The crucial role of atomic corrugation on the flat bands and energy gaps of twisted bilayer graphene at the "magic angle" $\theta\sim 1.08^\circ$
Procolo Lucignano, Dario Alf\`e, Vittorio Cataudella, Domenico Ninno,, Giovanni Cantele

TL;DR
This paper demonstrates that atomic corrugation significantly influences the flat bands and energy gaps in twisted bilayer graphene at the magic angle, emphasizing the importance of out-of-plane relaxations for accurate modeling.
Contribution
It combines large-scale first principles calculations with a continuum model to accurately describe flat bands, highlighting the role of atomic corrugation and relaxations.
Findings
Out-of-plane relaxations are essential for accurate flat band width and gap predictions.
Results align quantitatively with recent experimental data.
Atomic corrugation critically affects electronic properties at the magic angle.
Abstract
We combine state-of-the-art large-scale first principles calculations with a low-energy continuum model to describe the nearly flat bands of twisted bilayer graphene at the first magic angle . We show that the energy width of the flat band manifold, as well as the energy gap separating it from the valence and conduction bands, can be obtained only if the out-of-plane relaxations are fully taken into account. The results agree both qualitatively and quantitatively with recent experimental outcomes.
Click any figure to enlarge with its caption.
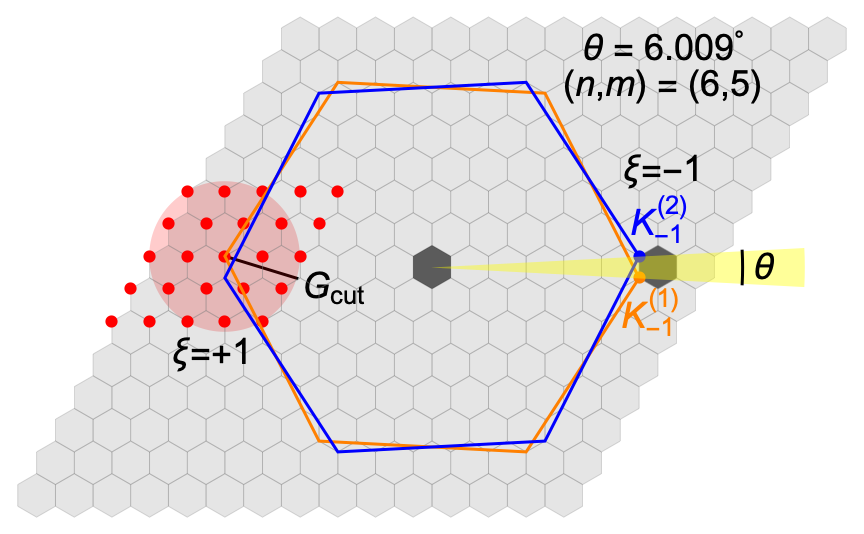 Figure 1
Figure 1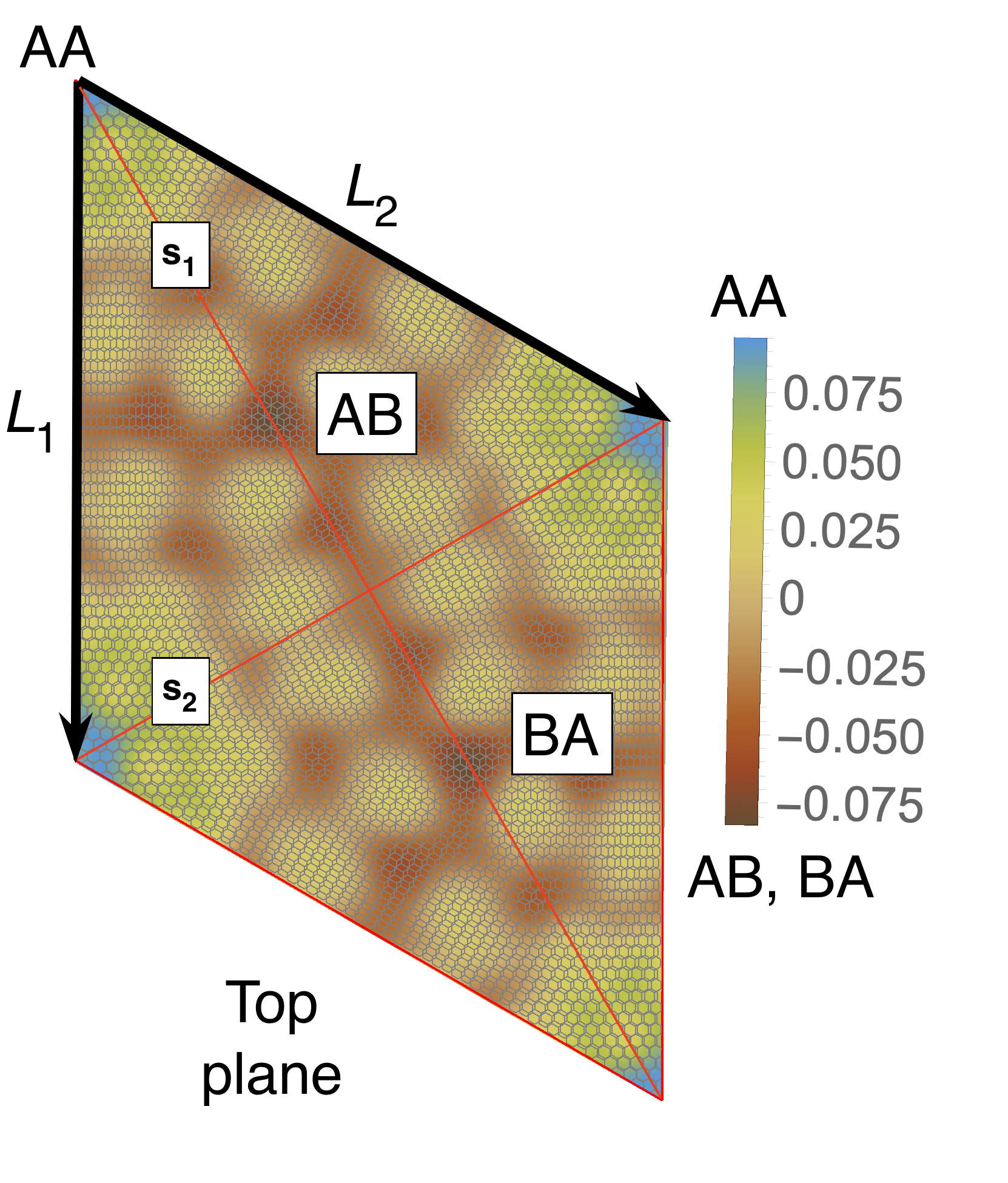 Figure 2
Figure 2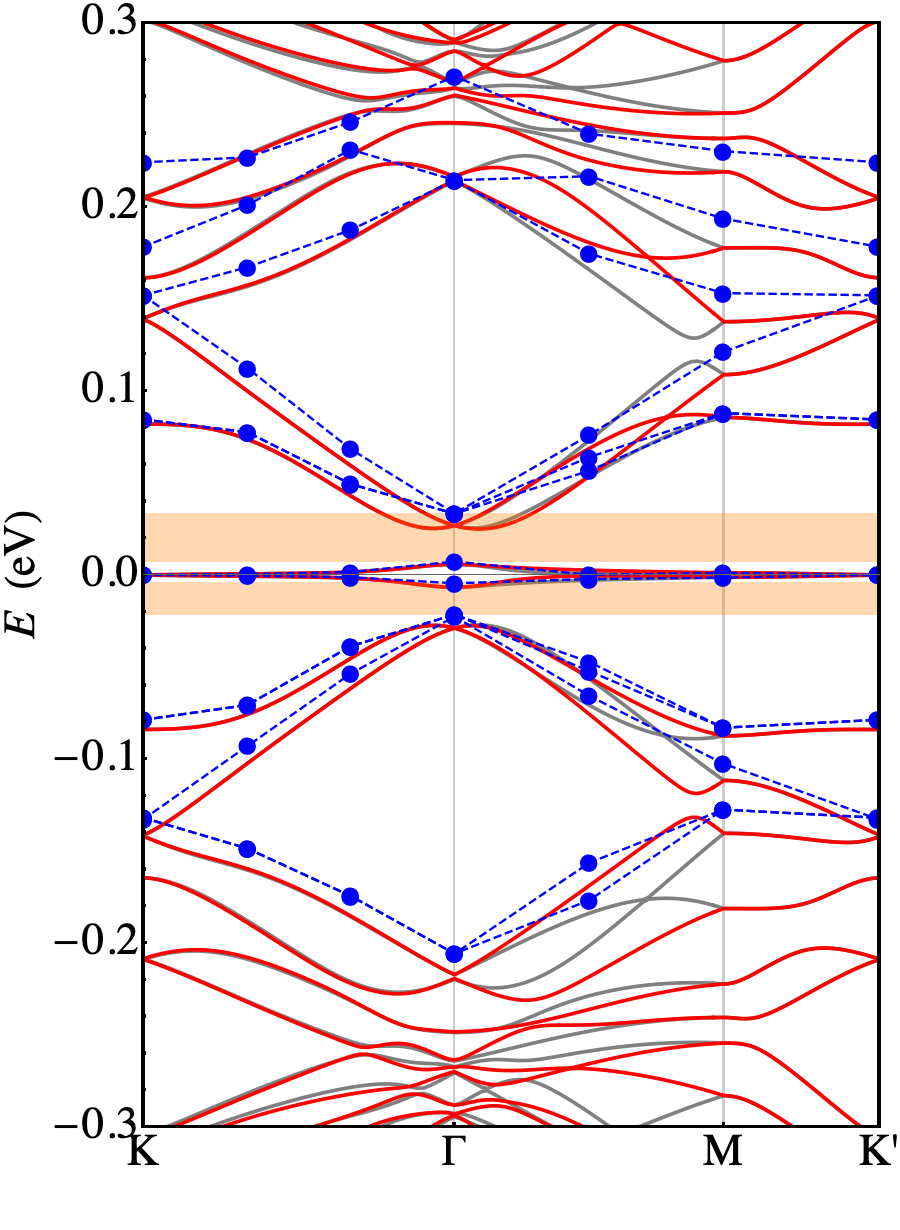 Figure 3
Figure 3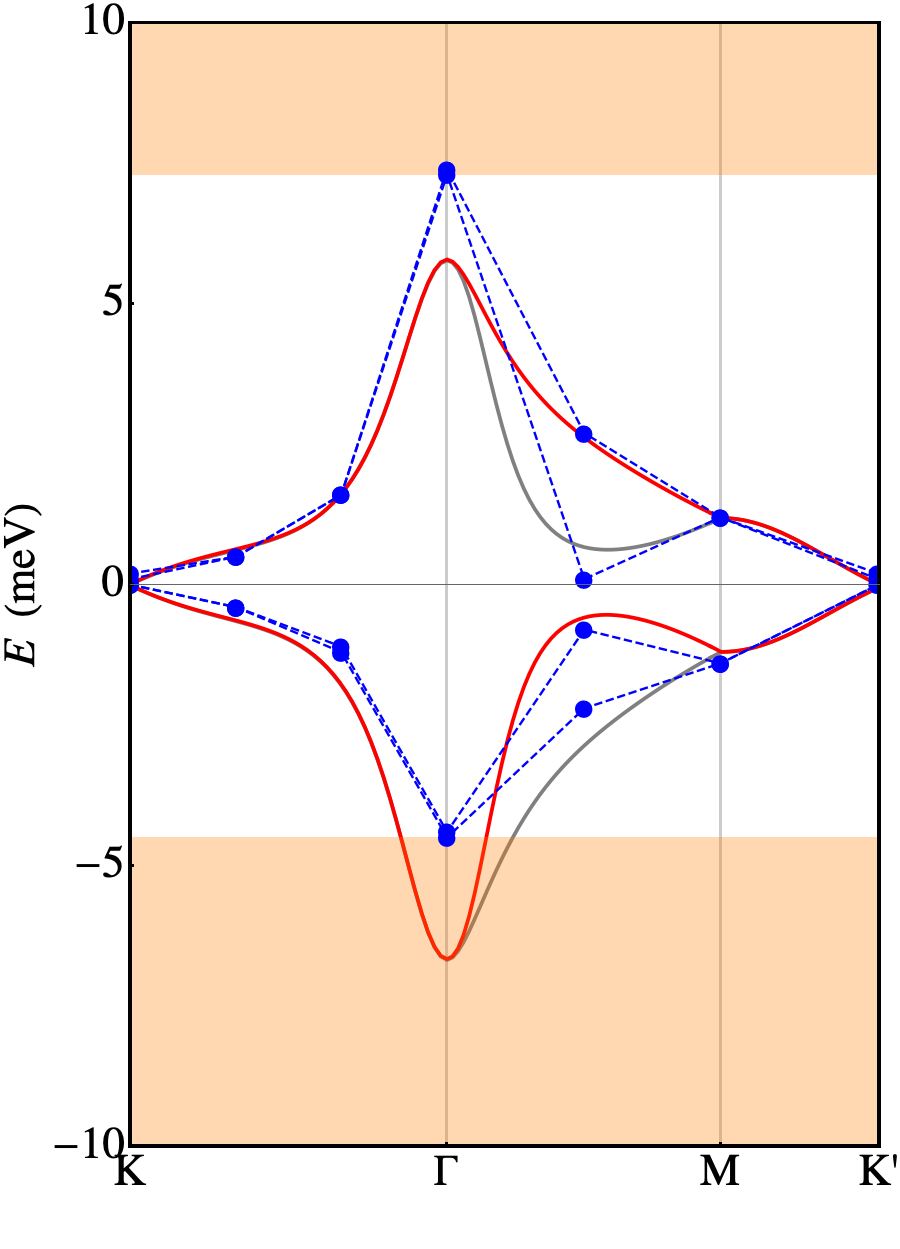 Figure 4
Figure 4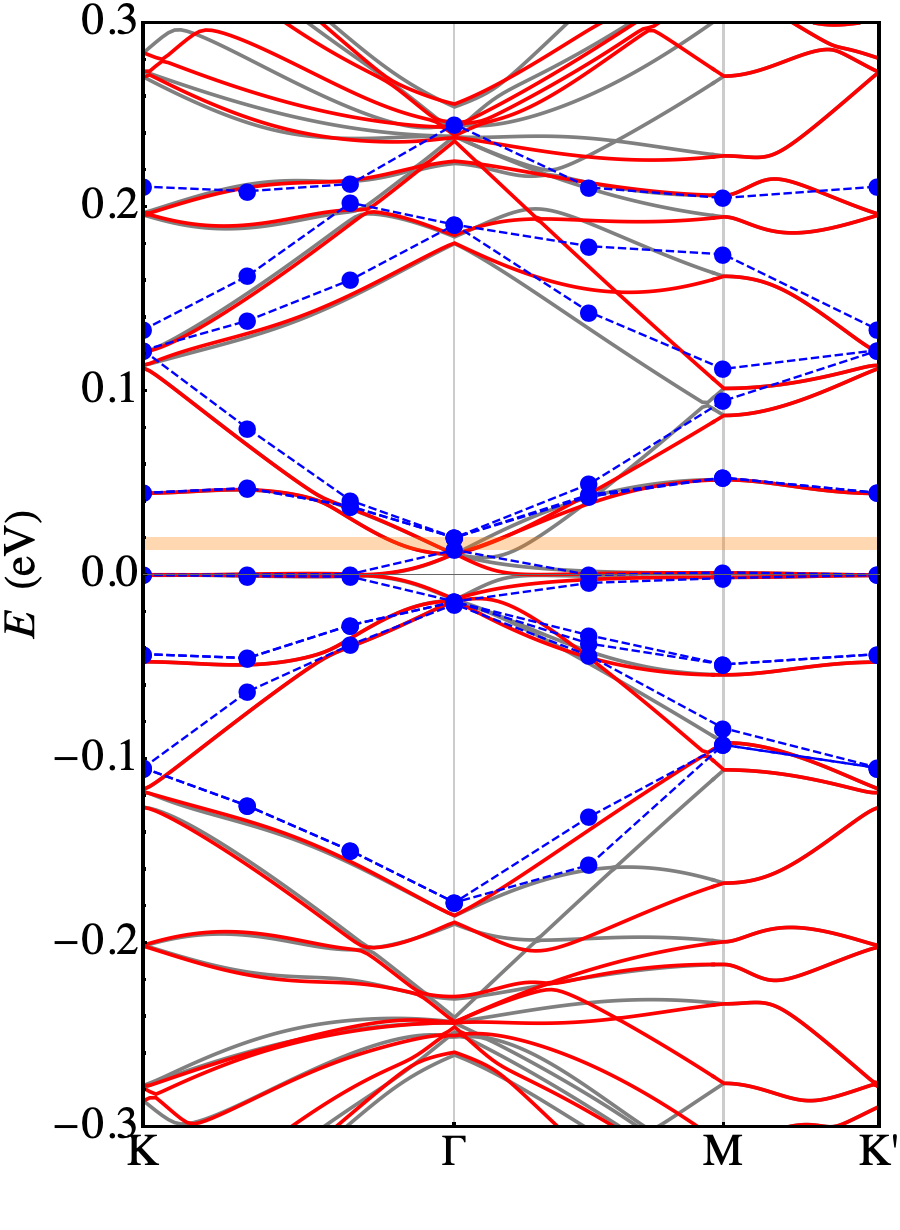 Figure 5
Figure 5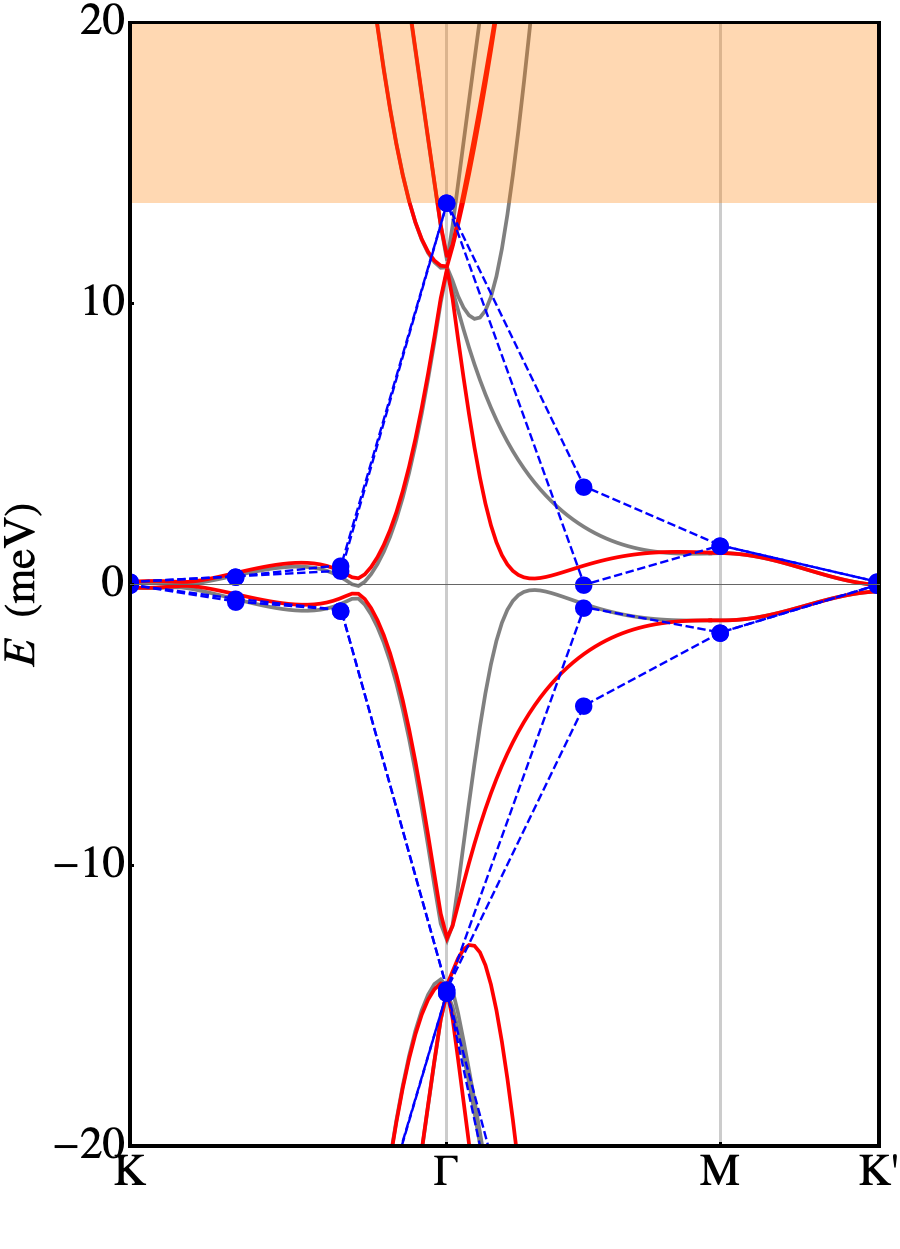 Figure 6
Figure 6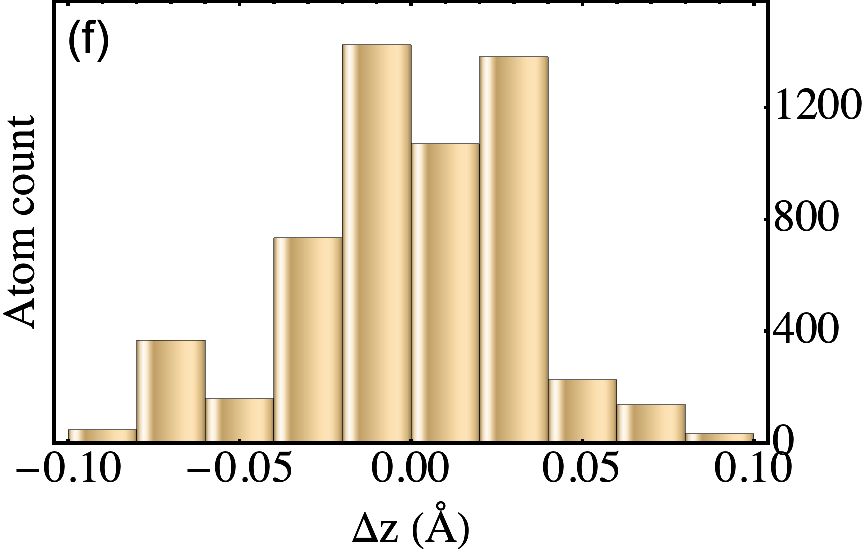 Figure 7
Figure 7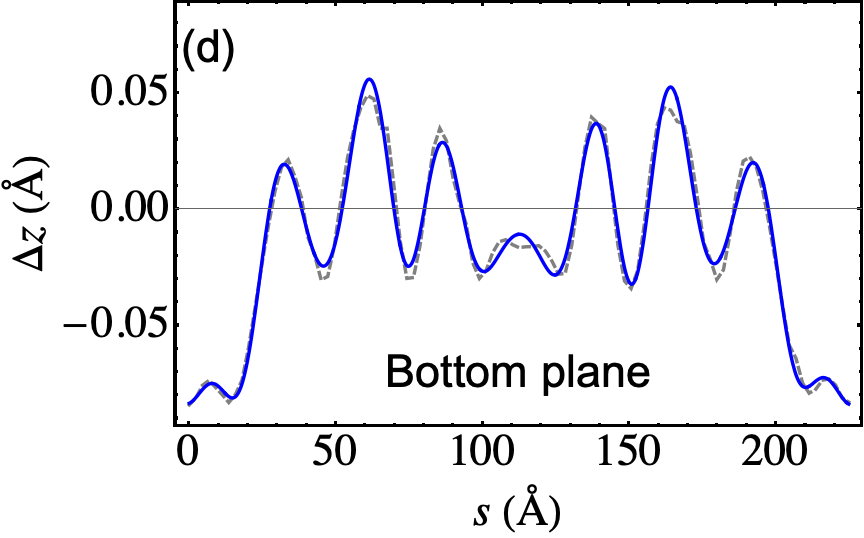 Figure 8
Figure 8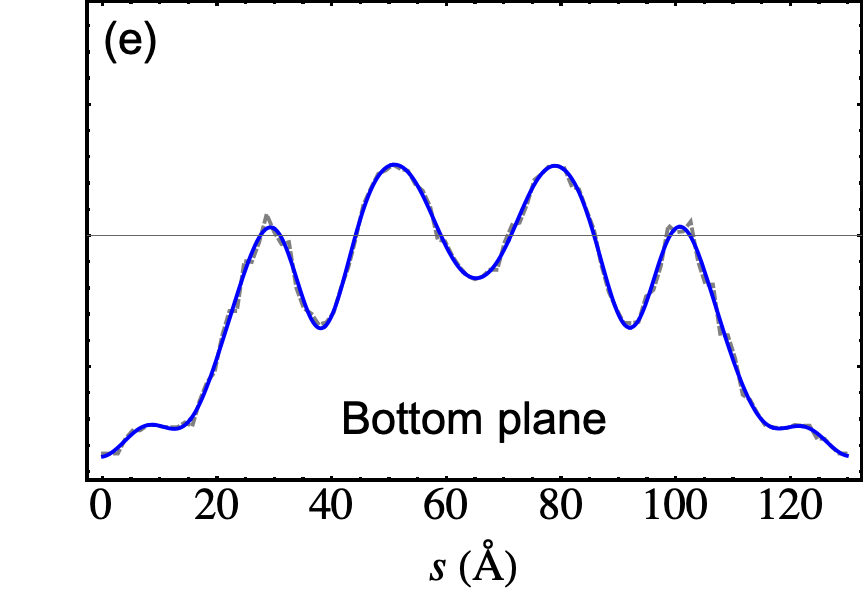 Figure 9
Figure 9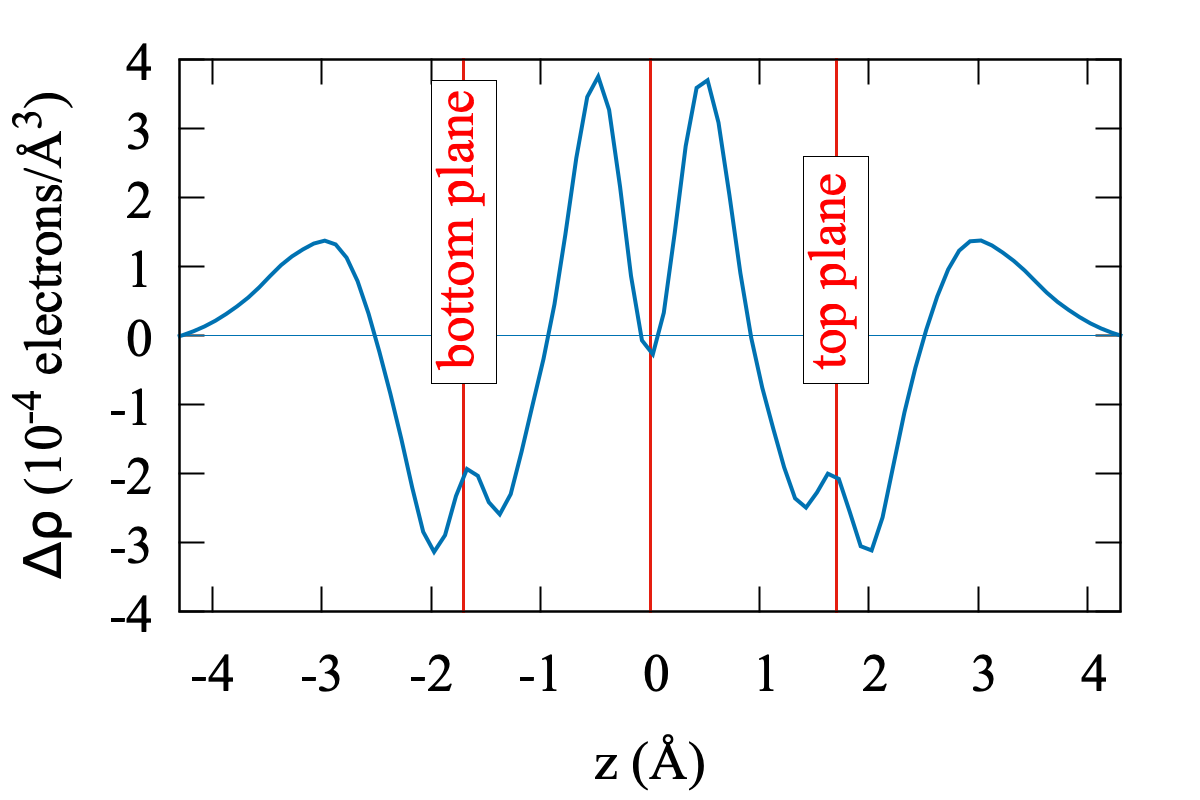 Figure 10
Figure 10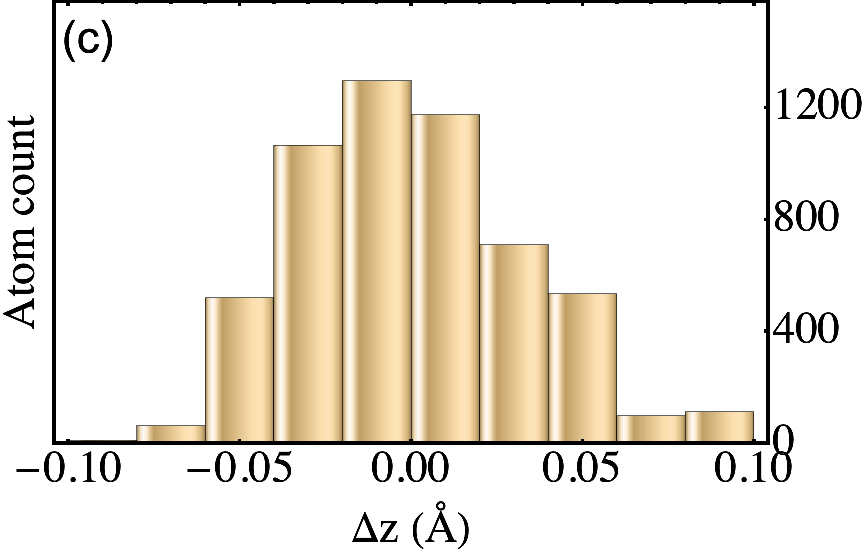 Figure 11
Figure 11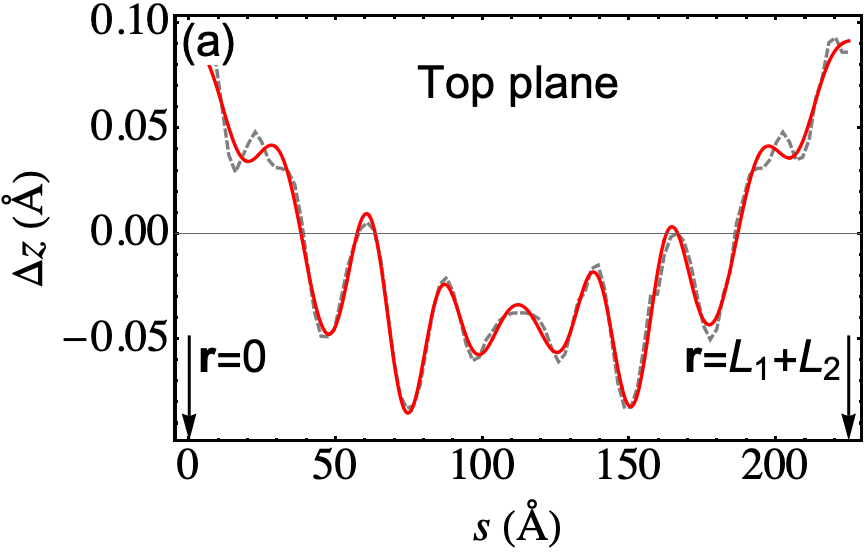 Figure 12
Figure 12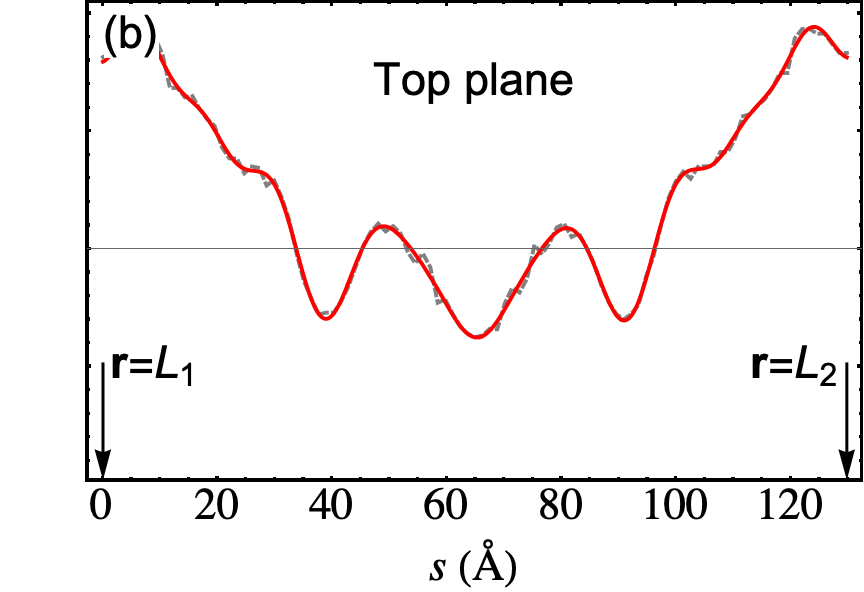 Figure 13
Figure 13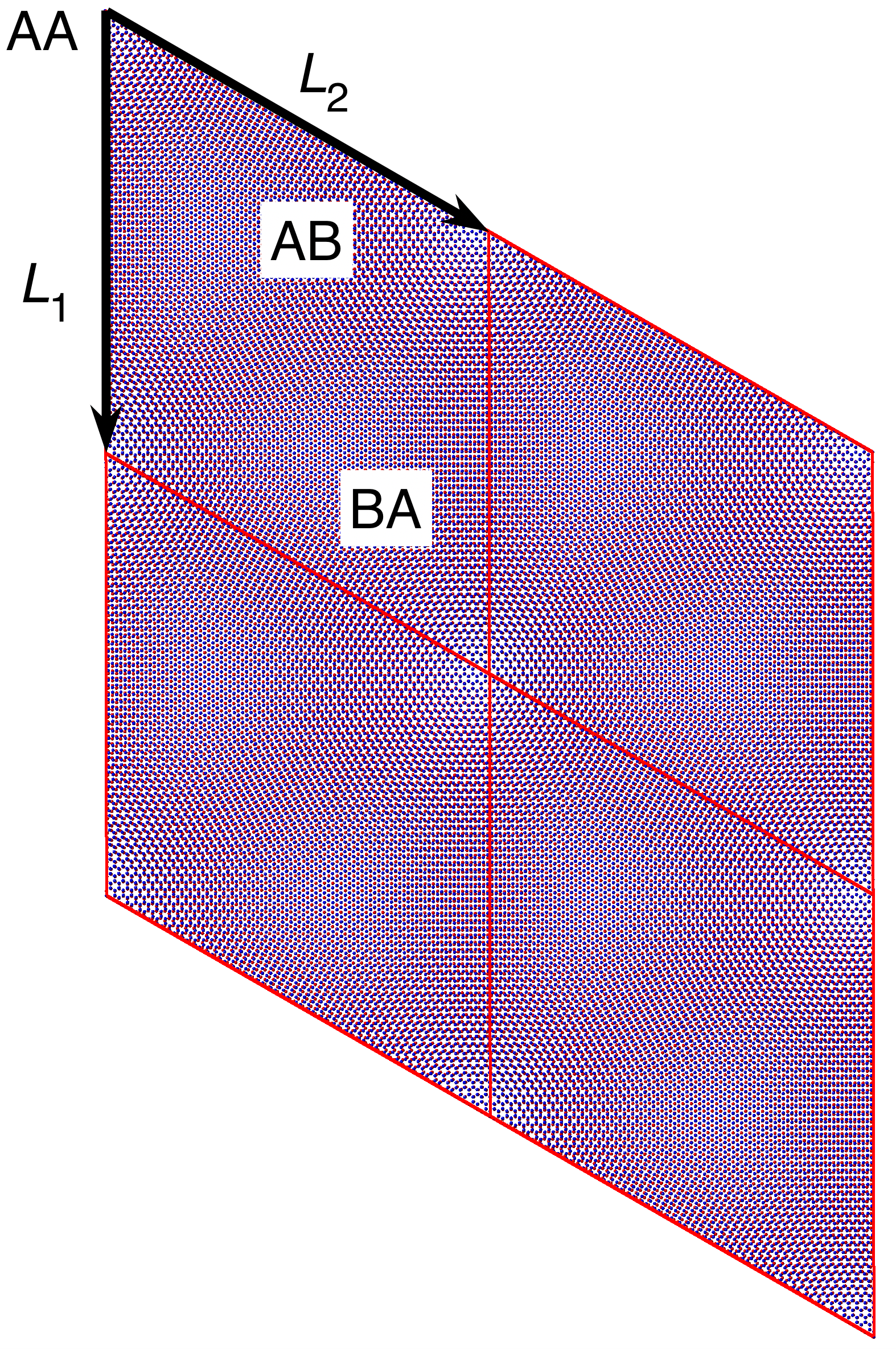 Figure 14
Figure 14Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
The crucial role of atomic corrugation on the flat bands and energy gaps of twisted bilayer graphene at the “magic angle”
Procolo Lucignano
CNR-SPIN, c/o Complesso di Monte S. Angelo, via Cinthia - 80126 - Napoli, Italy
Dario Alfè
Dept. of Earth Sciences and London Centre for Nanotechnology University College London, Gower Street, London, WC1E 6BT, UK
Università degli Studi di Napoli “Federico II”, Dipartimento di Fisica ”Ettore Pancini”, Complesso di Monte S. Angelo, via Cinthia - 80126 - Napoli, Italy
Vittorio Cataudella
Domenico Ninno
Università degli Studi di Napoli “Federico II”, Dipartimento di Fisica ”Ettore Pancini”, Complesso di Monte S. Angelo, via Cinthia - 80126 - Napoli, Italy
CNR-SPIN, c/o Complesso di Monte S. Angelo, via Cinthia - 80126 - Napoli, Italy
Giovanni Cantele
CNR-SPIN, c/o Complesso di Monte S. Angelo, via Cinthia - 80126 - Napoli, Italy
Abstract
We combine state-of-the-art large-scale first principles calculations with a low-energy continuum model to describe the nearly flat bands of twisted bilayer graphene at the first magic angle 1.08∘. We show that the energy width of the flat band manifold, as well as the energy gap separating it from the valence and conduction bands, can be obtained only if the out-of-plane relaxations are fully taken into account. The results agree both qualitatively and quantitatively with recent experimental outcomes.
pacs:
73.22.Pr,73.21.-b
I Introduction
The Moiré patterns originating from the commensurate rotation of two graphene layers with respect to each other have revealed, at small twist angles , that the Dirac cone picture breaks down Cao et al. (2018a, b); Codecido et al. (2019). Twisted Bilayer Graphene (TBG) at the ”magic angle” shows almost flat bands (FBs) at the Fermi energy, with a measured bandwidth as small as meV. The FBs manifold can host up to four electrons above the Fermi energy and four holes below it and is separated by an energy gap of meV from both higher and lower energy bands. When an external gate tunes the system chemical potential within these gaps, a clear band insulating phase appears. A second, unexpected, insulating phase shows up at half-filling of the FB manifold, both on the electron and on the hole side ( electrons with respect to charge neutrality). After electrostatic doping, achieved by gating the structure, unconventional superconductivity, with a 1.7 K critical temperature, appears in a strong pairing regime, with a phase diagram very similar to that of the underdoped cuprates. The latter two features are attributed to enhanced electron-electron or electron-phonon interaction within the FBs, respectively, and are currently under study Choi and Choi (2018); Sboychakov et al. (2018).
This remarkable scenario reveals how the twist angle can be used as a further degree of freedom Ribeiro-Palau et al. (2018) to combine two-dimensional (2D) materials exhibiting vertical stacking, to implement desired properties Geim and Grigorieva (2014); Cantele and Ninno (2017); Borriello et al. (2012); Cantele et al. (2009). The twisted lattice geometry gives rise to topological properties of TBG Song et al. (2018); Hejazi et al. (2019); Liu et al. (2018), by contrast to conventional topological materials Hasan and Kane (2010), where topological properties are mostly due to spin-orbit interactions Bercioux and Lucignano (2015); Lucignano et al. (2008) and Brillouin zone topology.
In this paper we focus on the band insulating phase, that requires an accurate description of the single-electron properties determining the band structure of the TBG. These electronic properties have been addressed mostly adopting continuum effective models, focusing only on low-energy states Lopes dos Santos et al. (2007, 2012); Mele (2010); Bistritzer and MacDonald (2011); Moon and Koshino (2013); Koshino et al. (2018), tight binding Jung and MacDonald (2013); Sboychakov et al. (2015); Fang and Kaxiras (2016); Lin and Tománek (2018); Kang and Vafek (2018); Angeli et al. (2018) or calculations using time-dependent Schrödinger approaches Nam Do et al. (2018); Le and Do (2018). The point is that the unit cell, at the first magic angle , contains 11164 atoms, and this makes it impossible to perform a full many body calculation (details about the lattice geometry and the reciprocal space can be found in appendix A). Moreover, even a full description of the system in the framework of ab-initio density functional theory (DFT), including atomic relaxations, remains challenging (the corresponding supercell is described by an hexagonal lattice with an in-plane lattice parameter of Å). To the date, there are very few first-principles calculations Kang and Vafek (2018); Song et al. (2018) on TBG at small twist angles. For example, in Ref. Kang and Vafek, 2018 the energy bands obtained from DFT calculations carried out on the unrelaxed structure, are reported. However, the ab initio results at the magic angle show no gap between the FB manifold and the closest lower band, although a band set consistent with the experimental outcomes is reproduced at a larger angle, , corresponding to a smaller unit cell (7804 atoms to be contrasted with 11164 at the first magic angle). However, since the experimental uncertainty over the measured angle in Ref. Cao et al., 2018a is of the order of , a thorough theoretical description at is demanding.
In this manuscript we present a fully ab initio DFT calculation of the electronic structure of TBG at showing that a band structure consistent with that measured in Ref. Cao et al., 2018a is obtained provided that the out-of-plane atomic relaxations are fully taken into account. In particular, the occurrence of energy gaps between the FBs and the lower and higher energy bands emerge as a direct consequence of the corrugation due to the out-of-plane displacements.
Such result is strictly related with the nature of the FBs close to the Fermi energy: indeed, we can infer that the appearance of these bands is the result of the inter-layer vdW interaction, whose effect can be tuned by controlling the twist angle. The out-of-plane relaxation of the atomic positions obviously modifies the strength of the interaction, as we are going to discuss in the following.
The paper is organized as follows. In sec. II we describe the ab-initio calculation with particular attention to the geometric optimization of the superlattice. In Sec. III we introduce the low energy effective continuum model of Ref.s Lopes dos Santos et al., 2007, 2012; Mele, 2010; Bistritzer and MacDonald, 2011; Moon and Koshino, 2013; Koshino et al., 2018 and specialize it to describe our optimized structures. In Sec. IV we show our numerical results. In Sec. V we summarize our findings. Appendices A, B and C describe details of the geometrical structure, of the DFT ab initio calculation and of the low energy continuum model, respectively.
II Geometric Optimization
Although previous studies have pointed out that the atomic corrugation due to inter-plane vdW interaction might have relevant effects on the band structure and on the effective point symmetries Uchida et al. (2014); van Wijk et al. (2015); Lin et al. (2018); Angeli et al. (2018), they were mainly based on molecular dynamics and classical interatomic potentials Fang and Kaxiras (2016); Gargiulo and Yazyev (2018); Jain et al. (2016), which, as such, can only give a partial answer to the problem posed.
Here, DFT calculations, using the vdW-DF2 exchange-correlation functional Hamada (2014), have been carried out using the Vienna Ab initio Simulation Package (VASP) Kresse and Furthmüller (1996). The atomic positions have been fully optimized, as detailed in appendix B. The calculation required from 2880 (distributed over 80 nodes) up to 5760 physical cores (distributed over 160 nodes) of a Cray XC-40 machine, over a period of about 30 days.
The outcome of the geometry optimization is depicted in Fig. 1, where we use a color map to show the out of plane deformation of the top layer. Inspection shows that atomic relaxation tends to increase the interplane distance in correspondence to the AA stacking regions, and to decrease it in the AB regions. Out-of plane displacements are modulated on length scale, intermediate between the Moiré and the graphene periodicity, and seem to be in agreement with the emergent D6 symmetry described in Ref. Angeli et al., 2018. As we will show in the following, these geometric properties have severe consequences on the electronic band structure of the system. Details on the optimized structure are shown in Fig. 2. In the top panels we illustrate the characteristics of the TBG top plane: in particular, in Fig.s 2(a)-(b) we show the corrugation profile respectively along the line and the line (both shown in Fig. 1). In Fig. 2(c) we show an histogram representing the atomic population binned according to the -coordinate of the atoms. The same features are shown in Fig.s 2(d)-(e)-(f) for the the TBG bottom plane. The corrugations show clear oscillations, along the lines and , however a simple analytical expression interpolating between the atomic position is not easily accessible, due to large harmonic content of the oscillatory behavior. The peaks of the histograms define an average -coordinate for the top and for the bottom plane. Their difference defines the average interlayer spacing Å that is half way between the equilibrium distances of 3.31 Å and 3.496 Å between consecutive planes in graphite with AB Bernal and AA stacking, respectively (both calculated using the same vdW-DF2 exchange-correlation functional). Within each plane, the atomic displacements occur within an interval of about 0.2 Å (0.1 Å with respect to the average in each plane).
III Effective Continuum model
For increasing the understanding of the ab initio results, we also describe a continuum model generalizing the model proposed in Ref.s Lopes dos Santos et al., 2007; Bistritzer and MacDonald, 2011; Moon and Koshino, 2013; Koshino et al., 2018, providing an effective low-energy band structure which shows a remarkable agreement with the DFT calculation. Our results can be viewed as an accurate single-particle description of TBG at the first magic angle and used as starting point for a full many-body calculation taking into account electronic correlations. In the following we shortly summarize the model, referring to Refs. Lopes dos Santos et al., 2012; Moon and Koshino, 2013; Koshino et al., 2018 for further details. At small twist angles, the Moiré period is much longer than the lattice constant . The superlattice Mini Brillouin Zone (MBZ) extends over a tiny small area of the graphene BZ, and it is an hexagon whose vertices are the two Dirac points and of the two layers after rotation (compare small and large hexagons in Fig. A2), where is the valley index. Close to these points the single-layer graphene spectrum can be safely assumed to be linear and a low energy (long wavelength) Hamiltonian of each layer can be used:
[TABLE]
where eV and are Pauli’s matrices. In the following we neglect the intervalley mixing, because of the huge distance between the two graphene valleys on the MBZ scale. Hence each valley can be studied separately. In the presence of inter-layer interaction the bilayer system can be described by the matrix Hamiltonian
[TABLE]
The off-diagonal coupling terms are expressed in terms of overlap integrals (see appendix C). The parameters and are calculated in Ref. Koshino et al., 2018 at and kept constant when calculating the band structure for all the points in the MBZ. As the MBZ is a small hexagon of side , it seems to be a reasonable approximation, in particular at small twist angles. In our calculations, in order to give a minimal model capable of describing (at least) the low energy properties of the ab-initio band structure, we do not calculate but use them as fitting parameters. In the following we will show that the fitted parameters are relatively close to (but quantitatively different from) those obtained performing the hopping integrals shown in the appendix C. Such discrepancy can be ascribed to the fact that a low energy effective model obtained expanding a tight-binding Hamiltonian around the point does not entail all the complexity of the full-ab-initio approach, but nevertheless can constitute a relevant tool to get closer and closer to the desired solution, with an accurate choice of the model parameters.
The wave function is calculated as a linear superposition of plane waves of momentum where are reciprocal lattice vectors. The point expansion extends, in principle, over the full (infinite) set of vectors. However, for numerical purposes this set has to be truncated. We choose a cutoff radius , and keep only the vectors inside the sphere of radius . It turns out that the number of required vectors to converge the lowest energy states is rather small. The low-energy continuum Hamiltonian matrix has the dimension , and (as in the example reported in the figure) allows for a good convergence in an energy shell of few hundreds on meV around the Fermi energy, whereas full convergence, i.e. band energies converged within less than 1 meV, is achieved with .
IV Results and Discussion
In Fig. 3 we represent the band structure calculated in the absence of structural relaxation. The blue dots are the outcome of a DFT ab-initio calculation, while the full curves are obtained diagonalizing the continuum model in the two valleys: . The parameters eV are obtained after the fitting procedure on the ab-initio points. The results show a reasonable agreement between these two approaches. Here we discuss some relevant features emerging from our numerical calculations.
First of all we may notice that the FB has a dispersion of 20 meV (calculated ab-initio) which is almost twice as the one measured in the experiment of Ref. Cao et al., 2018b. Another relevant issue is that the unrelaxed ab-initio calculation is not able to reproduce the gap between the FB and the first excited bands (both on the electron and on the hole side) that are responsible for the band insulating phases. Our calculation performed adopting the low-energy continuum model shows a good agreement with the ab-initio calculation and reproduces all its relevant features. Few discrepancies shows up, only when zooming in the very fine details of the FB (see the bottom panel of Fig. 3), that are not relevant as they do not change the way the data compare with experiments. However, these results point in the direction that it is not possible to interpret experimental data using an unrelaxed structure.
Relaxation of the structure drastically changes the scenario, with a substantial agreement with the experiments. Plots of the relaxed band structures are shown in Fig. 4. The parameters eV, eV are again obtained fitting the ab-initio band structure. The FB, now extends for meV around the Fermi level (calculations performed with larger supercells with -axis = 12 and 14 Å show that this number is subject to an error of approximately 3 meV). That bandwidth is in good agreement with the one measured in experiments ( 10 meV, see Ref. Cao et al., 2018a). It is separated by a gap of 26 meV (16 meV) from the highest occupied (lowest unoccupied) bands to be compared with the thermal activation gap of 40 meV measured in experiments. Such discrepancy is not much larger than the convergence error in the DFT calculations. It is clear that the vdW inter-plane interactions, despite being weak, play a crucial role in determining the details of the TBG at the meV level. This is confirmed by the computed charge transfer, shown in Fig. 5. It is defined as where is the total charge density of TBG, whereas are the charge densities of the top and bottom plane, respectively, calculated removing the other plane from the supercell at frozen atomic positions. Fig. 5 shows the average charge transfer over planes orthogonal to the axis. It turns out that an electronic charge depletion shows up in correspondence of both planes and most of the charge is redistributed in the interplane region.
V Conclusions
To summarize, our calculations can finally give a clear explanation of some of the most striking features of the electronic structure of TBG at the first magic angle . In particular, the extension of the FBs and the presence of band gaps separating them from excited states, both on the negative and positive energy side, can be explained successfully in terms of atomic out-of-plane displacements. By allowing a full ab initio structural optimisation, a non-negligible atomic corrugation shows up in both the graphene layers. As expected from simple electrostatic arguments, the interlayer distance gets larger (smaller) in correspondence of AA (AB/BA) stacking regions, with a maximum (minimum) distance of 3.68 Å (3.28 Å). Such corrugation is a direct consequence of the interplay between vdW interaction and twisting of the graphene layers. It implies a decrease of the FB bandwidth of 4 meV and induces gaps between the FB and the closer bands, in good agreement with the experimental findings. Our ab initio results can also be interpreted in terms of a simple continuum model in which interplane hopping potentials have been used as fitting parameters. This simple model reproduces with reasonable accuracy the electronic structure and could pave the way for further investigations, to better describe also the other relevant phases of the TBG at small twist angle, i.e. the superconducting and the correlated insulating one.
Acknowledgements.
We thank D. Bercioux, M. Fabrizio and A. Tagliacozzo for fruitful discussions. We acknowledge use of the Monsoon2 system, a collaborative facility supplied under the Joint Weather and Climate Research Programme, a strategic partnership between the UK Met Office and the Natural Environment Research Council.
G.C. and P.L. contributed equally to this work.
Appendix A Lattice Geometry and Reciprocal Space
Let , be the vectors defining the graphene primitive cell, where , and nm is the lattice constant. The corresponding reciprocal lattice vectors are , and . In the absence of geometric relaxation, let us consider, as starting point, the unrotated bilayer, with perfect AA stacking (each C atom in the first layer laying exactly on top of a C atom in the second layer). Choosing a pair of stacked C atoms, each belonging to one of the layers, the twisted bilayer at angle can be obtained by rotating the first and the second layer around the axis passing through these atoms (that indeed are fixed points of the rotation), by and respectively. After rotation, the Bravais direct lattices of the first and of the second layer are described by the vectors and the reciprocal lattice by the vectors where identifies the layer and is a two-dimensional matrix describing the rotation by . For an arbitrary rotation angle, the resulting structure shows a Moiré pattern but is aperiodic and cannot be described through a Bravais lattice because the periods of the two layers are, in general, incommensurate. However, periodic structures can be achieved when is the angle between two lattice vectors and with being an arbitrary pair of integers. The points at and merge after the rotation of the two planes and the lattice vectors of the Moiré supercell (MS) are thus given by and . The rotation angle can be expressed in terms of the integers as . The magic angle corresponds to , with the number of atoms in the unit cell given by . As , the lattice constant nm is coincident in this case with the Moiré pattern period . Moon and Koshino (2013)
The top panel of Fig. 1 shows the atomic structure of the TBG at (four unit supercells are shown). The Moiré patterns originating from regions with different stacking are highlighted: AA and AB/BA, AB (BA) corresponding to the stacking of a C atom in the top (bottom) layer and the center of an hexagon in the other layer.
The reciprocal lattice vectors for the Moiré pattern are obtained as . The resulting Mini Brillouin Zone (MBZ) is shown as a dark hexagon in the center of Fig. A2. It should be noticed that the Brillouin zones (BZs) of the two graphene layers are rotated with respect to each other by the same angle as the graphene layer themselves, as shown in Fig. A2 (blue and orange bigger hexagons). As such, each graphene layer has its Dirac points at where labels the valley index. For example, in the same figure we show the points at the valley for both layers. It turns out that the coincides with one of the sides of the MBZ.
Appendix B DFT Ab-initio calculations
Density Functional Theory calculations, using the vdW-DF2 exchange-correlation functional Hamada (2014), have been carried out using the Vienna Ab initio Simulation Package (VASP) Kresse and Furthmüller (1996). We used a PAW potential Blöchl (1994); Kresse and Joubert (1999) for carbon with the 2p orbitals in valence, and the 1s orbitals frozen in the core. The single particle Bloch waves were expanded with a plane wave basis set, using a cutoff energy of 400 eV. Sampling of the BZ for the self-consistent (SCF) calculations was restricted at the point. Single-particle energies at other points in the BZ were obtained by non-SCF calculations. Because of the size of the simulation cell, we could only compute one -point at a time, and the reported single-particle energies were therefore referred to the Fermi energy computed as the energy at the point. The size of the supercell in the direction orthogonal to the layers (-axis) was initially fixed at 10 Å, corresponding to about 6.5 Å vacuum space, introduced to prevent periodic replicas of the TBG supercell from interacting with each other. Full relaxation of the atomic positions was carried out until the residual forces were smaller than 0.002 eV/Å. Additional calculations were repeated using supercells with -axis of 12 Å and 14 Å. A small residual (maximum) relaxation of less than 0.002Å was observed as the -axis was increased to 12 Å, but no further relaxation was detectable with the largest 14 Å vacuum space. The initial relaxation was carried out using 2880 physical cores distributed over 80 nodes of a Cray XC-40 machine, over a period of about 30 days. Calculations with 14 Å vacuum required 5760 cores on 160 nodes to accommodate the extra memory requirements. All symmetries were turned off.
Appendix C Effective Continuum model
In the presence of inter-layer interaction the bilayer system can be described by the matrix Hamiltonian
[TABLE]
The interlayer Hamiltonian is
[TABLE]
with . It couples each point of the layer to a point of the layer according to the selection rules . The coefficients are given in Ref. Koshino et al., 2018:
[TABLE]
where is the unit cell area of the pristine graphene and is the transfer integral between two sites at distance , originating from the Slater-Koster tight binding parametrization for carbon atoms:
[TABLE]
Here is the decay length of the transfer integral, is the first-neighbor distance in graphene, nm is the intralayer distance, chosen in agreement with that of graphite. and are the in-plane and out of plane nearest-neighbours hopping energy as from Ref. Moon and Koshino, 2013. We seek for solutions of the kind:
[TABLE]
The point expansion extends, in principle, over the full (infinite) set of vectors. However, for numerical purposes this set has to be truncated. We choose a cutoff radius , and keep only the vectors inside the sphere of radius . This is schematically shown in Fig. A2, where the shadowed circle of radius includes the subset of vectors, represented by the red dots. It turns out that the number of vectors to converge the lowest energy states is rather small. The low-energy continuum Hamiltonian matrix has the dimension , and (as in the example reported in the figure) allows for a good convergence in an energy shell of few hundreds on meV around the Fermi energy, whereas full convergence, i.e. band energies converged within less than 1 meV, is achieved with .
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cao et al. (2018 a) Y. Cao, V. Fatemi, A. Demir, S. Fang, S. L. Tomarken, J. Y. Luo, J. D. Sanchez-Yamagishi, K. Watanabe, T. Taniguchi, E. Kaxiras, R. C. Ashoori, and P. Jarillo-Herrero, Nature Publishing Group 556 , 80 (2018 a).
- 2Cao et al. (2018 b) Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras, and P. Jarillo-Herrero, Nature Publishing Group 556 , 43 (2018 b).
- 3Codecido et al. (2019) E. Codecido, Q. Wang, R. Koester, S. Che, H. Tian, R. Lv, S. Tran, K. Watanabe, T. Taniguchi, F. Zhang, M. Bockrath, and C. N. Lau, (2019), ar Xiv:1902.05151 [cond-mat.mes-hall] .
- 4Choi and Choi (2018) Y. W. Choi and H. J. Choi, Phys. Rev. B 98 , 241412(R) (2018) . · doi ↗
- 5Sboychakov et al. (2018) A. O. Sboychakov, A. V. Rozhkov, A. L. Rakhmanov, and F. Nori, ar Xiv (2018), arxiv:1807.08190 v 1 .
- 6Ribeiro-Palau et al. (2018) R. Ribeiro-Palau, C. Zhang, K. Watanabe, T. Taniguchi, J. Hone, and C. R. Dean, Science 361 , 690 (2018).
- 7Geim and Grigorieva (2014) A. K. Geim and I. V. Grigorieva, Nature 499 , 419 (2014).
- 8Cantele and Ninno (2017) G. Cantele and D. Ninno, Phys. Rev. Materials 1 , 014002 (2017) . · doi ↗