Momentum space calculations of the binding energies of argon dimer
Taghi Sahraeian, M. R. Hadizadeh

TL;DR
This paper uses momentum space calculations to accurately determine the binding energies of argon dimer, confirming known vibrational levels and predicting additional states with high precision, extending to higher rotational states.
Contribution
It introduces a momentum space approach to calculate argon dimer energies, confirming previous results and providing precise predictions for higher vibrational and rotational states.
Findings
Confirmed eight vibrational levels of ground state
Predicted the ninth vibrational level with high accuracy
Calculated energies for all 174 bound states
Abstract
The binding energies of argon dimer are calculated by solving the homogeneous Lippmann-Schwinger integral equation in momentum space. Our numerical analysis using two models of argon-argon interaction developed by Patkowski {\it et al.} confirms not only the eight argon dimer vibrational levels of the ground state of argon dimer (i.e. for ) predicted by other groups but also provides a very precise means for determining the binding energy of the ninth state which its value is a matter of discussion. Our calculations have been also extended to states with higher rotational quantum number and we have calculated the energy of all 174 bound states for both potential models. Our numerical results for vibrational levels of the ground state of argon dimer are in excellent agreement with other theoretical calculations and available experimental data.
Click any figure to enlarge with its caption.
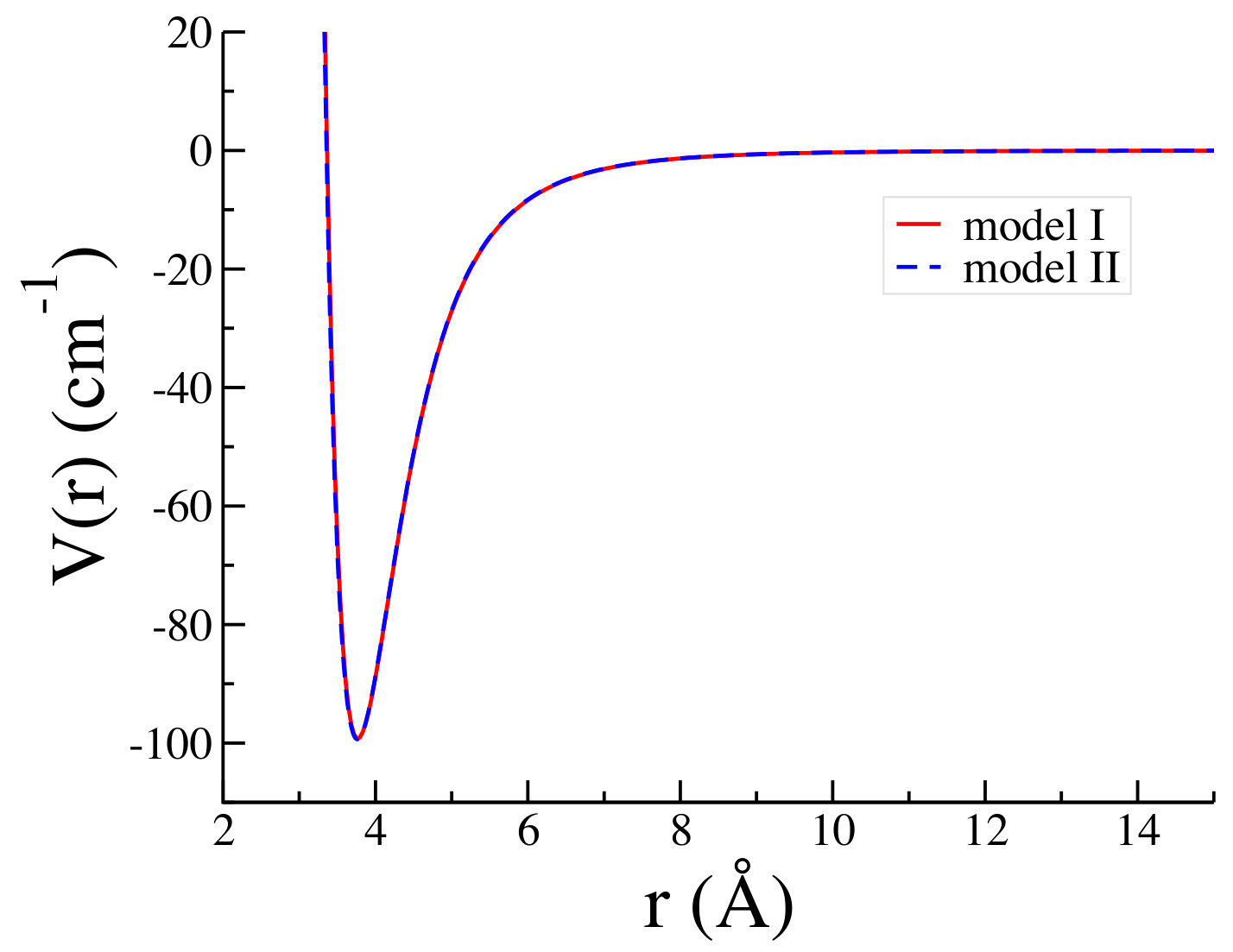 Figure 1
Figure 1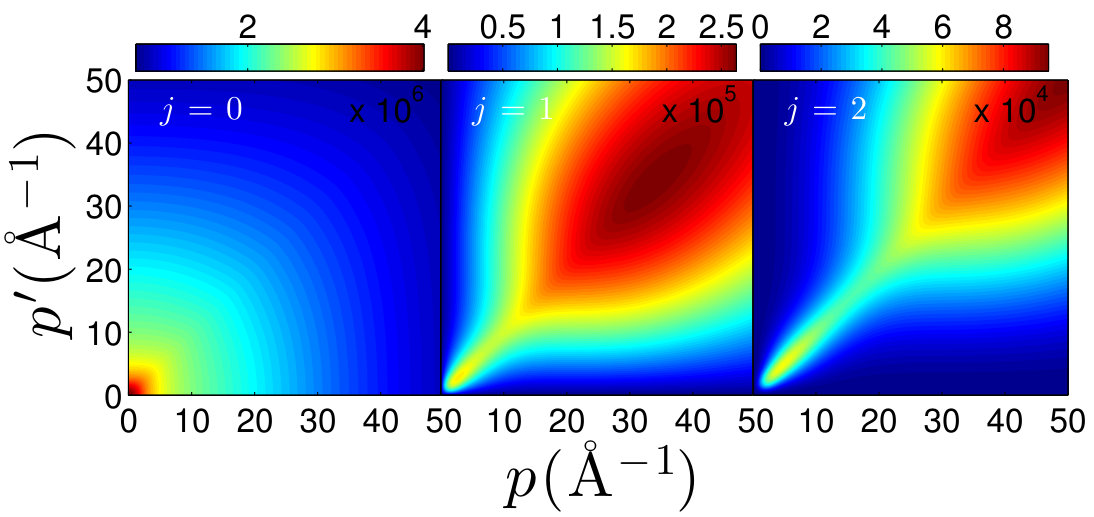 Figure 1
Figure 1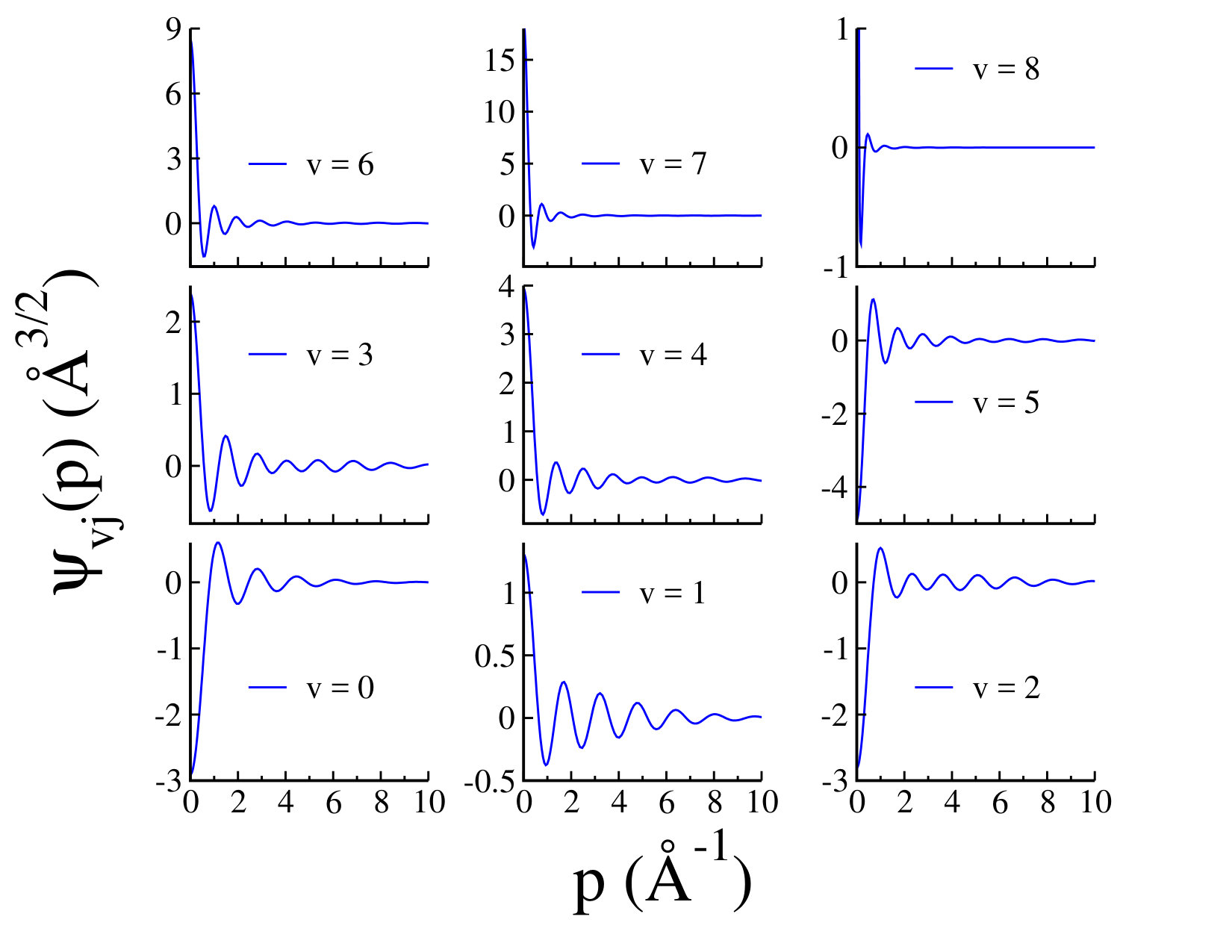 Figure 2
Figure 2 Figure 2
Figure 2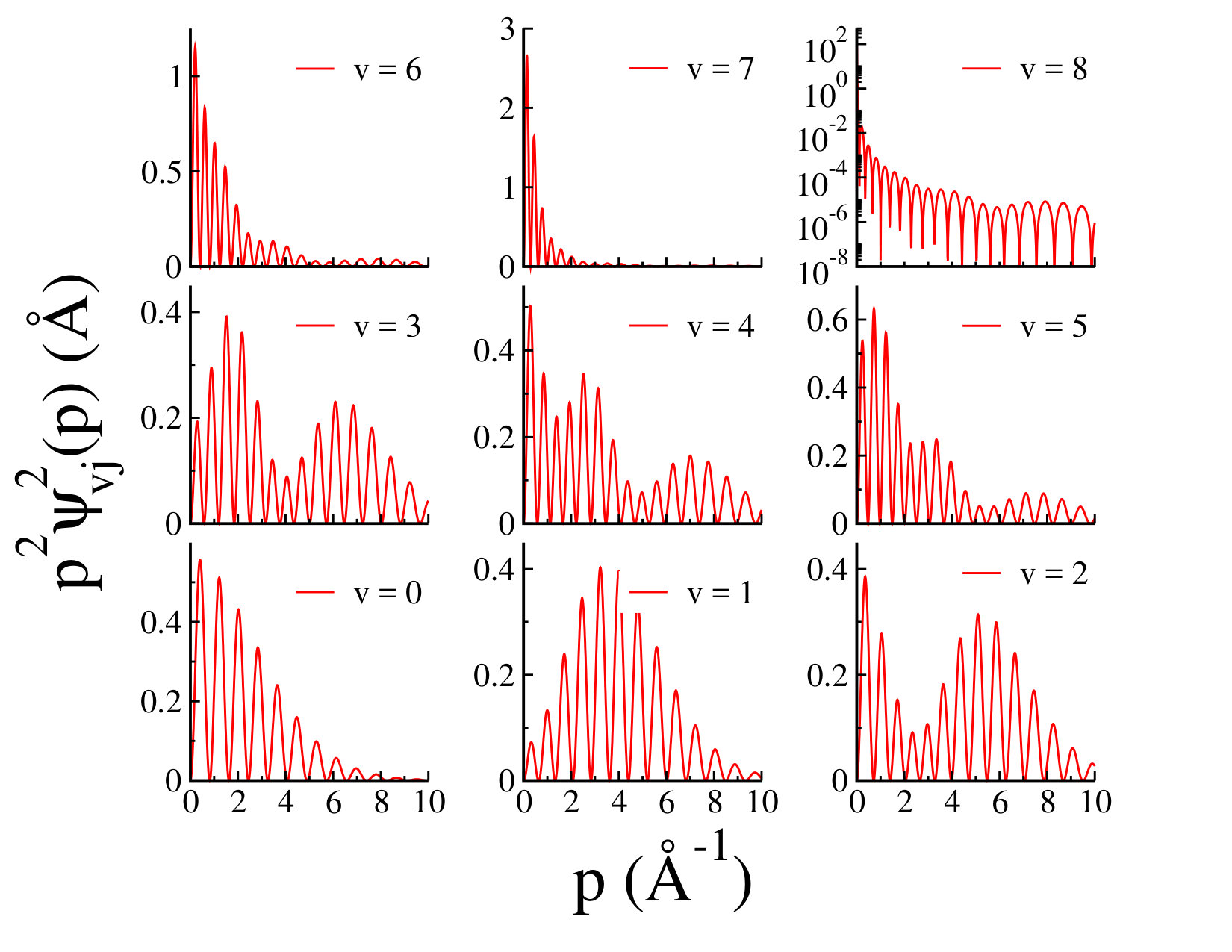 Figure 3
Figure 3 Figure 3
Figure 3| State | Model I | |||
| Present | Ref. 3 | |||
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Momentum space calculations of the binding energies of argon dimer
Taghi Sahraeian
Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio, 43210, USA
M. R. Hadizadeh
College of Science and Engineering, Central State University, Wilberforce, OH 45384, USA
Abstract
The binding energies of argon dimer are calculated by solving the homogeneous Lippmann-Schwinger integral equation in momentum space. Our numerical analysis using two models of argon-argon interaction developed by Patkowski et al. confirms not only the eight argon dimer vibrational levels of the ground state of argon dimer (i.e. for ) predicted by other groups but also provides a very precise means for determining the binding energy of the ninth state which its value is a matter of discussion. Our calculations have been also extended to states with higher rotational quantum number and we have calculated the energy of all 174 bound states for both potential models. Our numerical results for vibrational levels of the ground state of argon dimer are in excellent agreement with other theoretical calculations and available experimental data.
1 Introduction
The argon dimer has been the subject of different theoretical studies and experimental research in the fields of physics and chemistry. The development of accurate argon-argon (Ar-Ar ) interactions has been decisive for decades and different Ar-Ar interatomic interactions have been developed by different groups, from HFDID empirical potential of Aziz 1 to semi-empirical potentials of Tang and Toennies 2. Significant improvements in computational power and numerical techniques in the last decade have led to the development of highly accurate ab initio potentials advanced by Patkowski et al. 3, 4 and Slavicek et al. 5. Recently a new empirical potential energy function for Ar2 has been also developed by Myatt et al. 6 using a critical re-analysis of all available spectroscopic and virial coefficient data for Ar2.
In this paper, we have solved the Lippmann-Schwinger equation in momentum space to calculate the binding energies of argon dimer, using two models of Ar-Ar interaction of Refs. 3, 4. Both potential models support nine bound states for ; however, defining the shallowest state which is very close to dissociation is numerically challenging. This has been discussed in details. Moreover, we extend the solution of Lippmann-Schwinger equation to higher partial wave channels and present the numerical results for 174 vibrational and rotational energy levels of argon dimer.
2 Lippmann-Schwinger integral equation in momentum space
The nonrelativistic bound state of two particles with masses and , and the relative momentum , interacting by an arbitrary central force can be described by homogeneous Lippmann-Schwinger equation
[TABLE]
where is two-body (2B) wave function and is free propagator. is the binding energy of 2B system ( is the mass of dimer) and is the reduced mass of 2B system. In a partial wave representation Eq. 1 can be presented in momentum space as the following integral equation 7, 8
[TABLE]
where and are the vibrational and rotational quantum numbers, respectively. The input of the integral equation 2 is the two-body interaction which depends on the magnitude of the initial and final 2B relative momenta and . is the projection of the interaction in the partial wave channel
[TABLE]
The matrix elements of the interaction in momentum space can be obtained by Fourier transformation of 2B interaction in configuration space , which depends on the relative distance , as
[TABLE]
and are the magnitudes of initial and final 2B relative momentum vectors, is the angle between them, and is the magnitude of the momentum transfer.
3 Argon-argon interatomic potentials
In this study for Ar-Ar interatomic interaction, we have used the fitted analytical potential functions of Refs. 3, 4 with the following general form
[TABLE]
where , and are the Tang-Toennies damping functions
[TABLE]
In our calculations, we have used the potential parameters of Refs. 3, 4 which are given in Table 1. The potential models I and II predict a minimum at Å and Å with a depth of and , respectively. In Fig. 1 we have shown both potential models I and II of Ar-Ar interaction in configuration space as a function of the interatomic distance . We have also presented few examples of the matrix elements of the potential model I in momentum spaces, as a function of the relative momenta and , for the rotational quantum numbers and 2.
4 Numerical Results and Discussion
The first step toward the numerical solution of the integral equations (2), (3), and (4) is discretization of the continuous momentum, angle, and configuration variables and to this aim, we have used Gauss-Legendre quadratures. The momentum integration interval is covered by a combination of hyperbolic and linear transformations of Gauss-Legendre points from the interval to the intervals as
[TABLE]
The typical values for , and in our calculations are , and Å*-1*. For the angle integration in Eq. (3) as well as configuration integration of the Fourier transformation of the potential in Eq. (4), a linear transformation is used. The configuration integration domain is transformed to the interval , where we have used Å. The number of grid points for momentum, angle, and configuration variables are , and , respectively.
The integral equation (2) can be written schematically as eigenvalue equation , where the physical dimer binding energies are corresponding to the eigenvalue . The eigenvalue equation is solved by direct method. For calculation of argon dimer binding energies we have solved the integral equation (2) by searching in a wide range of energies in the region and we have extracted the physical bound states for with a relative error of .
Our numerical results for all 174 vibrational and rotational energy levels of argon dimer using potential models I and II are listed in Tables 2 and 3, respectively. In Tables 4 and 5, however, our vibrational levels of argon dimer, for the rotational states , are compared with those of previous studies by Tennyson et al. using R-matrix method 9, 10 and also with the findings of Ref. 11. The comparisons indicate that our results for both potential models I and II are in an excellent agreement with the results of other groups with a relative percentage difference estimated to be at most , , and for , , and , respectively. It should be pointed out that the argon mass used in our calculations and also in Refs. 9, 10 corresponds to pure 40Ar mass of amu, whereas the results of Ref. 11 are obtained by 40Ar mass of amu.
In Table 4, we have also listed the experimental data of Herman et al. for the binding energies of six of the nine vibrational levels for , determined by high-resolution VUV emission from three excited electronic states of Ar2.
As it is shown in Table 6, there is an evidence in Ref. 12 suggesting the potential model I should support a ninth vibrational level for argon dimer ground state for with a very small binding energy in the microKelvin range. Our numerical analysis confirms the existence of this extremely shallow state with a binding energy of K which is in good agreement with the reported energy of K in Ref. 12. Although the R-matrix method 9 has been able to detect only eight of the nine vibrational weakly bound states, some recent improvements in this method 10 have provided the possibility to generate the ninth vibrational state for both potential models I and II, however, at this point, the exact value of the energy level of the ninth state is unstable and that is why it is not listed in Table 4.
We should point out that Myatt et al. have recently performed a full rotational treatment using a new empirical potential energy function for Ar2 and have reported 173 vibrational and rotational energy levels 6. In their numerical analysis, the employed potential energy supports only eight, rather than nine vibrational levels. For the numerical implementation, the authors have used a computer code called "LEVEL", developed by R. J. Le Roy to solve the radial Schrödinger equation for diatomic systems and calculate the eigenvalues of the bound and quasibound levels of any smooth one-dimensional or radial potential 13. The numerical analysis of Ref. 10 confirms that not only the R-matrix method of Ref. 9, but also LEVEL code has been unable to detect the ninth vibrational weakly bound state, whereas our momentum space method provides a superior solution to the Schrödinger equation with the capability to achieve the extremely weakly bound states. By having dimer binding energies one can calculate the argon dimer wave functions using Eq. (2). In Fig. 2 we have shown dimer wave functions for all nine vibrational states of the ground state of argon dimer, for , obtained from the potential model I, as a function of the relative momentum . The dimer wave functions are normalized as As we expect, the structure of dimer wave function is expanded to higher momentum for lower bound levels, whereas for the higher bound levels the wave functions are more compact. As it is shown in Fig. 2, for the ninth vibrational state for which is weakly bound and very close to dissociation, the wave function is significant at very small values of the relative momentum . Our numerical analysis indicates that the binding energy of this weakly bound state is quite sensitive to the distribution of the grid points close to zero momentum. In order to be able to reasonably identify this state, it is crucial to consider a hyperbolic mapping with enough number of mesh points for the relative momentum in the domain . In Fig. 3, we have shown few examples of the vibrational radial probability densities of argon dimer ground state for , calculated for potential model I. We have also shown few examples of the rotational probability densities for and , , and . As we can see the argon dimer radial probability densities for higher states have been squeezed to smaller momenta which as we expect leads to larger expectation values for the relative distance between Ar-Ar pair.
5 Conclusion
We have presented the numerical solution of Lippmann-Schwinger integral equation in momentum space to calculate argon dimer binding energies. We have provided full rotational and vibrational energy levels of argon dimer using two models of argon-argon interaction. We have shown in this paper that the solution of Lippmann-Schwinger integral equation in momentum space has the advantages of catching the weakly bound states and numerically less challenging than the solution of Schrödinger equation in configuration space, where the two-body wave function has been expanded to a very large relative distance. Instead, in momentum space, the behavior of the wave function is reversely and for shallow bound states, the wave function is squeezed to a very small momentum which can be numerically treated much more efficiently. Our numerical results for the vibrational levels of the ground state of argon dimer are in excellent agreement with other theoretical predictions, especially with those reported by Patkowski et al. and also by recent results obtained by R-matrix method. The comparison of our numerical results for dimer binding energies with other theoretical results and experimental data indicates that our method is technically feasible, reliable, and a highly efficient method to determine the dimer binding energies. In the next step, we are going to extend a direct integration method called "three-dimensional" approach 14, 15, which has been successfully applied to nuclear bound and scattering systems and avoids the traditional partial wave representation and its complexity, to atomic three- and four-body bound states.
ACKNOWLEDGMENTS
The authors thank Konrad Patkowski for providing dimer binding energy values of the potential models I and II, and also to Tom Rivlin for helpful discussions and for providing dimer binding energies obtained by R-matrix method. This work is performed under the auspices of the National Science Foundation under Contract No. NSF-HRD-1436702 with Central State University. M. R. H. acknowledges the partial support from the Institute of Nuclear and Particle Physics at Ohio University.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aziz 1993 R. A. Aziz, The Journal of Chemical Physics 99 , 4518 (1993), https://doi.org/10.1063/1.466051 , URL https://doi.org/10.1063/1.466051 . · doi ↗
- 2Tang and Toennies 2003 K. T. Tang and J. P. Toennies, The Journal of Chemical Physics 118 , 4976 (2003), https://doi.org/10.1063/1.1543944 , URL https://doi.org/10.1063/1.1543944 . · doi ↗
- 3Patkowski et al. 2005 K. Patkowski, G. Murdachaew, C.-M. Fou, and K. Szalewicz, Molecular Physics 103 , 2031 (2005), URL https://doi.org/10.1080/00268970500130241 . · doi ↗
- 4Patkowski and Szalewicz 2010 K. Patkowski and K. Szalewicz, The Journal of Chemical Physics 133 , 094304 (2010), URL https://doi.org/10.1063/1.3478513 . · doi ↗
- 5Slavicek et al. 2003 P. Slavicek, R. Kalus, P. Paska, I. Odvarkova, P. Hobza, and A. Malijevský, The Journal of Chemical Physics 119 , 2102 (2003), https://doi.org/10.1063/1.1582838 , URL https://doi.org/10.1063/1.1582838 . · doi ↗
- 6Myatt et al. 2018 P. T. Myatt, A. K. Dham, P. Chandrasekhar, F. R. W. Mc Court, and R. J. L. Roy, Molecular Physics 116 , 1598 (2018), https://doi.org/10.1080/00268976.2018.1437932 , URL https://doi.org/10.1080/00268976.2018.1437932 . · doi ↗
- 7Hadizadeh and Khaledi-Nasab 2016 M. Hadizadeh and A. Khaledi-Nasab, Physics Letters B 753 , 8 (2016), ISSN 0370-2693, URL http://www.sciencedirect.com/science/article/pii/S 0370269315009284 .
- 8Hadizadeh and Tomio 2010 M. R. Hadizadeh and L. Tomio, AIP Conference Proceedings 1296 , 334 (2010), URL http://aip.scitation.org/doi/abs/10.1063/1.3523199 .