Quantum information-based analysis of electron-deficient bonds
Jan Brandejs, Libor Veis, Szil\'ard Szalay, Gergely Barcza, Ji\v{r}\'i, Pittner, and \"Ors Legeza

TL;DR
This paper applies quantum information theory to analyze electron-deficient bonds, successfully describing complex bonding in molecules like diborane and novel compounds with unusual bonding patterns.
Contribution
It extends the correlation theory of chemical bonds to electron-deficient bonds, demonstrating its effectiveness on complex and recently synthesized molecules.
Findings
Successfully characterized three-center two-electron bonds in diborane(6)
Described bonding in diborane(4) and beryllium complexes with unusual stability
Validated the correlation theory as a tool for analyzing exotic chemical bonds
Abstract
Recently, the correlation theory of the chemical bond was developed, which applies concepts of quantum information theory for the characterization of chemical bonds, based on the multiorbital correlations within the molecule. Here for the first time, we extend the use of this mathematical toolbox for the description of electron-deficient bonds. We start by verifying the theory on the textbook example of a molecule with three-center two-electron bonds, namely the diborane(6). We then show that the correlation theory of the chemical bond is able to properly describe bonding situation in more exotic molecules which have been synthetized and characterized only recently, in particular the diborane molecule with four hydrogen atoms [diborane(4)] and neutral zerovalent s-block beryllium complex, whose surprising stability was attributed to a strong three-center two-electron bond…
Click any figure to enlarge with its caption.
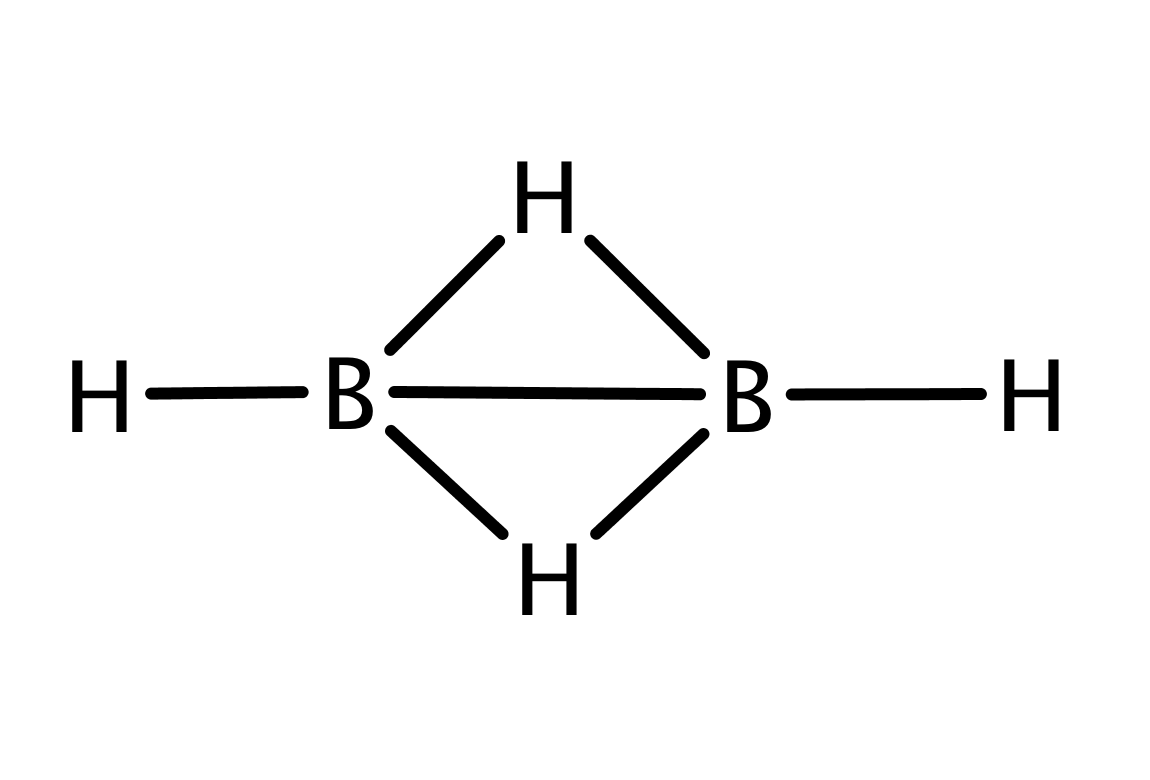 Figure 1
Figure 1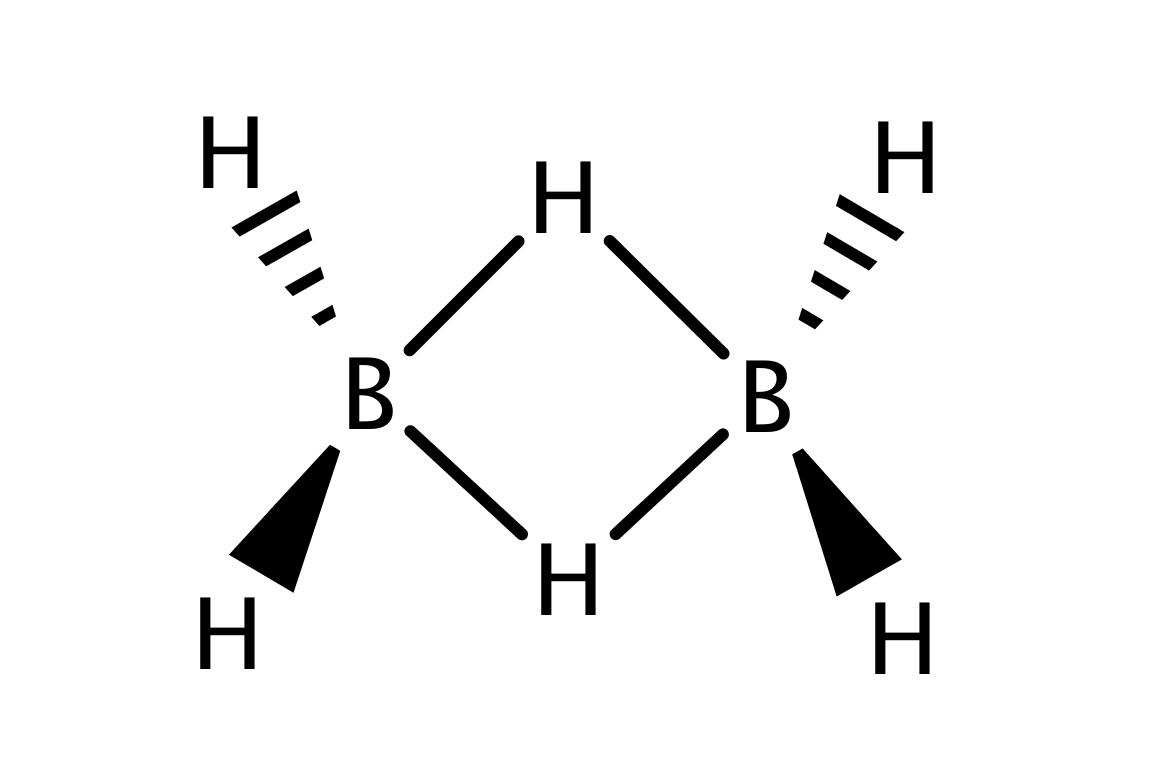 Figure 2
Figure 2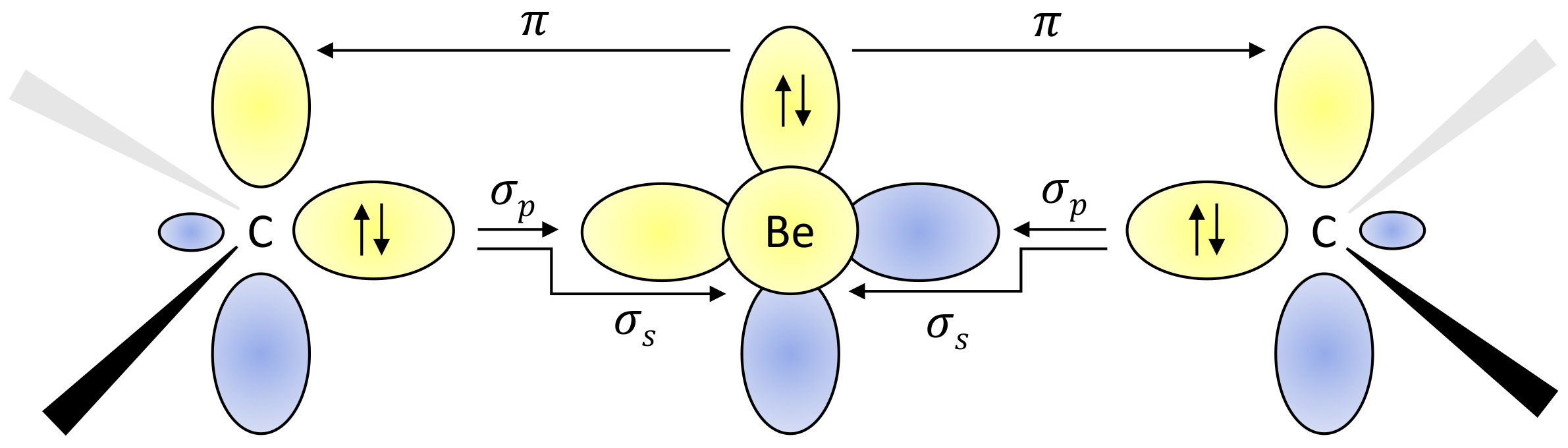 Figure 3
Figure 3 Figure 4
Figure 4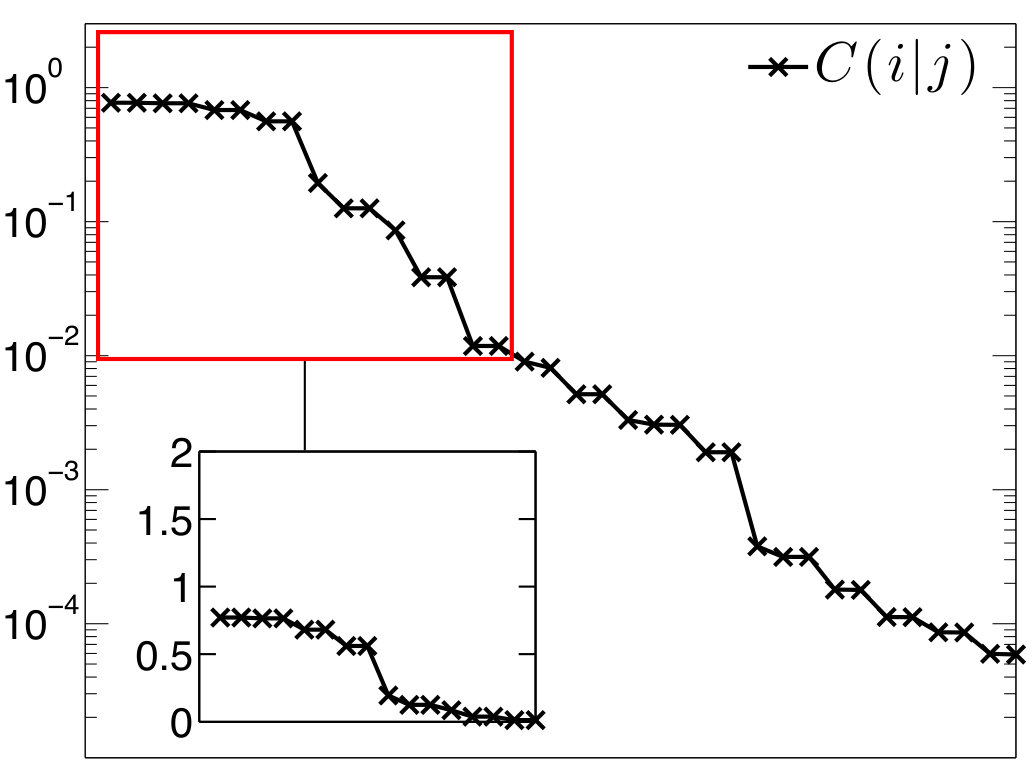 Figure 5
Figure 5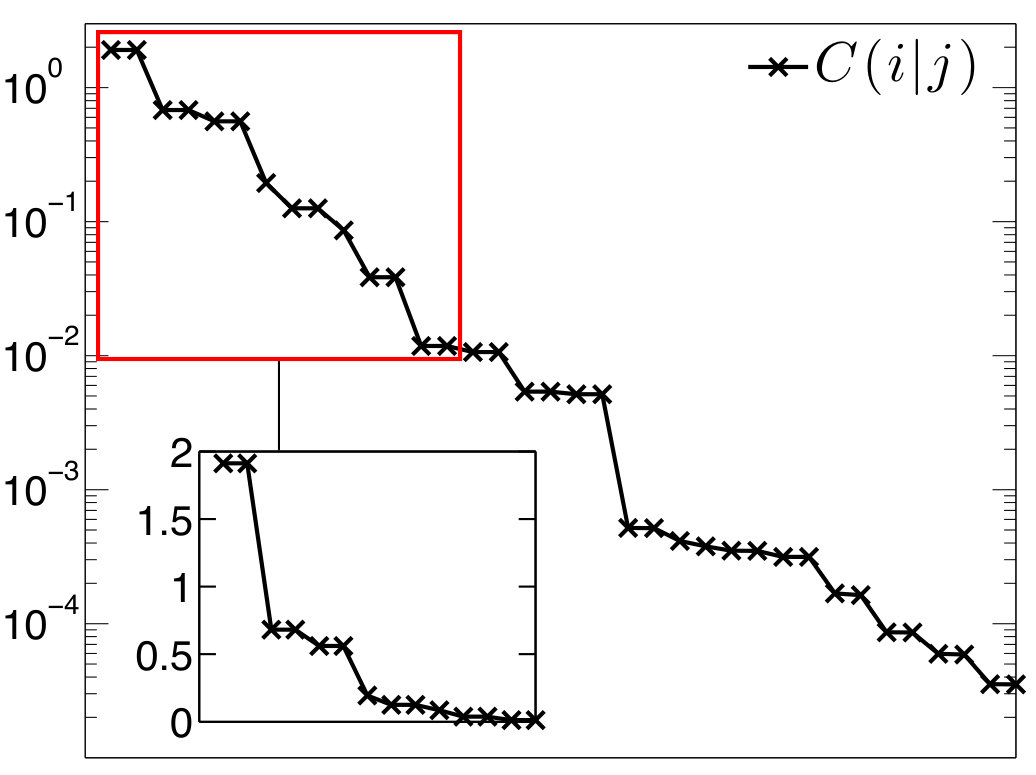 Figure 6
Figure 6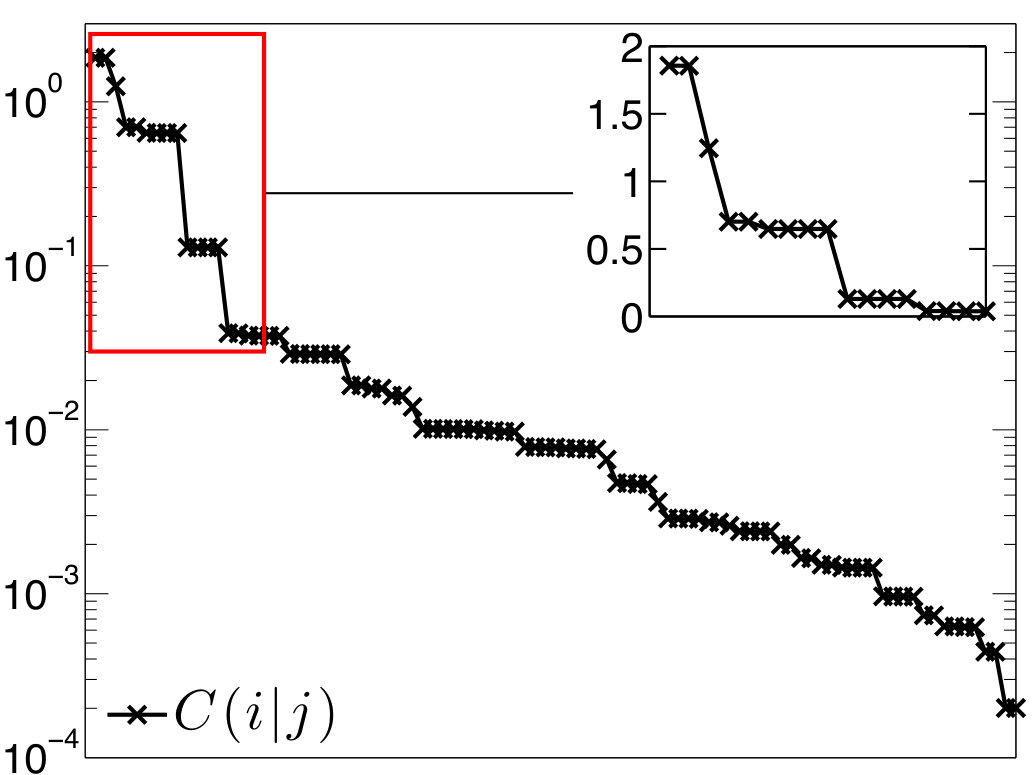 Figure 7
Figure 7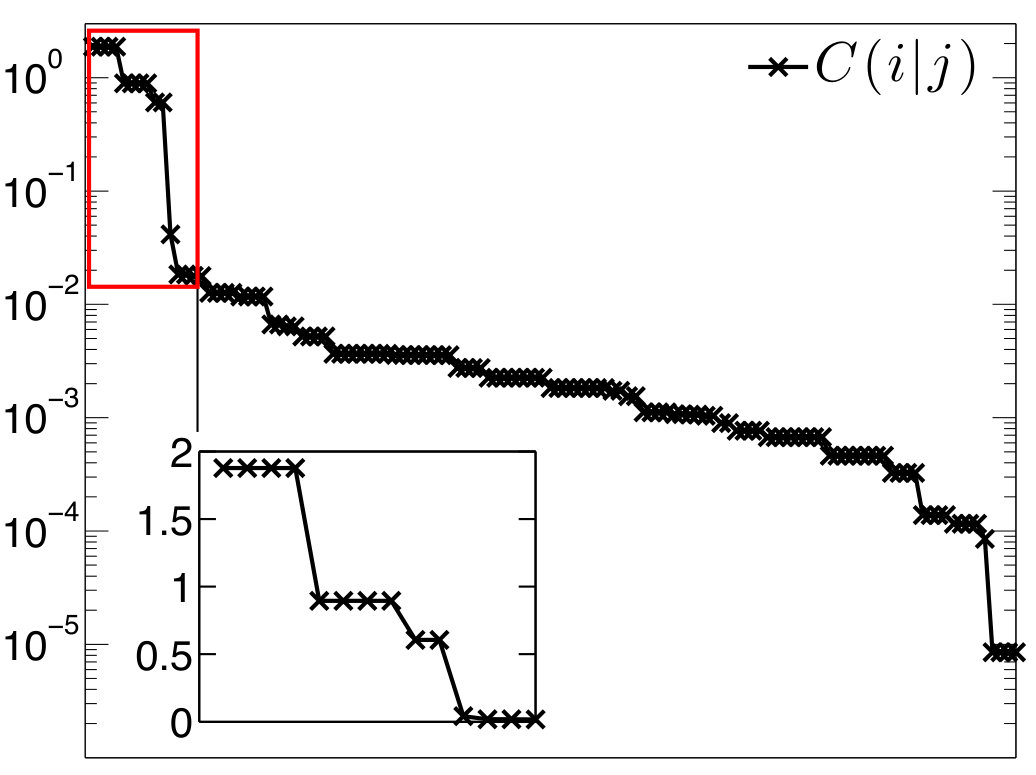 Figure 8
Figure 8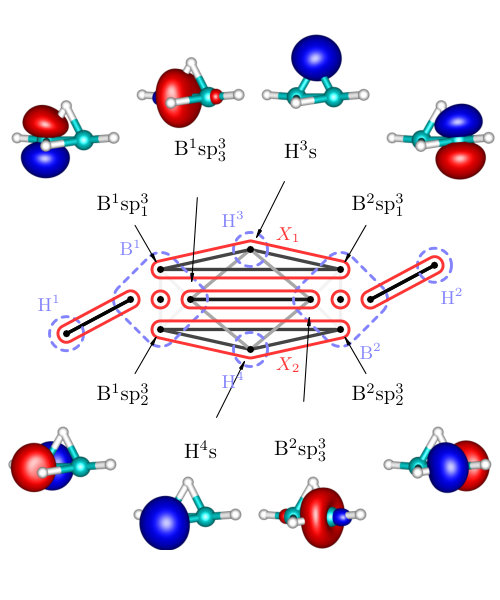 Figure 9
Figure 9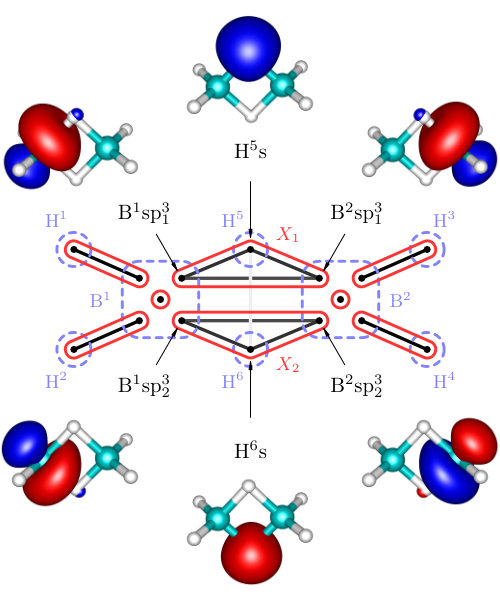 Figure 10
Figure 10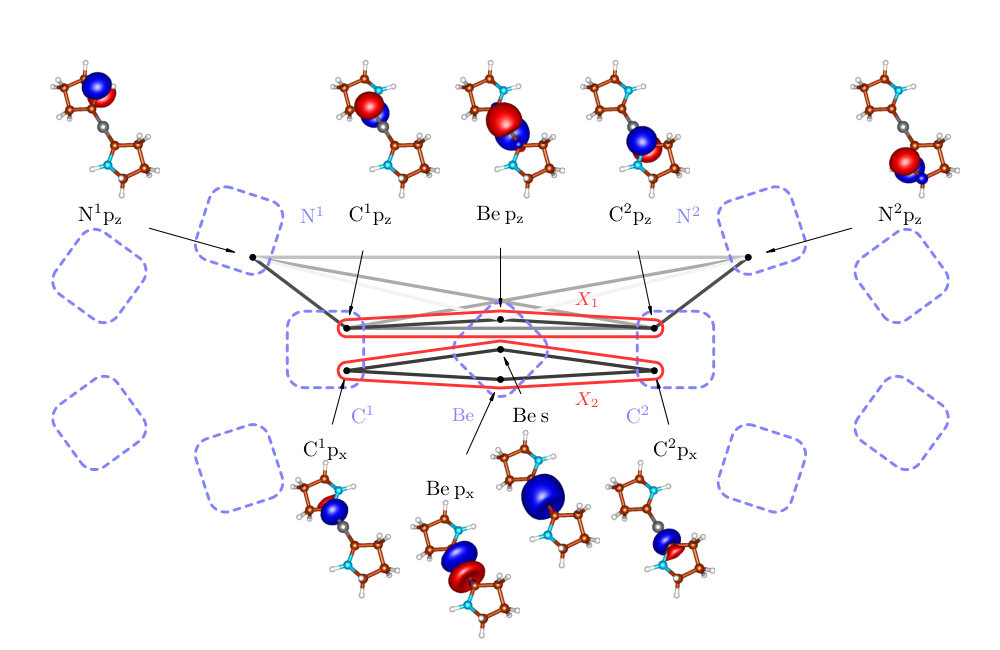 Figure 11
Figure 11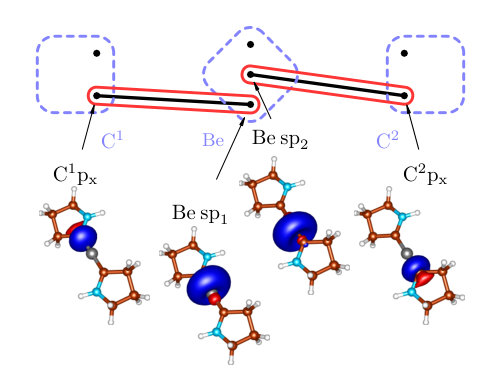 Figure 12
Figure 12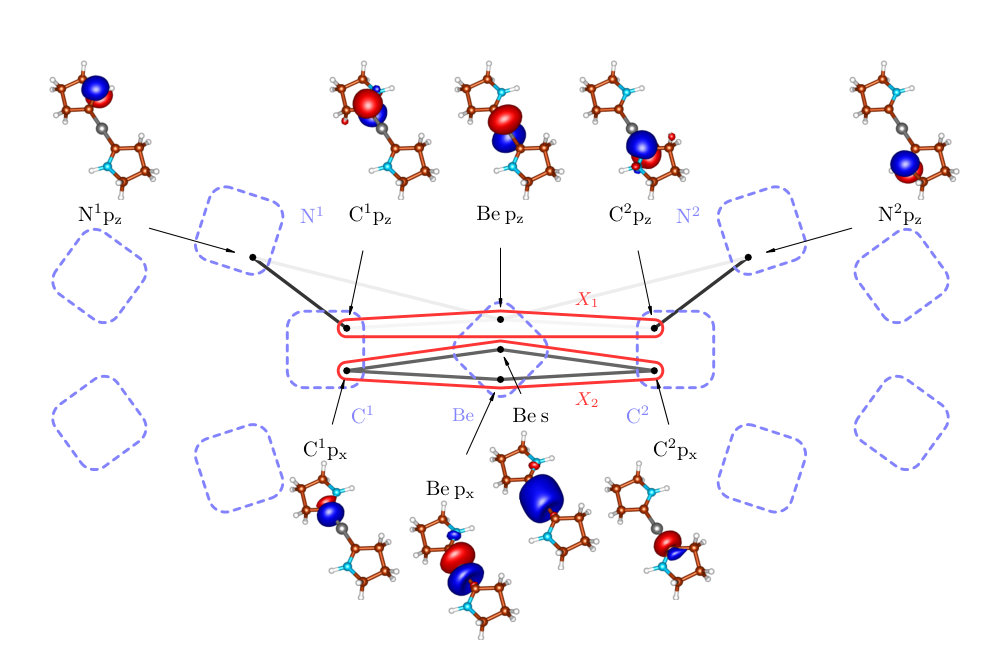 Figure 13
Figure 13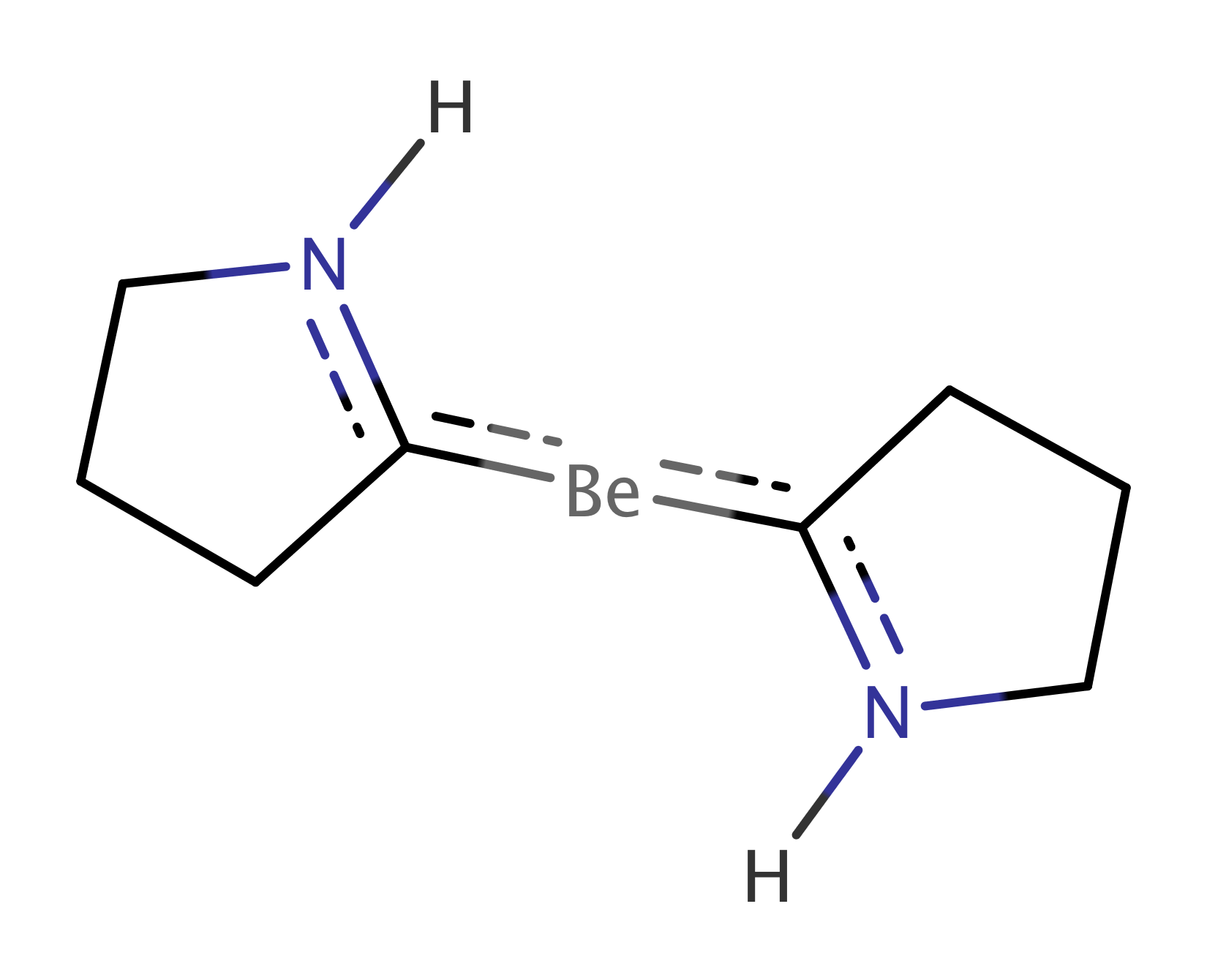 Figure 14
Figure 14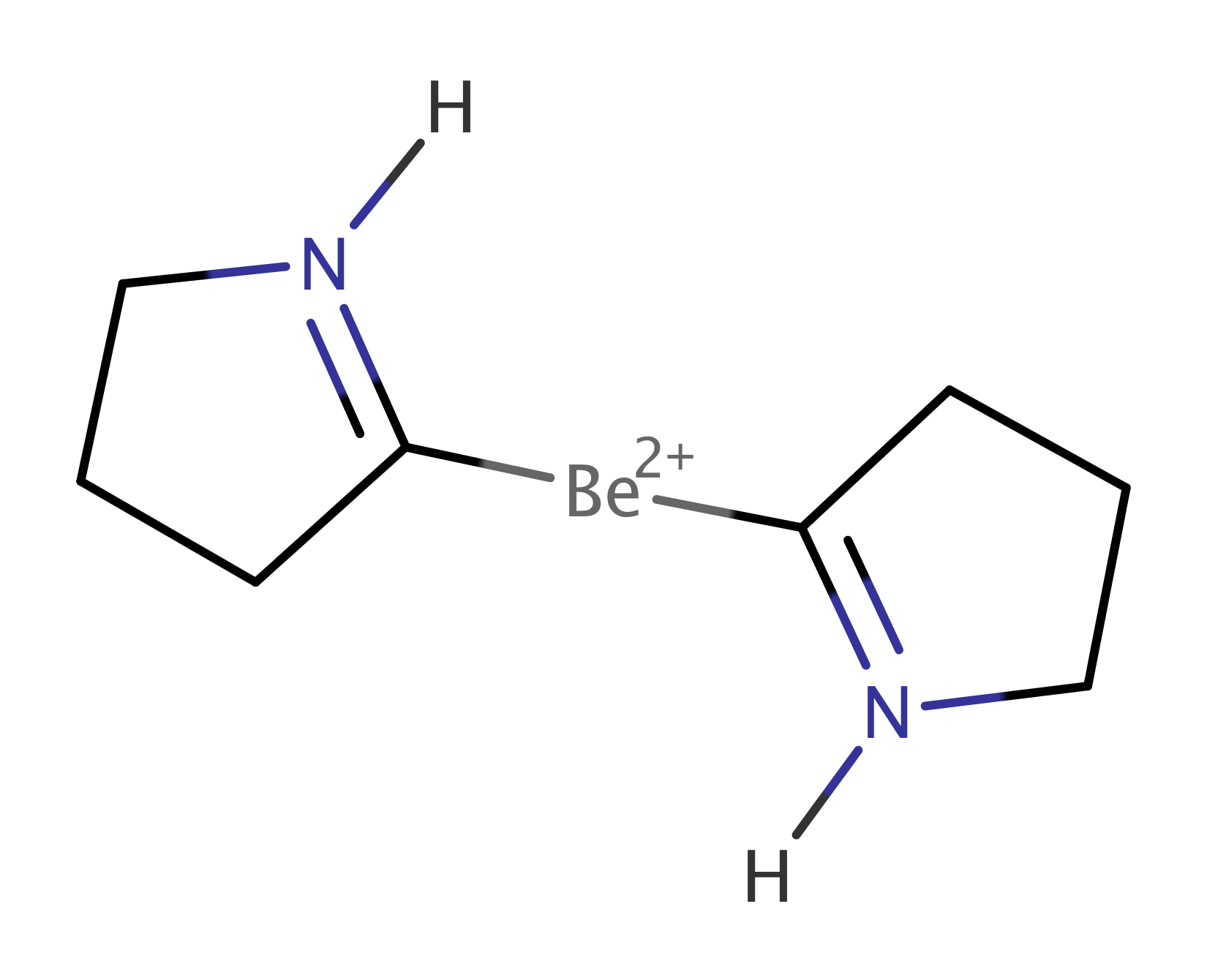 Figure 15
Figure 15| correlation | abs. value | rel. value |
|---|---|---|
| 0.515 | 8.6% | |
| 0.412 | 3.4% | |
| 1.852 | 46% | |
| 1.852 | 46% | |
| 2.114 | 53% | |
| 0.894 | 45% | |
| 0.894 | 45% | |
| 0.605 | 30% | |
| 1.500 | 75% | |
| 2.394 | 60% | |
| 0.309 | 5.2% | |
| 0.042 | 2.1% |
| correlation | abs. value | rel. value |
|---|---|---|
| 1.328 | 22% | |
| 0.647 | 32% | |
| 0.701 | 35% | |
| 1.388 | 69% | |
| 2.089 | 52% | |
| 1.438 | 36% | |
| 1.245 | 62% | |
| 0.130 | 6.5% | |
| 0.535 | 6.7% | |
| 0.066 | 1.1% | |
| 0.639 | 16% |
| correlation | abs. value | rel. value |
|---|---|---|
| 3.424 | 86% | |
| 2.432 | 61% | |
| 2.032 | 34% | |
| 0.194 | 9.7% | |
| 0.681 | 34% | |
| 1.153 | 58% | |
| 1.834 | 46% | |
| 0.915 | 46% | |
| 0.209 | 2.6% | |
| 1.737 | 43% | |
| 0.560 | 28% | |
| 0.209 | 2.6% | |
| 0.765 | 38% | |
| 0.771 | 39% | |
| 1.936 | 97% | |
| 1.912 | 96% |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Quantum information-based analysis of electron-deficient bonds
Jan Brandejs
J. Heyrovský Institute of Physical Chemistry, Academy of Sciences of the Czech Republic, v.v.i., Dolejškova 3, 18223 Prague 8, Czech Republic
Faculty of Mathematics and Physics, Charles University, Prague, Czech Republic
Libor Veis
J. Heyrovský Institute of Physical Chemistry, Academy of Sciences of the Czech Republic, v.v.i., Dolejškova 3, 18223 Prague 8, Czech Republic
Szilárd Szalay
Strongly Correlated Systems “Lendület” Research Group, Institute for Solid State Physics and Optics, MTA Wigner Research Centre for Physics, H-1121 Budapest, Konkoly-Thege Miklós út 29-33, Hungary
Gergely Barcza
J. Heyrovský Institute of Physical Chemistry, Academy of Sciences of the Czech Republic, v.v.i., Dolejškova 3, 18223 Prague 8, Czech Republic
Strongly Correlated Systems “Lendület” Research Group, Institute for Solid State Physics and Optics, MTA Wigner Research Centre for Physics, H-1121 Budapest, Konkoly-Thege Miklós út 29-33, Hungary
Jir̆í Pittner
J. Heyrovský Institute of Physical Chemistry, Academy of Sciences of the Czech Republic, v.v.i., Dolejškova 3, 18223 Prague 8, Czech Republic
Örs Legeza
Strongly Correlated Systems “Lendület” Research Group, Institute for Solid State Physics and Optics, MTA Wigner Research Centre for Physics, H-1121 Budapest, Konkoly-Thege Miklós út 29-33, Hungary
Abstract
Recently, the correlation theory of the chemical bond was developed, which applies concepts of quantum information theory for the characterization of chemical bonds, based on the multiorbital correlations within the molecule. Here for the first time, we extend the use of this mathematical toolbox for the description of electron-deficient bonds. We start by verifying the theory on the textbook example of a molecule with three-center two-electron bonds, namely the diborane(6). We then show that the correlation theory of the chemical bond is able to properly describe bonding situation in more exotic molecules which have been synthetized and characterized only recently, in particular the diborane molecule with four hydrogen atoms [diborane(4)] and neutral zerovalent s-block beryllium complex, whose surprising stability was attributed to a strong three-center two-electron bond stretching across the C-Be-C core. Our approach is of a high importance especially in the light of a constant chase after novel compounds with extraordinary properties where the bonding is expected to be unusual.
Introduction
Recent years have witnessed remarkable interest in application of tools of quantum information theory in chemistry Legeza and Sólyom (2003, 2004); Huang and Kais (2005); Rissler et al. (2006a); Pipek and Nagy (2009); Barcza et al. (2011); McKemmish et al. (2011); Boguslawski et al. (2012a, b, 2013); Kurashige et al. (2013); Fertitta et al. (2014); Duperrouzel et al. (2015); Murg et al. (2015a); Knecht et al. (2014); Boguslawski and Tecmer (2015); Szalay et al. (2015); Boguslawski and Tecmer (2015); Freitag et al. (2015); Zhao et al. (2015); Szilvási et al. (2015); Molina-Espíritu et al. (2015); Krumnow et al. (2016); Stein and Reiher (2016, 2017); Kovyrshin and Reiher (2017); Szalay et al. (2017); Stemmle et al. (2018). As a prominent example, the performance of state-of-the-art tensor product methods for electronic structure calculations Kurashige and Yanai (2009); Murg et al. (2010); Nakatani and Chan (2013); Szalay et al. (2015); Chan et al. (2016); Keller et al. (2015); Wouters and Van Neck (2014); Gunst et al. (2018) heavily relies on proper manipulation of entanglement Legeza and Sólyom (2003); Rissler et al. (2006a); Barcza et al. (2011); Fertitta et al. (2014); Murg et al. (2015a); Szalay et al. (2015); Krumnow et al. (2016). These include density matrix renormalization group (DMRG) method White (1992, 1993), which variationally optimizes wave functions in the form of matrix product states (MPS).Schollwöck (2011)
Other important examples represent characterization of electron correlation into its static (strong) and dynamic contributions Boguslawski et al. (2012b), automatic (black-box) selection of the active spaces Legeza and Sólyom (2003); Barcza et al. (2011); Szalay et al. (2015); Stein and Reiher (2016, 2017); Faulstich et al. (2018), or the self-adaptive tensor network states with multi-site correlators Kovyrshin and Reiher (2017), all of which harness single- and two-orbital entanglement entropies. Last but not least, correlation measures based on the single- and two-orbital entanglement entropies have also been employed for the purposes of bond analysis Boguslawski et al. (2013); Szilvási et al. (2015).
In the preceding work Szalay et al. (2017), we have presented the very general correlation theory of the chemical bond based on multiorbital correlation measures which goes beyond the scope of two-orbital picture. It is able to properly describe multiorbital bonds, and we have demonstrated its performance on a representative set of organic molecules (aliphatic as well as aromatic).
In the present article, we apply this theory to systems with electron-deficient bonds, i.e., to compounds which have too few valence electrons for the connections between atoms to be described as covalent bonds, and which have always fascinated chemists. First we apply the theory to the notoriously known textbook example of the diborane(6)111The number in parentheses denotes the number of hydrogen atoms. molecule (B2H6) with two-electron three-center bridge bonds and then also to recently characterized diborane(4) Chou et al. (2015) (B2H4) and zero-valent complexes of beryllium Arrowsmith et al. (2016); Brabec et al. (2018). The neutral form of the latter compound exhibits surprising stability, which was attributed to a strong three-center two-electron bond stretching across the C-Be-C core Arrowsmith et al. (2016). Unlike in the previous study Szalay et al. (2017), here we work in the bigger detail in a sense that we also employ eigenstates of multiorbital reduced density matrices, which give us additional insights into the character of bonding.
The article is organized as follows: in Sec. II, we briefly present the studied systems, in Sec. III we review the main concepts of the theory of multiorbital correlations, Sec. IV presents the computational details and Sec. V the results of our calculations which are followed by their discussion, the final Section closes with conclusions.
Studied systems
Diboranes
In its ground state, diborane(6) (Figure 1a) adopts its most stable conformation with two bridging B-H-B bonds, and four terminal B-H bonds. Its structure was first correctly measured in 1943 from infrared spectra of gaseous samples by an undergraduate student, Longuet-Higgins.Longuet-Higgins (1946); Longuet-Higgins and Bell (1943) Subsequent measurements with electron diffraction confirmed his conclusionsEberhardt et al. (1954), and X-ray diffraction detected further systems with bridging hydrogen bond.Lammertsma and Ohwada (1996) The B-H-B bridging was considered an atypical electron-deficient covalent chemical bond.Lipscomb (1973) Diborane(6) is a prominent example of a molecule with three-center two-electron bonds.Neeve et al. (2016) As it is a well studied system, we use its B-H-B linkage as a reference to compare with bond strengths and properties of more complex systems featuring three-center two-electron bonds.
According to quantum-chemical calculations Vincent and Schaefer (1981); Mohr and Lipscomb (1986); Curtiss and Pople (1989a, b); Demachy and Volatron (1994); Alkorta et al. (2011), different species of diborane with less than six hydrogens should exist, also featuring the bridging B-H-B bonds. However, all candidates are short-lived reaction intermediates, difficult to prepare and to identify. Hence, no neutral species has been identified experimentally until 2015, when Chou irradiated diborane(6) dispersed in neon at with far-ultraviolet light, detecting diborane(4), B2H4 (Figure 1b).Chou et al. (2015) This new species with two terminal hydrogen atoms possesses two bridging hydrogen atoms, and so it became the simplest neutral boron hydride identified with such a structural feature.
Beryllium complexes
Complexes of metal atoms of the s-block of the periodic table are often found in their zero oxidation state due to their exceptional electron donation. For their interesting reactivities, these became frequent synthetic targets, competing with traditional transition metal complexes.Power (2010, 2011); Giffin and Masuda (2011) We follow the recent experimental work of Arrowsmith, who isolated, for the first time, neutral compounds with zero-valent s-block metal, beryllium. Arrowsmith et al. (2016) These brightly coloured molecules have very short Be-C bonds and beryllium in linear coordination geometries.Niemeyer and Power (1997); Naglav et al. (2015); Arnold et al. (2015); Lerner et al. (2003) This indicates strong multiple Be–C bonding. According to the theoretical and spectroscopic results, the molecules adopt a closed-shell singlet configuration with a Be(0) metal centre. Arrowsmith et al. (2016) The complexes are surprisingly stable, and this was ascribed to an unusually strong three-center two-electron bond stretching across the C–Be–C unit. Two bonding mechanisms depicted in Figure 2 are taking place, namely donation from the carbon doubly occupied sp2 hybrid orbital to empty s and p orbitals on the central Be atom and back donation from the beryllium p orbital to p orbitals located on C atoms.
We studied two of the proposed systems. First the Be(CAC)2 complex (Figure 6a), where CAC corresponds to cyclic amino carbene donors, which stabilize the compound due to their -acidity. Mondal et al. (2013); Li et al. (2013) We performed a multireference calculation, in order to verify the proposed singlet configuration with a Be(0) metal centre and to provide a deeper insight into the bonding scheme. Next we studied dication [Be(CAC)2]2+ (Figure 6b), in which the removal of two electrons disrupts the bridging C–Be–C bond. This allowed us to compare with the former system and to determine the stabilization effect of the bridging bond.
Methodology
Recently, the correlation theory of the chemical bond Szalay et al. (2017) was developed, characterizing bonds based on the correlations among orbitals localized on individual atoms. Simply put, if we think of a simple covalent bond and localize the bonding and antibonding molecular orbitals into their atomic contributions, these localized orbitals will be highly correlated. Therefore, standard two-orbital bonds can be characterized by pairs of strongly correlated localized orbitals, and the strength of the correlation characterizes the strength of the bond from the quantum information theoretical point of view.
The correlation theory of the chemical bond can also be used for the characterization of bonds more involved than the covalent bonds. The concept in general is to find the finest possible correlation based clustering of the localized orbitals into clusters, so that the clusters are weakly correlated with each other, and the orbitals inside the clusters are strongly correlated.Szalay et al. (2017) These clusters then form independent bonds of a Lewis structure of a given molecule and the strength of the correlation with respect to this clustering refers to the validity of such a representation. The weaker the correlation is, the better the Lewis structure represents bonding.
In order to review the correlation measuresSzalay (2015), which will be used in our analysis, let us denote the set of (the labels of) localized orbitals with . We aim at investigating the correlations in an set of orbitals (cluster). The state of the full electronic system of the cluster is given by the density operator , while the reduced state of a (sub)cluster is given by the reduced density operator in general.Ohya and Petz (1993); Araki and Moriya (2003); Wilde (2013) If the cluster of orbitals can be given by a state vector (for example, when a given eigenstate of the whole molecule is considered), then its density operator is of rank one, , called a pure state. Its reduced density operator is usually mixed (not of rank one), which is the manifestation of entanglementHorodecki et al. (2009) between (sub)cluster and the rest of the cluster . In general, a density operator can be decomposed in infinitely many ways into state vectors with mixing weights as . The spectral decomposition (where the weights are the eigenvalues, and the -s are eigenvectors, being orthogonal) is a special one, in the sense that its weights are the least mixed.Schrödinger (1936); Hughston et al. (1993) Each eigenvector can be expanded in the occupation number basis, the square of the absolute value of the coefficients are the weights of the given occupations in that given eigenvector of weight .
On the first level, the correlation is defined with respect to a partitionDavey and Priestley (2002) of the set of the orbitals,Szalay and Kökényesi (2012); Szalay (2015); Szalay et al. (2017); Szalay (2018) denoted with , where the clusters , called parts, are disjoint subsets of the cluster , and . The measure of correlation among the parts is the -correlation,Szalay (2015); Szalay et al. (2017)
[TABLE]
Here is the von Neumann entropy.Ohya and Petz (1993); Wilde (2013) (Note that we use the logarithm to the base , which is the dimension of the Hilbert space of an orbital. The resulting numerical values are then the same as of the original measures with natural logarithmSzalay et al. (2017) given in the units of . Note that , where is the number of orbitals in cluster .) As a special case, the correlation of two single orbitals,
[TABLE]
is the well-known (two-orbital) mutual information,Ohya and Petz (1993); Wilde (2013); Adesso et al. (2016) which has already been considered in chemistry.Legeza and Sólyom (2006); Rissler et al. (2006b); Barcza et al. (2011); Boguslawski et al. (2013); Kurashige et al. (2013); Mottet et al. (2014); Fertitta et al. (2014); Duperrouzel et al. (2015); Boguslawski and Tecmer (2015); Freitag et al. (2015); Szilvási et al. (2015); Murg et al. (2015b); Barcza et al. (2015); Zhao et al. (2015); Szalay et al. (2017) (For convenience, we omit the curly brackets in the cases when this does not cause confusion.) For a general partition , we have the boundSzalay et al. (2017)
[TABLE]
Note that is zero for the trivial split , and it takes its maximum, , for the finest split . The latter quantity is also called total correlation,Lindblad (1973); Horodecki (1994); Legeza and Sólyom (2004); Legeza et al. (2006); Herbut (2004)
[TABLE]
(Note that if cluster is described by a pure state then , and the correlation is entirely quantum entanglement.Horodecki et al. (2009); Modi et al. (2010); Szalay (2015) Moreover, the correlation in a pure state with respect to a bipartition is just two times the usual entanglement entropyBennett et al. (1996); Nielsen and Chuang (2000); Wilde (2013)
[TABLE]
because of the Schmidt decomposition of pure states.Schmidt (1907); Nielsen and Chuang (2000); Wilde (2013))
On the second level, the correlations can be defined in an overall sense, that is, without respect to a given partition.Szalay et al. (2017); Szalay (2015, 2018) The -partitionability correlation and the -producibility correlation, areSzalay et al. (2017)
[TABLE]
for . These characterise the strength of two different (one-parameter-) notions of multiorbital correlations; those which cannot be restricted inside at least parts, and those which cannot be restricted inside parts of size at most , respectively.Szalay et al. (2017)
For the cluster , as special cases, C_{\text{\absolutevalue{L}-part}}=C_{\text{1-prod}}=C(\bot) grabs all the correlations, it is zero if and only if there is no correlation at all in the cluster . On the other hand, C_{\text{2-part}}=C_{\text{(\absolutevalue{L}-1)-prod}} is sensitive only for the strongest correlations, it is nonzero if and only if the cluster is globally correlated. Note also that C_{\text{1-part}}=C_{\text{\absolutevalue{L}-prod}}=C(\top)=0, by definition. Beyond these, there are no such coincidences among the partitionability and producibility correlations for other values of , however, the relation C_{\text{k-part}}\geq C_{\text{(\absolutevalue{L}-k+1)-prod}} holds.Szalay et al. (2017) Also, the following (non stricht) bounds hold Szalay et al. (2017)
[TABLE]
Computational details
In case of diborane molecules, the ground state geometries were optimized with the B3LYP/cc-pVDZ method. For the multiorbital correlation studies, the Pipek-Mezey Pipek and Mezey (1989) localized HF/STO-3G molecular orbitals (MOs) were employed Szilvási et al. (2015); Szalay et al. (2017) and they were manually hybridized (rotated) to better reflect the chemical environment. The quantum chemical (QC-) DMRG method was applied to study the multiorbital correlations in the full orbital following the procedure outlined in Ref. (26).
The ground state geometries of both forms of the beryllium complex, namely Be(CAC)2 and [Be(CAC)2]2+ were taken from Ref. (41) and they correspond to the BP86/def2-TZVPP level of theory. Due to the size of the problem, multiorbital correlation studies by means of QC-DMRG are not feasible in the full orbital space. We have rather chosen a different strategy. Since we were interested only in the bonding of the C-Be-C atomic core, we have selected the complete active space (CAS) of relevant orbitals participating or influencing these bonds. In particular, 2p orbitals on both C and Be atoms and 2s orbital on Be, all of them contributing to the bonds and 2p orbitals on both C and neighbouring N atoms and Be, forming or directly influencing the bonds. The CAS orbitals were optimized by means of the CASSCF(10,9)/cc-pVDZ method in case of the neutral complex and CASSCF(8,9)/cc-pVDZ method in case of the dication and again localized using the Pipek-Mezey Pipek and Mezey (1989) procedure. They were not hybridized in order to directly compare with the previous work Arrowsmith et al. (2016) making conclusions about atomic-like orbitals.
All the quantum chemistry calculations except the QC-DMRG ones were performed with the MOLPRO package Werner et al. (2010). The QC-DMRG calculations were carried out using the Budapest QC-DMRG code Legeza et al. . Molecular orbitals were visualized with Charmol Chalupsky .
Results
The results on diborane(6) are summarized in Figure 3 and Table 1, whereas results on diborane(4) are presented in Figure 4 and Table 2. All the Figures depict mutual information of pairs of localized orbitals, defined in 2, while the Tables contain numeric values of measures of the relevant kinds of multiorbital correlations, which are discussed below.
In a similar fashion, the results on beryllium complexes are presented in Figures 5, 7, and 8 and Table 3.
In the Figures, individual localized orbitals are represented as black dots and dashed blue lines encircles orbitals belonging to one atom. The mutual information is plotted as grayscaled edges between the orbitals. Black lines correspond to the strongest correlations, while light gray lines connect the weakly correlated orbitals. Based on the mutual information structure, the orbitals are grouped into strongly correlated clusters, which in our examples correspond to either core orbitals or chemical bonds. The clusters are encircled by red borders.
Discussion
Diborane(6)
Figure 3 shows that our results fit well the established bonding picture of diborane(6) with two bridging B-H-B bonds. Let us now discuss in detail how the analysis of correlations leads to the clustering and to the bonding picture presented in Figure 3. We will discuss only the bridging bonds, the core orbitals as well as terminal B-H bonds are well separated, i.e. not correlated with the rest. (This is confirmed by the weak correlation in Table 1.)
First we consider the cluster containing sp3 hybrid orbitals on B atoms and 1s orbital on the bridging H atom. Because of the point group symmetry, the same results hold for cluster . As can be seen in Table 1, the correlation (entanglement) of with the remaining orbitals is very weak, only 8.6% of the maximum value, which indicates that forms an independent three-center bond.
However, to confirm this conclusion, we have to show that cannot be split further. If we take separate pairs of orbitals from and measure the correlation with the remaining orbitals, we obtain significantly higher values, in particular 46% and 53% of the maximum values (see Table 1), which justifies existence of the three-center bond.
The mutual information of pairs of orbitals within reach rather small relative values (45%, 30%, see Table 1), however according to our numerical experience, even strong multicenter bonds typically yield low percentage, never approaching near the theoretical maxima.Szalay et al. (2017) Intuitive perspective suggests that the correlation of one orbital with the others can be thought of as a resource shared among the orbitals. In other words, all the pairs inside cannot reach the maximum simultaneously, bounded by entanglement monogamy.Osborne and Verstraete (2006); Coffman et al. (2000) The formulation of an inequality bounding the mutual correlations inside orbital clusters still remains an open problem, to our best knowledge. The smaller value of the mutual information between sp3 hybrid orbitals on B atoms reflects their larger internuclear distance.
We employ -partitionability in order to quantify and benchmark the strength of the diborane(6) three-center bonds (in terms of correlation). As can be seen in Table 1, C_{\text{2-part}}(X_{1}) reaches 75% of the upper bound and C_{\text{3-part}}(X_{1}) 60%, which points at a strong bond in .
The very weak correlation (entanglement) of with the remaining orbitals also indicates that the state of the cluster is close to a pure state. Indeed, the eigenstate analysis of the reduced density operator shows that there is the following two-electron eigenstate with a corresponding eigenvalue (probability) of
[TABLE]
where the ordering of orbitals in a ket corresponds to B1sp3, H51s, and B2sp3. Other eigenstates have probabilities below . The principal two-electron eigenstate together with the above discussion on correlations imply that the three orbitals of form a three-center two-electron bond. Note that the electron pair exhibits a preferred occupation on H atom, which is due to its higher electronegativity when compared to B, as we can see from the principal eigenstate. It is in agreement with the expectation values of particle-number-operators () which for B1sp3, H51s and B2sp3 equal 0.53, 0,95 and 0.53, respectively.
As one can observe in Table 1, the main source of correlation between and the remaining orbitals is the correlation with the other three-center bond, . Specifically, the correlation between two bridging H atoms is the strongest, which is caused by higher electron density on these atoms.
Diborane(4)
In case of diborane(4), the two terminal H atoms are missing and instead a direct covalent bond connecting both B atoms is present Chou et al. (2015). This is also the picture resulting from our analysis and depicted in Figure 4. In comparison to diborane(6), we have the similar three-orbital clusters and , but also the two-orbital cluster containing sp3 hybrid orbitals on B atoms and corresponding to the aforementioned B-B bond.
Considering the cluster , one can observe in Table 2 that it is more correlated with the remaining orbitals than in case of diborane(6). The value of is more than two times larger, but the picture of as a standalone chemical bond is still justifiable. Consequently, the reduced density operator is more mixed, with the principal eigenvalue . The remaining eigenstates share low probabilities (below ), and therefore, the picture of as a standalone chemical bond is still a reasonable qualitative description. The principal eigenstate is again two-electron, i.e. electron-deficient, and it has the following form
[TABLE]
Similarly to diborane(6), higher electron density is on the bridging H atom, which is due to its higher electronegativity.
Comparing the two-orbital correlations inside with diborane(6) (Tables 1 and 2), one can see a weaker correlation between sp3 hybrid orbitals on B atoms and H 1s orbital, but a slightly stronger correlation between both B-atom-orbitals. This stronger correlation can be certainly assigned to a shorter distance of B atoms (1.477Å vs. 1.784Å). Based on the values of C_{\text{2-part}}(X_{1}) and C_{\text{3-part}}(X_{1}), the covalent bond corresponding to the cluster is slightly weaker (in terms of correlation) than the same bond in diborane(6).
For the cluster , correlation with the remaining orbitals is stronger than for and , which in turn weakens the internal two-orbital correlation. The major contribution to the correlation of with the remaining orbitals originates from , which is still very weak compared to other correlations in the molecule (see Table 2).
The overall correlation of the three bonding clusters , , and with the rest of the system is similarly weak as in diborane(6) so the considered bonding can be described independently of the rest of the molecule.
Beryllium complexes
In order to check how well our bonding picture of Be(CAC)2 fits the one proposed by Arrowsmith et al. Arrowsmith et al. (2016), we consider the clusters and from Figure 5. The cluster contains p orbitals on C and Be atoms and corresponds to the suggested three-center two-electron bond, whereas the cluster contains C p orbitals and Be s and p orbitals and corresponds to the bonds.
Let us start with . In Table 3, one can see that the correlation of with the remaining orbitals is higher than 30% of the maximum value, which means that the picture of the three-orbital C-Be-C bond might be good as a qualitative description, but for a quantitatively adequate description, we might seek to include further orbitals into X1, as shown below. This is also demonstrated by weaker pairwise correlations within , especially , than in the three-orbital bonds discussed above. The correlations of the internal pairs in with the remaining orbitals are large (61% and 86%) and clearly cannot be considered as standalone bonds.
The inaccuracy of the picture of a standalone three-orbital bond is also demonstrated by the reduced density operator (see in Appendix A), which is much more mixed than in previous cases. It has three dominant eigenvalues, instead of just one. The most significant, nevertheless, corresponds to the two-electron state, which is in agreement with the overall picture of the three-orbital two-electron bond.
As can be seen in Figure 5, the strongest external correlation of is . It results from a conjugation of p orbitals and has a stabilization effect. Notice that the N1-C1-Be-C2-N2 group of atoms form perfectly planar structure (the dihedral angle ) enabling an efficient overlap of all p orbitals, which is necessary for the aforementioned conjugation.
The more accurate bonding picture can thus be obtained by considering the enlarged cluster
[TABLE]
The corresponding structure of Be(CAC)2 is depicted in Figure 6a. The cluster is independent of the rest of the molecule. This follows from the negligible correlation of with the remaining orbitals (see Table 3). Employing the standard notation, the electron bond can be denoted as , i.e. six-electron (two electrons from the Be atom and two from each N atom lone pair) five-center bond, which is confirmed by the particle number expectation value of .
In order to verify the suggested back-donation mechanism Arrowsmith et al. (2016), or in other words probe the local electronic configuration of the Be atom, we have also performed the correlation analysis for the dication [Be(CAC)2]2+. As can be seen in Figure 7, the difference between the correlation picture of Be(CAC)2 and [Be(CAC)2]2+ are almost missing correlations inside the cluster , which is for example demonstrated by the negligible value of . Also the reduced density operator is highly mixed and without the dominating two-electron eigenstate (see in Appendix A). On the other hand, the correlations in the cluster remain practically unchanged.
This is in agreement with the picture of Be atom having originally two electrons in the pz orbital. When the C-Be-C bond is formed, they are shared with C-atom pz orbitals through the back donation mechanism, as is depicted in Figure 2. These two electrons are missing in case of the dication and the aforementioned bond is clearly not formed.
Another feature of the correlation picture from Figure 7 is that there are considerably stronger pairwise correlations between C and N-atom pz orbitals []. They are indeed of the strength of donor-acceptor bonds Szilvasi et al. (2015). We thus assign double bonds between N and C atoms to the dication, as is depicted in Figure 6b. The bonds are formed from the originally doubly filled N p orbitals and empty C p orbitals. The existence of these bonds is also confirmed by almost perfectly planar environment with the dihedral angle . Note that in Figure 7, we can see only the part of the double bond corresponding to the bond - the bonds along the rings are excluded from the active space.
Let us now turn to the cluster of Be(CAC)2, i.e. to the bonding. The correlation of with the remaining orbitals is insignificant, however splitting of the four-orbital cluster into two bonds is not possible in this basis. It may therefore seem that the correct bond is also multiorbital. It is, however, only the artefact of the atomic-like basis, which was used in order to directly compare with Ref. (41). By simple rotation of Be s and px orbitals (forming the sp hybrids as in case of diboranes), one can form two independent bonds, essentially without influencing the rest of the system, which can be seen in Figure 8. Note that in the correlation theory of the chemical bond, superposing orbitals is allowed if this does not affect their locality too much. So superposing orbitals on different atoms is usually forbidden, while doing the same on a given atom is allowed.
Last but not least, we would like to compare the strength of both contributions to the bonding of the C-Be-C core, namely the bond (clusters and ) and the bond (cluster ). In the previous study Arrowsmith et al. (2016), it was shown by means of the energy decomposition analysis combined with natural orbitals for chemical valence (EDA-NOCV) Krapp et al. (2007) that the bonding channel is considerably more energy stabilizing than the channel. Let us now check the strength of both contributions by means of the correlations.
When using the more rough (three-orbital two-electron) description of the bond (cluster ) and by looking at Table 3, one can see on the values of that bonds are considerably stronger than the bond. The situation, however, dramatically changes when we use more accurate conjugated description (cluster ). Note that since we are not interested in a split dissecting N and C, (because the N-C bond on the ring is stabilized by another bond, not visible on the plots), we have to consider the pz orbitals together on N and C atoms of the same ring, and calculate , which turns out to be . Also note that in this paragraph we compared the absolute values of the correlation measures, because the clusters are of different sizes.
In Table 3, one can see that the more accurate description of the bond () makes it of a similar strength as the bonds (1.737 vs 1.936). We believe that our results describe the nature of a Be(CAC)2 bonding reliably, especially because we have used the genuine multireference description unlike in Ref. (41), where the analysis was based on the density functional theory (BP86 functional). We would also like to note that we have studied slightly different system than in Ref. (41). In our case all substituents were replaced by hydrogen atoms. This, however, should not influence the electronic structure of the C-Be-C core. Also note that using the s and p orbitals on beryllium in was only for the purpose of comparison with the previous studyArrowsmith et al. (2016). For having a more physical picture, we should use the hybridized orbitals (Figure 8), by which consists of two simple covalent bonds. Then, in order to characterize the strength of the bond, we would wave to consider the Be sp1 and sp2 orbitals together and calculate . Nevertheless, since this value is nearly identical to , the conclusion is the same.
Conclusions
In this article, we have reviewed the recently developed correlation theory of the chemical bond Szalay et al. (2017) and applied it on molecules with multicenter electron-deficient bonds. We have demonstrated the usefulness of our methodology in characterizing molecular bonding properties by fingerprints of correlations among individual orbitals which form these types of bonds.
We have verified the computational procedure on a textbook molecule with electron-deficient bonds, namely diborane(6), and further characterized bonding in diborane(4) and zero-valent complexes of beryllium with intricate bonding patterns. In all the cases, our results fit well with known bonding pictures or previous theoretical predictions. We have therefore proved capabilities of our new method to reliably describe bonding in complex molecular systems.
In case of the Be(CAC)2 molecule, we have also compared both contributions to the C-Be-C bonding ( and ), finding, in contrast to the previous study Arrowsmith et al. (2016), the and contributions of a similar strength, in the sense of correlational quantities. We believe that our result is reliable and attribute the discrepancy with the previous study to the single reference description employed in Ref. (41), which may not be accurate enough in this multireference case.
Finally, we would like to note that, despite employing the DMRG method White (1992, 1993) for calculations of subsystem reduced density matrices, the theory presented in this article is general and other correlated methods can in principle be employed as well Boguslawski and Tecmer (2015). Especially in cases of large molecules with the electronic structure dominated by the dynamical correlation for which the DMRG description may be unnecessary and computationally prohibitive.
Acknowledgements
This work has been supported by the Czech Science Foundation (grants no. 16-12052S, 18-24563S, and 18-18940Y), Czech Ministry of Education, Youth and Sports (project no. LTAUSA17033), and the Hungarian-Czech Joint Research Project MTA/16/05. G.B., Sz.Sz. and Ö.L. are supported by the National Research, Development and Innovation Fund of Hungary (NRDIFH) within the Researcher-initiated Research Program (project Nr: NKFIH-K120569) and the “Lendület” Program of the Hungarian Academy of Sciences (HAS). Sz.Sz. and Ö.L. are supported by the Quantum Technology National Excellence Program (project Nr: 2017-1.2.1-NKP-2017-00001) of NRDIFH. G.B. and Sz.Sz. are also supported by the “Bolyai” Research Scholarship of HAS. Ö.L. also acknowledges financial support from the Alexander von Humboldt foundation.
Appendix A Eigenvectors of the reduced density operators
A.1 Be(CAC)2
The (reduced) density operator of the orbitals consists of the following eigenstates of the three highest eigenvalues (probabilities).
Probability :
[TABLE]
Probability :
[TABLE]
Probability :
[TABLE]
all the other eigenvalues are less than .
A.2 [Be(CAC)2]2+
As mentioned earlier in the text, the reduced density operator for is highly mixed in this this case, with no dominant state. Therefore we only list the highest eigenvalues to show this:
[TABLE]
A.3 Distribution of the two-orbital correlations
Figure 9 shows the distribution of the two-orbital correlations for diborane and beryllium complexes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Legeza and Sólyom (2003) Ö. Legeza and J. Sólyom, Phys. Rev. B 68 , 195116 (2003) . · doi ↗
- 2Legeza and Sólyom (2004) Ö. Legeza and J. Sólyom, Phys. Rev. B 70 , 205118 (2004) . · doi ↗
- 3Huang and Kais (2005) Z. Huang and S. Kais, Chemical Physics Letters 413 , 1 (2005) . · doi ↗
- 4Rissler et al. (2006 a) J. Rissler, R. M. Noack, and S. R. White, Chem. Phys. 323 , 519 (2006 a) . · doi ↗
- 5Pipek and Nagy (2009) J. Pipek and I. Nagy, Phys. Rev. A 79 , 052501 (2009) . · doi ↗
- 6Barcza et al. (2011) G. Barcza, Ö. Legeza, K. H. Marti, and M. Reiher, Phys. Rev. A 83 , 012508 (2011) . · doi ↗
- 7Mc Kemmish et al. (2011) L. K. Mc Kemmish, R. H. Mc Kenzie, N. S. Hush, and J. R. Reimers, The Journal of Chemical Physics 135 , 244110 (2011), 10.1063/1.3671386 . · doi ↗
- 8Boguslawski et al. (2012 a) K. Boguslawski, K. H. Marti, O. Legeza, and M. Reiher, J. Chem. Theory Comput. 8 , 1970 (2012 a).