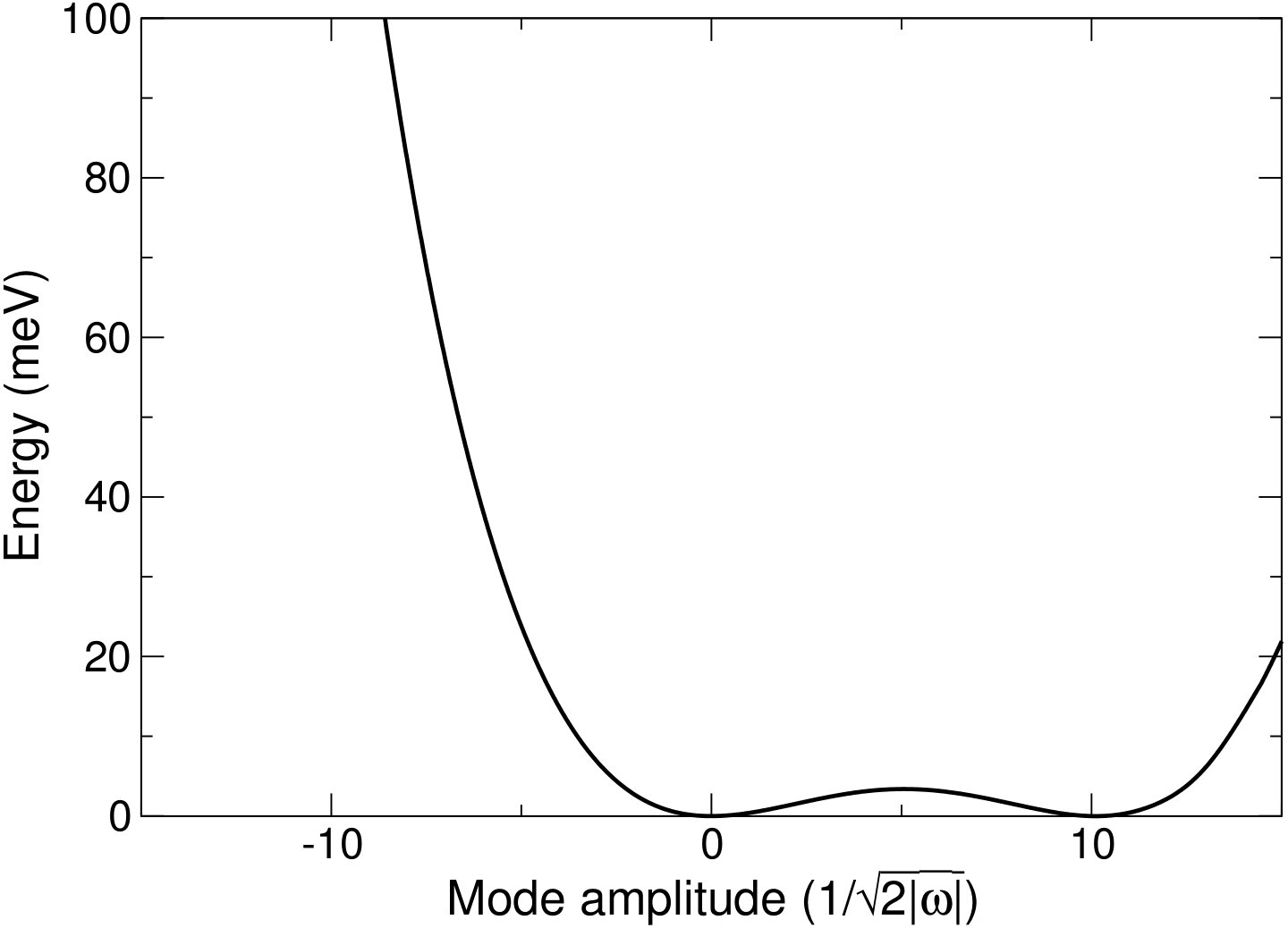 Figure 4
Figure 4| (meV per f.u.) | ||||||
| BZ point | Space group | GPa | GPa | GPa | GPa | |
| P4/mbm | ||||||
| C2/m | ||||||
| I4/mcm | ||||||
| I4cm | ||||||
| I4/mcm | ||||||
| (eV per f.u.) | ||||||
| Post-perovskite | Cmcm | |||||
| (eV per f.u.) | |||||||||||||
| Pressure (GPa) | |||||||||||||
| Temperature (K) | |||||||||||||
| Structure | I4/mcm | ||||||||||||
| C2/m | |||||||||||||
| Pressure (GPa) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Temperature (K) | ||||||||||||
| (Å) | ||||||||||||
| Increase (%) | – | – | – | – | ||||||||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
First-principles anharmonic vibrational study of the structure of calcium silicate perovskite under lower mantle conditions
Joseph C. A. Prentice
Departments of Materials and Physics, and The Thomas Young Centre for Theory and Simulation of Materials, Imperial College London, London SW7 2AZ, United Kingdom
TCM Group, Cavendish Laboratory, University of Cambridge, J. J. Thomson Avenue, Cambridge CB3 0HE, United Kingdom
Ryo Maezono
School of Information Science, JAIST, 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan
R. J. Needs
TCM Group, Cavendish Laboratory, University of Cambridge, J. J. Thomson Avenue, Cambridge CB3 0HE, United Kingdom
Abstract
Calcium silicate perovskite (CaSiO3) is one of the major mineral components of the lower mantle, but has been the subject of relatively little work compared to the more abundant Mg-based materials. One of the major problems related to CaSiO3 that is still the subject of research is its crystal structure under lower mantle conditions – a cubic Pmm structure is accepted in general, but some have suggested that lower-symmetry structures may be relevant. In this work, we use a fully first-principles vibrational self-consistent field (VSCF) method to perform high accuracy anharmonic vibrational calculations on several candidate structures at a variety of points along the geotherm near the base of the lower mantle, in order to investigate the stability of the cubic structure and related distorted structures. Our results show that the cubic structure is the most stable throughout the lower mantle, and that this result is robust against the effects of thermal expansion.
I Introduction
Of the various layers that make up the internal structure of the Earth, the lower mantle is by far the largest by volume, constituting around 55% of the volume of the entire EarthDziewonski and Anderson (1981). Its upper boundary is marked by the ‘transition zone’ between the upper and lower mantle at depths of - km, whilst its lower edge lies at the core-mantle boundary (CMB) at a depth of km.Jordan (1979); Burns (1993) The composition of the lower mantle and the structures of the minerals within it is still subject to debateMattern et al. (2005); Bovolo (2005).
The main elements present in the mantle as a whole are magnesium, iron, calcium, silicon, aluminium and oxygen, with the upper mantle, transition zone, and lower mantle differentiating themselves through the minerals that these elements form. Olivine ((Mg,Fe)2SiO4), which dominates the upper mantle, undergoes a series of phase transitions within the transition zone, ending with the minerals Mg-perovskite and magnesiowüstite, which have chemical formulae (Mg,Fe)SiO3 and (Mg,Fe)O respectively. These two minerals constitute over 80% of the lower mantle between themIrifune (1994). More recent work has also identified a further km thick layer directly above the CMB, known as the D*′′* layer, which exhibits more complex behaviour. The D*′′* layer has been the subject of considerable study, and is thought to arise from a further phase transition, from Mg-perovskite to the ‘post-perovskite’ phaseOganov and Ono (2004); Murakami et al. (2004); Tsuchiya et al. (2004). This is a layered CaIrO3-type structure with orthorhombic Cmcm symmetry, whereas the perovskite structure has cubic Pmm symmetry.
Beyond Mg-perovskite and magnesiowüstite, the third most significant mineral by volume in the lower mantle is calcium silicate perovskite, CaSiO3Irifune (1994); Caracas and Wentzcovitch (2006). Much work has been focused on the magnesium and iron-based minerals, and the behaviour of this less common but still significant material has been relatively neglected, although it is thought to have an effect on the shear velocities of seismic waves as they travel through the Earth, potentially having significance for understanding earthquakesGréaux et al. (2019); Caracas et al. (2005); Mattern et al. (2005); Karki and Crain (1998). However, the most basic property of calcium silicate – its crystal structure – is still the subject of research efforts. Understanding the structure of calcium silicate in the lower mantle is vital, as changing the structure may have significant effects on the seismic velocity in the materialKarki and Crain (1998); Uchida et al. (2009). In particular, it has been shown that a tetragonal distorted structure of CaSiO3 has a significantly lower S-wave speed than the cubic structureStixrude et al. (2007); Kudo et al. (2012); Gréaux et al. (2019).
The fundamental structure of any perovskite material is cubic, and contains a standard motif of corner-sharing oxygen octahedra. In many perovskite-class materials, however, this perfect cubic structure may be unstable with respect to various symmetry-breaking distortions, depending on the identities of the ions involved. These instabilities reveal themselves as soft modes of the cubic stucture, often involving partial rotations of the octahedra. Previous work on the existence of such instabilities in calcium silicate has produced mixed results, both from a theoretical and an experimental perspective. Although it was thought initially that the cubic structure was the ground state Wentzcovitch et al. (1995); Hemley et al. (1987); Liu and Ringwood (1975); Wang et al. (1996), subsequent theoretical and experimental work demonstrated the existence of soft modes in the cubic structureStixrude et al. (1996); Caracas and Wentzcovitch (2006); Stixrude et al. (2007); Jung and Oganov (2005), leading to experimental demonstrations that the ground state is a distorted cubic structureShim et al. (2002); Kurashina et al. (2004); Ono et al. (2004). Unfortunately, however, the distortions are small enough that there is a degree of experimental uncertainty in the structure of the lowest energy distorted state, with both tetragonalShim et al. (2002); Stixrude et al. (2007); Komabayashi et al. (2007); Caracas and Wentzcovitch (2006) and orthorhombicUchida et al. (2009); Magyari-Köpe et al. (2002); Adams and Oganov (2006) phases proposed. Distorted structures with several different symmetries have been studied, including those with space groups I4/mcm, Imma, P4/mbm, I4/mmm, Im3, P42/nmc and PnmaCaracas and Wentzcovitch (2006). It is believed that the cubic Pmm phase will be stabilised at some temperature and pressure, but different groups have obtained different results for these transition conditions – although it is generally accepted that calcium silicate is cubic in most of the lower mantleNestola et al. (2018); Sun et al. (2014); Komabayashi et al. (2007); Stixrude et al. (2007); Adams and Oganov (2006); Noguchi et al. (2012), there have been suggestions that distorted structures are also relevant in some regionsUchida et al. (2009); Ono et al. (2004); Li et al. (2006). Intriguingly, it has even been suggested that a phase transition from a cubic structure into a distorted structure may occur at very high pressures, seen at the very base of the mantleHemley et al. (1987). Previous studies have suggested that anharmonic vibrational effects are significant in this systemSun et al. (2014); Hemley et al. (1987), implying that high accuracy anharmonic calculations will be necessary to discover the correct structure of calcium silicate in the lower mantle. In the present work, we concentrate on the bottom of the lower mantle for several reasons: the presence of the D*′′* layer, the previous suggestion of a cubic to distorted phase transition, and the difficulty of performing experimental measurements at relevant conditions, meaning first-principles calculations have a vital role to play.
The conditions within the layers in the Earth vary significantly with depth. In particular, with increasing depth, the pressure and temperature increase, although the relationship between the two is not necessarily simple. The geotherm, shown in Fig. 1, describes the relationship between temperature and pressure (or depth) within the EarthNomura et al. (2014). A given depth corresponds to a set of temperature and pressure () conditions on the geotherm. To obtain the correct structure of a mineral at this depth, the free energy of the system must be minimised under this set of conditions. Experimentally, it is often difficult to control both temperature and pressure simultaneously if conditions deep within the Earth are of interest, making accurate first-principles calculations extremely valuable for this purpose.
In this work, we go beyond previous work on this system by approaching it from a fully ab initio standpoint – we use highly accurate first-principles density functional theory (DFT) calculations, together with a vibrational self-consistent field method for anharmonic vibrational calculations. Using these methods, we examine the relative stability of three possible crystal structures of calcium silicate, including the cubic perovskite structure, at four points on the geotherm, including the effects of anharmonic vibrations. This investigation constitutes the most in-depth and accurate investigation of this important topic yet attempted. Our results suggest that, although the cubic phase of calcium silicate is unstable to distortions at zero temperature, calcium silicate takes up the cubic structure throughout the lower part of the lower mantle. The effects of thermal expansion are also considered, and found not to affect the conclusions of this work.
The rest of this work is organised as follows: in Sec. II we outline the computational methods employed in this work, and give technical details of the calculations. In Sec. III we present the main results of our study, detailing the structures considered at different points along the geotherm, their relative stabilities, and the effect of anharmonicity on these results. In Sec. IV we give a brief summary of our results and some concluding remarks.
II Calculational methods
In this work, as we are studying systems along the geotherm, the effect of both temperature and pressure must be included when comparing different structures. This means that the appropriate quantities to compare are the enthalpy rather than the energy at zero temperature, and the Gibbs free energy rather than the Helmholtz free energy at finite temperature, where and .
II.1 DFT calculations
All DFT calculations in this work were performed using version 16.1 of the castep codeClark et al. (2005), with the corresponding ‘on-the-fly’ ultrasoft pseudopotentialsVanderbilt (1990) and the PBE exchange-correlation functionalPerdew et al. (1996). The PBE functional was chosen as it has been used in previous calculations under lower mantle conditionsJung and Oganov (2005); Oganov and Ono (2004); Pickard and Needs (2015). A cut-off energy of eV was used in all calculations, and Monkhorst-Pack gridsMonkhorst and Pack (1976) with spacings as close to as possible were used throughout – this corresponded to a grid in the unit cell of all the undistorted cubic structures considered. The total energy was always converged to within eV, to ensure accurate forces for the vibrational calculations. Within geometry optimisation calculations, the atomic positions were adjusted until the root mean square of the forces on all the atoms was below eV/Å.
II.2 Vibrational calculations
In this work a vibrational self-consistent field (VSCF) method, described in Ref. Monserrat et al., 2013 and used successfully several times sinceAzadi et al. (2014); Monserrat et al. (2014); Engel et al. (2015); Prentice et al. (2017), is used to include anharmonic effects in our results. This method has been used successfully in high pressure and temperature systemsMonserrat et al. (2014, 2018), but this study consitutes the first time such a method has been applied to lower mantle materials. The method uses a basis of harmonic normal mode co-ordinates to describe the Born-Oppenheimer (BO) surface that the nuclei move in, but does not enforce the harmonic approximation itself. Instead, we go beyond the harmonic representation of the BO surface by using a principal axes approximation,Jung and Gerber (1996) resulting in a many-body term expansion of the BO surface:
[TABLE]
Here, is a collective vector containing the normal mode amplitudes , Latin indices are collective labels for the quantum numbers where is a phonon wavevector and a phonon branch, and is the energy of the BO surface when the atomic nuclei are in configuration . Anharmonicity is already included in the terms, as they are not constrained to the harmonic form. In this work, this expansion is truncated to only include the terms, as corrections due to higher order terms in this expansion have been found to be less importantMonserrat et al. (2013); Prentice and Needs (2017). The various terms are found by mapping the BO surface as a function of the amplitude of each normal mode, typically using DFT calculations with the normal mode frozen in, and then fitting a spline to the results. This provides a truly first-principles description of the BO surface, without assuming a functional form, and provides significant amounts of information about the surface the nuclei move in. Mapping the BO surface is by far the most computationally expensive part of the VSCF method, as a large number of DFT calculations are required to achieve an accurate fit. Once a form for the BO surface is obtained, the resulting nuclear Schrödinger equation is solved numerically (and self-consistently) to obtain the anharmonic energies and wavefunctions for both ground and excited vibrational states. These can then be used to calculate the anharmonic vibrational free energy.
In order to obtain the basis of harmonic normal modes required for the VSCF method, a harmonic vibrational calculation was performed. This used a finite displacement method to calculate the matrix of force constants, which was then Fourier transformed to obtain the dynamical matrix.Kunc and Martin (1982) This was diagonalised to obtain the harmonic vibrational frequencies and eigenvectors. Atomic displacements of Å were used. To reduce the computational cost of the mapping of the BO surface within the VSCF method itself, the non-diagonal supercells method was used to reduce the size of the supercell needed to sample a given point in the vibrational Brillouin zoneLloyd-Williams and Monserrat (2015), and DFT force data was used to give a more accurate fit for a given number of mapping calculationsPrentice and Needs (2017). 21 different amplitudes per mode were used to map the BO surface. For the purpose of self-consistently solving the nuclear Schrödinger equation, the vibrational wavefunction is written as a Hartree product of the normal modes, .
The states are represented in a basis of one-dimensional harmonic oscillator eigenstates. To be able to get accurate results for the extreme temperatures considered in this work, it is necessary to have a good description of the high energy vibrational states and their vibrational wavefunctions, which itself requires a large basis set. To ensure that we have a sufficiently large basis set for this purpose, basis functions were used for each normal mode in this work.
II.3 Thermal expansion
The high temperatures present in the lower mantle make it important to consider the effects of thermal expansion. This is most often done using the quasiharmonic method, where the free energy is calculated as a function of temperature within the harmonic approximation for several different values of the lattice parameters. Fitting to these results then allows the minimum of free energy at a given temperature to be found as a function of these lattice parameters. It is also possible to go beyond the quasiharmonic approximation within the VSCF method used in this work; this involves using the calculated nuclear wavefunctions to compute the vibrational average of the stress tensor, adjusting the lattice parameters accordingly, and repeating the anharmonic calculation until convergence of the lattice parameters is reached. However, using the VSCF method in this way involves mapping the BO surface several times at different volumes, making it significantly more expensive than the quasiharmonic method, which itself can be expensive. In this work, we instead use previous experimental dataNoguchi et al. (2012) to estimate the thermal expansion along the geotherm, and consider the results in the light of the results of the full anharmonic calculations.
III Results
III.1 Structures investigated
To obtain a picture of how CaSiO3 behaves across the bottom of the lower mantle, four points on the geotherm were considered, corresponding to external pressures of , , , and GPa. GPa was chosen as a representative point in the main lower mantle, GPa as corresponding to the top of the D*′′* layer, GPa as a point in the middle of the D*′′* layer, and GPa as corresponding to the CMB. As shown in Fig. 1, each of these pressures is associated with a range of temperatures on the geotherm: -, -, -, and - K correspond to , , , and GPa respectively. In this work, the vibrational free energy and relative stabilities are calculated at the maximum and minimum temperature on the geotherm for each pressure, as well as at zero temperature. Fig. 1 also shows that there are no discontinuities in the gradient of the geotherm, associated with changes in phase or composition, in the upper part of the lower mantle. This implies that the results for GPa should be qualitatively applicable to the rest of the lower mantle, but further first-principles work would be required to obtain a quantitative description.
To obtain appropriate lattice constants for each pressure, the perfect cubic perovskite structure was allowed to relax whilst maintaining its symmetry. This led to zero temperature lattice constants of , , , and Å for , , , and GPa respectively. These compare well with experimental results for the unit cell volume at high pressures and temperatures, which imply a cubic lattice constant of Å at GPa and KNoguchi et al. (2012). The slight overestimation is typical for GGA-based DFT functionals such as PBEHaas et al. (2009).
Once the relaxed lattice constants had been obtained, the next task was to find appropriate distorted structures to compare against the perfect cubic structure. Many different structures have been considered in previous workShim et al. (2002); Stixrude et al. (2007); Komabayashi et al. (2007); Caracas and Wentzcovitch (2006); Uchida et al. (2009); Magyari-Köpe et al. (2002); Adams and Oganov (2006), and it is not feasible to do a full anharmonic calculation on every one of these. Instead, we obtained distorted structures via a more rigorous method – by following the soft modes present in the cubic structure. A harmonic vibrational calculation was conducted for each of the cubic structures, sampling the vibrational Brillouin zone (BZ) with a grid using the non-diagonal supercells methodLloyd-Williams and Monserrat (2015). This size of grid was chosen as previous work has shown that the strongest soft modes lie at the and symmetry points, which have vibrational BZ co-ordinates and respectively. Soft modes were indeed found at the and points, as expected, as well as along the line between the and points at co-ordinates and . For each pressure considered, the cubic structure was distorted to follow each of these soft modes and the atomic positions and lattice parameters were allowed to relax. In each case, this resulted in the same set of distorted structures, each associated with a high symmetry point in the vibrational BZ, which are summarised in Table 1. Visualisations of the structures are also presented in Fig. 2. At all pressures, the two lowest enthalpy structures were the C2/m and I4/mcm structures originating from the point. These structures were therefore selected for a full anharmonic calculation and comparison with the cubic structure, and will be the only distorted structures discussed in the following sections.
One addition was made to this rigorous method of obtaining alternative structures, in order to investigate the stability of the Cmcm post-perovskite structure of MgSiO3 when applied to CaSiO3. CaSiO3 in this structure was allowed to relax, whilst maintaining symmetry, at the different pressures selected. The properties of the resulting structures are also noted in Table 1 and presented in Fig. 2. As can be seen, the post-perovskite structure was found to be more than eV higher in enthalpy than the cubic perovskite phase across all phases, making it energetically uncompetitive. From this, we can conclude that CaSiO3 does not undergo a post-perovskite phase transition like MgSiO3, and we therefore do not mention this structure in the rest of this work. The structures used in this work, not including the post-perovskite structure, can be found in .cif format in the Supplemental Materialsup .
III.2 Anharmonic calculations
III.2.1 Vibrational Brillouin zone sampling
A key consideration when undertaking vibrational calculations such as the ones presented here is the sampling of the vibrational Brillouin zone. The finer this sampling is, the more accurate the calculations should be, but they will also increase significantly in expense. Although we use the non-diagonal supercells methodLloyd-Williams and Monserrat (2015) to reduce this, as mentioned previously, it is still necessary to find a balance between accuracy and cost. In this work, we use an grid sampling of the vibrational Brillouin zone for the cubic structures, and a grid for the distorted structures.
III.2.2 Mapping amplitudes
The very high temperatures present in the lower mantle mean that the nuclei will have enough energy to explore the Born-Oppenheimer surface out to large amplitudes, and therefore in order to obtain accurate results our first-principles mapping of the BO surface must do so too. In order to ensure this, the BO surface was mapped along each normal mode up to a maximum amplitude of at least , where is the harmonic expectation value of the mode amplitude squared. In several cases this was insufficient to reach energies at least higher than the reference state, and in these cases the mapping was extended until this energy was obtained. This allowed us to ensure that the most thermally relevant part of the BO surface was mapped from directly from first-principles.
Although this part of the BO surface is the most important to get correct, it is not the only part that participates in the calculations. A key part of the VSCF method is the integral over the mode amplitudes of the product of the nuclear probability density (the modulus squared of the wavefunction) and the BO surface. As we are describing the nuclear wavefunction of each normal mode using harmonic oscillator basis functions, some of which may extend significantly beyond the amplitude already mapped explicitly, it is necessary to extend the limits of the integration when integrating over these basis functions. This means it is also necessary to have an expression for the BO surface at these amplitudes. To this end, we extend the BO surface by assuming it has a quadratic form beyond the explicitly mapped region, as we observe that the mapped BO surface has a quadratic form at the extremes of the mapping. A different quadratic fit is used for extending the BO surface in the negative and positive directions, and for each fit the parameters are found by fitting to the three mapped points with the most negative/positive amplitude.
With this extension of the BO surface in hand, the limits of integration for an integral over a particular harmonic oscillator basis function, quantum number and frequency , are taken to be either
- •
the amplitudes that have already been explicitly mapped, or
- •
the maximum amplitude of a classical harmonic oscillator with the same energy as the quantum harmonic oscillator state associated with the basis function, given by ,
whichever is greater. This is because the correspondence principle tells us that as increases, the quantum harmonic oscillator states describe probability distributions that look more and more like the classical trajectory, which is bounded at the amplitude given above.
III.3 Relative stabilities
Table 2 presents the main results of this work – that is, the relative stabilities of the structures considered at several different points on the geotherm. Figure 3 presents the same information graphically.
The key result here is that at the temperatures and pressures found in the lower part of the lower mantle, the cubic structure of CaSiO3 is always more stable, and in fact becomes more stable the deeper into the mantle one goes, from meV – at GPa – to meV – at GPa – lower in free energy than the lowest energy distorted structure (at the lowest temperature considered for each pressure). This supports previous work that suggests that calcium silicate is cubic in the lower mantleSun et al. (2014); Komabayashi et al. (2007); Stixrude et al. (2007); Adams and Oganov (2006); Noguchi et al. (2012), utilising a novel high accuracy first-principles method to complement previous work. The distortion that is lowest in free energy changes as the pressure and temperature increase, going from C2/m at GPa to I4/mcm at an estimated crossover pressure of GPa.
At zero temperature, the zero point vibrational energy has the effect of closing the gap between the cubic structure and the distorted structures, which would be significantly lower in free energy if this contribution were not included. At GPa, this effect is actually strong enough to alter the relative free energies by more than eV and make the distorted structures higher in free energy than the cubic structure, making the cubic structure thermodynamically stable (although still dynamically unstable due to the presence of soft modes).
III.4 Mapped phonon modes
Figure 4 shows two examples of phonon modes mapped as part of these calculations. Figure 4(a) shows one of the soft modes present in the cubic structure at GPa, whilst Fig. 4(b) shows the same mode, but mapped with the I4/mcm distorted structure as the reference structure at GPa. Both show the characteristic double well potential of a soft mode. Both wells in Fig. 4(b) correspond to distortions with the same symmetry, whilst the structure corresponding to the maximum is cubic. This presents the possibility of the system being able to ‘hop’ between these wells if it has enough energy, resulting in the structure becoming cubic on average, although instantaneously the structure is distorted, in a similar way to the dynamic Jahn-Teller effectPrentice et al. (2017). At the very high temperatures seen in the lower mantle, this is unlikely to be the root cause of the stability of the cubic structure, as the depth of these wells will become negligible compared to the thermal energy, but at intermediate temperatures this may be an important effect in stabilising the cubic structure.
III.5 Thermal expansion
Previous experimental work by Noguchi et al.Noguchi et al. (2012) has provided several different models for a thermal equation of state for cubic calcium silicate perovskite under lower mantle conditions. Using these models allows us to link pressure, volume and temperature, and find an estimate for the volume at a given pressure and temperature, including the effects of thermal expansion. Here, we use the thermodynamic thermal pressure model (model 4 in Ref. Noguchi et al., 2012), which gives the equation of state as
[TABLE]
where GPa, Å3, K*-1*, GPa K*-1*, and K. K is used as the reference temperature, as they found this was above the transition temperature to the cubic phase. is the reference volume at this temperature and bar MPa, but to account for the error in the volume calculated by DFT compared to the experimental volume, we solve this equation for instead of .
To estimate the thermal expansion, we solved this equation for each pressure at the high and low temperatures on the geotherm, as well as at [math] K. Although the model uses a reference temperature of K, we found that the variation in the DFT volumes at [math] K with pressure matched the prediction of the model very well, giving us confidence that the model was still applicable at [math] K (at least theoretically, as the cubic phase is unstable at zero temperature). We can then use these results to estimate the effect of thermal expansion on the DFT lattice constant for each set of conditions, using the formula
[TABLE]
The results of this are presented in Table 3. It can be seen that there is an expansion of between 1 and 2% in all cases. However, with the exception of the data for GPa, all the lattice constants remain within the range of lattice constants already considered in this work. As Fig. 3 and Table 2 show, the cubic phase is stable across the whole range, meaning that the main conclusion of this work is still valid – CaSiO3 takes up the cubic structure throughout the lower region of the lower mantle. To quantify the stability of the cubic structure more exactly, a harmonic or full anharmonic calculation could be performed at these expanded lattice parameters, and a correction made to the non-thermally expanded result.
III.6 Effect of impurities
The calculations presented here focuses on pure calcium silicate, neglecting the effect of impurities. This is the assumption made by most previous theoretical work, and is justified by experimental results on both naturally occurring calcium silicateNestola et al. (2018) and laboratory samplesOno et al. (2004); Kesson et al. (1998); Irifune (1994). These experimental results imply that naturally occurring calcium silicate perovskite is very pure (90%), as impurities tend to reside in the magnesium silicate perovskite that exists alongside CaSiO3 in the lower mantle. At the very highest pressure present in the lower mantle, however, previous work has suggested that there is a limited amount of magnesium silicate perovskite in solid solution within calcium silicate (up to 20% per mole)Kesson et al. (1998). Although this could potentially cause the structure of CaSiO3 to distort, comparisons to similar mixtures of perovskites suggest it is more likely that this limited amount of mixing would not be quite enough to result in a change of structureČeh et al. (1987), resulting in the structure of CaSiO3 remaining cubic.
IV Conclusions
In summary, we have presented, to the best of our knowledge, the most in-depth first-principles study of calcium silicate perovskite and its structure yet. We have thoroughly mapped the Born-Oppenheimer surface of this key mantle material in both the high symmetry cubic phase and in two competing distorted phases, and used this data to conduct high accuracy anharmonic vibrational calculations of the free energy. These calculations show that, even down in the very depths of the mantle, calcium silicate takes up the high symmetry cubic structure. This supports previous work that suggests the structure is cubicSun et al. (2014); Komabayashi et al. (2007); Stixrude et al. (2007); Adams and Oganov (2006); Noguchi et al. (2012), and further demonstrates the validity of the first-principles VSCF method. Although our calculations do not explicitly include any thermal expansion, we suggest that this result is robust against the effect of thermal expansion, which was calculated using previous experimental data. Future work on this area could include the effect of impurities or thermal expansion fully from first principles, using either a quasiharmonic or fully anharmonic method. Anharmonic first-principles calculations could also be applied to other materials in the interior of Earth, and potentially other planets as well. This would both help improve the understanding of geophysical phenomena, and inform future experiments on these materials.
V Acknowledgements
J.C.A.P. and R.J.N. are grateful to the Engineering and Physical Sciences Research Council (EPSRC) of the UK for financial support [EP/P034616/1]. Computational resources were provided by:
- •
the Cambridge Service for Data Driven Discovery (CSD3) operated by the University of Cambridge Research Computing Service, provided by Dell EMC and Intel using Tier-2 funding from the Engineering and Physical Sciences Research Council (capital grant EP/P020259/1), and DiRAC funding from the Science and Technology Facilities Council,
- •
the ARCHER UK National Supercomputing Service, for which access was obtained via the UKCP consortium [EP/K014560/1],
- •
the Research Center for Advanced Computing Infrastructure at JAIST,
- •
and the UK Materials and Molecular Modelling Hub, which is partially funded by EPSRC (EP/P020194/1), for which access was obtained via the UKCP consortium and funded by EPSRC grant ref EP/P022561/1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Dziewonski and Anderson (1981) A. M. Dziewonski and D. L. Anderson, “Preliminary reference Earth model,” Phys. Earth Planet. Inter. 25 , 297 (1981) . · doi ↗
- 2Jordan (1979) T. H. Jordan, “Structural geology of the Earth’s interior,” Proc. Natl. Acad. Sci. U.S.A. 76 , 4192 (1979) .
- 3Burns (1993) R. G. Burns, Mineralogical Applications of Crystal Field Theory , 2nd ed. (Cambridge University Press, Cambridge, 1993). · doi ↗
- 4Mattern et al. (2005) E. Mattern, J. Matas, Y. Ricard, and J. Bass, “Lower mantle composition and temperature from mineral physics and thermodynamic modelling,” Geophys. J. Int. 160 , 973 (2005) . · doi ↗
- 5Bovolo (2005) C. I. Bovolo, “The physical and chemical composition of the lower mantle,” Phil. Trans. R. Soc. A 363 , 2811 (2005) . · doi ↗
- 6Irifune (1994) T. Irifune, “Absence of an aluminous phase in the upper part of the Earth’s lower mantle,” Nature 370 , 131 (1994) . · doi ↗
- 7Oganov and Ono (2004) A. R. Oganov and S. Ono, “Theoretical and experimental evidence for a post-perovskite phase of Mg Si O 3 in Earth’s D ′′ layer,” Nature 430 , 445 (2004) . · doi ↗
- 8Murakami et al. (2004) M. Murakami, K. Hirose, K. Kawamura, N. Sata, and Y. Ohishi, “Post-perovskite phase transition in Mg Si O 3 ,” Science 304 , 855 (2004) . · doi ↗